
Abstract
Heavily urbanized areas can hinder dispersal and gene flow between amphibian populations. Given the growth potential of urbanization, it is important to examine how this specific environment shapes their genetic patterns at the local scale. The ability of the European green toad to successfully colonize large human settlements has been convincingly confirmed in the recent past, but little is known about its population genetics under these new conditions. In this study, we examined the effects of the environment on genetic variation, population structure, and the level of gene flow in populations of this amphibian in the city of Košice and the adjacent rural area (eastern Slovakia) using a set of neutral genetic markers. We found that urban populations had lower genetic variability than populations in adjacent rural areas; however, the degree of inbreeding was relatively low in all samples. Genetic differentiation was higher, and gene flow was more restricted in urban area, although geographic distances between sites were significantly less than in rural area (2–4 km versus 6–13 km). Our analyses suggested genetic isolation of urban populations at sites with less suitable habitat for green toads. In contrast, admixture of the population inhabiting the large city park, established on a former floodplain, with all rural populations was likely the result of an intense historical gene flow. The densely developed environment of the other urban sites likely presents a strong barrier to gene flow. The lack of suitable wetland habitat prior to development suggests that these sites were only recently colonized by a limited number of founders. Thus, we found differential effects of the city on the population structure of the green toad. Understanding current local genetic variation and structure is important for future conservation plans in urban environments.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Since the 1950s, urbanization has become one of the most important processes altering natural landscapes worldwide, associated with degradation, fragmentation, and loss of habitats, as well as declines in many wildlife populations (Hamer and McDonnell 2008; Faeth et al. 2011; Elmqvist et al. 2013). At the global scale, an apparent decline in amphibians with aquatic larval development has been documented, significantly impacted by human-induced physical barriers between breeding pools and terrestrial habitats of adults (Stuart et al. 2004; Becker et al. 2007). Unidirectional movement from the natal pond to new breeding sites is important for recolonization or persistence of metapopulations, especially in fragmented urban landscapes (Semlitsch 2008). Therefore, maintaining viable populations requires sufficient quality of both habitat types and connectivity between local populations (Trenham and Shaffer 2005; Harper et al. 2008).
Depending on the life history traits, mobility, and habitat preferences of amphibians, the landscape surface may provide resistance to dispersal and thus gene flow (Garcia et al. 2017; Cayuela et al. 2020). For example, forested areas resulted in the lowest resistance surface to gene flow for populations of long-toed salamander (Ambystoma macrodactylum), whereas it had the opposite effect for Columbia spotted frogs (Rana luteiventris). However, for both species, the highest landscape resistance resulted from urban and highly modified rural land areas (Goldberg and Waits 2010). Several studies have identified anthropogenic landscape features as barriers that reduce gene flow among amphibian populations and cause their isolation, which can lead to inbreeding, higher genetic differentiation, and increased genetic drift (e.g., Hitchings and Beebee 1998; Safner et al. 2011; Lourenço et al. 2017). Roads, fences, and buildings have been considered the most typical dispersal barriers in the urban matrix (Hamer and McDonnell 2008). In addition to the hindering effect of roads as impermeable surfaces which impact population connectivity, they can also reduce population sizes and local densities of anurans due to higher mortality following collisions with cars (Fahrig et al. 1995; Holderegger and Di Gulio 2010).
In highly urbanized areas, the viability of fragmented populations may benefit from habitat corridors that promote gene flow. For example, for populations of the natterjack toads (Epidalea calamita) living in urbanized coastal areas, dunes and beaches serve such a function (Cox et al. 2017). On the other hand, some pond-breeding species with higher dispersal abilities or larger home-ranges can maintain viable populations in urban areas (Richardson 2012; Furman et al. 2016). Thus, the same landscape features can have different effects on the population genetics of even co-occurring species (Brede and Beebee 2004; Homola et al. 2019). In addition to the average ability of species to move, occasional vagrants in particular, which can travel further distances of several kilometres, can shape patterns of gene flow between amphibian populations and thus play an important role in overall population survival (Smith and Green 2005; Ronce 2007; Sinsch et al. 2012). Therefore, anthropogenic landscape features may determine genetic structure in different ways, depending on the urbanization potential of the species and the specific circumstances of the urban area (Miles et al. 2019; Schmidt and Garroway 2021).
The European green toad (Bufotes viridis) is an amphibian species that has successfully colonized human settlements in the recent past. Despite its limited movement ability (~ 800 m, Sinsch and Leskovar 2011) and small home range size (~ 0.25 ha, Indermaur et al. 2009), it is capable of colonizing diverse habitats (Degani et al. 2013) and it is a persistent urban dweller even in large European cities, for example, Rome (Ensabella et al. 2003), Kraków (Budzik et al. 2013), Cologne (Vences et al. 2019), or Warsaw (Mazgajska and Mazgajski 2020). However, there is limited information on how the urban environment shapes the genetic patterns of local populations (Vences et al. 2019). In the present study, we aimed to investigate the effects of different landscapes on the genetic patterns of populations of this species using a set of neutral genetic markers. We selected two contrasting landscapes, characterised either by native habitats or by urban structures to better understand population-genetics limits of this species to colonize novel environment. We examined genetic variation, population genetic structure, the level of gene flow, and signatures of demographic bottleneck in two groups of neighbouring populations living in the city of Košice and in rural areas of eastern Slovakia, respectively. Since the species is generally considered to be a successful urban colonizer, we hypothesized that genetic variation and the level of gene flow between nearby populations would be similar regardless of the environment. However, an urban environment is highly site-specific in terms of habitat proportions and spatial structure, so we alternatively hypothesized that isolated urban populations will demonstrate signatures of demographic bottleneck and will have reduced variation and stronger genetic structuring due to restricted gene flow.
Material and methods
Study populations
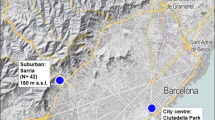
The green toad is widespread in the lowlands and submontane regions of eastern Slovakia. There, six local populations were selected from two contrasting landscapes (Fig. 1). Three urban sites (Mestský park, ‘city park’—uMSP, 48.724° N, 21.264° E, 205 m a.s.l.; Sídlisko KVP—uKVP, 48.715° N, 21.212° E, 298 m a.s.l.; Zuzkin park—uZUZ, 48.719° N, 21.238° E, 243 m a.s.l.) were located in the agglomeration of Košice, the second largest city in Slovakia (ca. 240 km2; ca. 239,000 inhabitants). These sites were parks or recreational areas with varying amounts of tree vegetation (approximate size of this habitat: uMSP—13 ha, uKVP—0.8 ha, uZUZ—2.2 ha) separated by settlements of buildings, roads, and other human constructions. Artificial ponds—fountains with periodic or permanent water serving as breeding pools—occurred at each urban site. Geographic distance between urban populations ranged from 2 to 4 km. Three rural sites were located 15–23 km SW from the periphery of the city. Two of them (Cestice—rCES, 48.589° N, 21.100° E, 203 m a.s.l.; Mokrance—rMOK, 48.592° N, 21.022° E, 194 m a.s.l.) were located in farmland with various shallow periodic pools, while one rural site (Drienovec—rDRI, 48.620° N, 20.932° E, 192 m a.s.l.) represented a limestone quarry with an artificial lake in the middle of cultivated fields. In contrast to the urban area, the average geographic distance between rural populations was much greater, ranging from 6 to 13 km. Twenty adults of B. viridis were sampled at each site during 2018–2022.

Location of six study populations of Bufotes viridis in eastern Slovakia (a, populations in the city of Košice; b, populations in the adjacent rural area) with landscape structures representing potential barriers to gene flow
Genotyping
In total, we successfully genotyped 120 individuals using 11 previously described microsatellite loci (Vences et al. 2019). A tissue sample was obtained by toe clipping (the last phalange of the third digit of the right forelimb) and stored in 96% ethanol until analysis. Total genomic DNA was extracted with GeneJET Genomic DNA Purification Kit (Thermo Fisher Scientific). Microsatellite markers were amplified by three multiplex PCR protocols (Table 1) using Qiagen Multiplex PCR kit (Qiagen). Each PCR protocol required 1 µl of genomic DNA (~ 100 ng/µl), forward and reverse primers of each locus (molar concentrations in Table 1), a 1 × Qiagen Master Mix and a 0.5 × Q-solution in a 12 µl reaction volume. A thermocycler program for the PCR amplification involved an initial cycle of denaturation at 95 °C for 5 min, 35 cycles of denaturation at 95 °C for 30 s, annealing at 60 °C for 45 s, extension at 72 °C for 90 s, and a final elongation step at 72 °C for 5 min. The fluorescent-labelled PCR products (Table 1) were separated by capillary electrophoresis in an ABI 3730XL genetic analyser, and fragment lengths estimated relative to the size standard LIZ600 were determined using Geneious Prime 2019 software (Biomatters). Scoring of peaks was performed by a single observer.
Microsatellite loci were checked for the Hardy–Weinberg equilibrium and frequency of null alleles using the package ‘PopGenReport’ 3.0.4 (Adamack and Gruber 2014) of the R 4.1.2 software (R Core Team 2021). In all populations but uMSP, 1–3 different loci deviated from the Hardy–Weinberg equilibrium (p < 0.05). Similarly, some linkage disequilibrium occurred in all populations. Because the linkage values differed from each other and no consistent pattern between any two loci was observed, loci were thus considered to be unlinked in the genome (see Vences et al. 2019). Loci exhibited in general low frequency of null alleles (up to 8% in Bvir3022) across all locations when tested using the Chakraborty et al.’s (1994) and Brookfield’s (1996) methods.
Genetic variation and differentiation
Basic characteristics of genetic variation in sampled populations i.e., the number of alleles (NA), observed (HO) and expected (HE) heterozygosity, and inbreeding coefficient (FIS) were calculated using the R package ‘adegenet’ 2.1.3 (Jombart 2008). Differences among groups were determined by the non-parametric Kruskal–Wallis ANOVA with post-hoc Dunn test for multiple comparisons in the R package ‘FSA’ 0.8.30 (Ogle et al. 2020). An individual- and a population-based pairwise estimates of genetic distances among sampled populations were calculated using DPS (Bowcock et al. 1994) and G′ST (Hedrick 2005), respectively, in the R package ‘graph4lg’ 1.2.0 (Savary et al. 2020).
Bottleneck analysis
For testing demographic bottleneck, we defined the null hypothesis as being presence of mutation-drift equilibrium under a two-phased mutation model which combines the stepwise mutation model and the infinite allele model. First, we tested for a recent reduction in effective size, thus an excess in heterozygosity. In addition, we controlled for signatures of a bottleneck by the deficiency in M-ratio as the total number of alleles (k) divided by overall range in allele size (R) (Garza and Williamson 2001). Supposing that k decreases faster than R when the population size is reduced, M-ratio is expected to be smaller in recently reduced populations compared to populations in mutation-drift equilibrium. Thus, the heterozygosity test assumes that a bottleneck eliminates rare alleles, whereas the M-ratio test is based on the idea that drift removes alleles randomly with respect to allele size (Williamson-Natesan 2005). According to suggestion by Peery et al. (2012), proportion of multi-step mutations (pg) was set to 0.22, and average multi-step mutation size (δg) was 3.1 during 100,000 coalescent simulations. Using the software INEST 2.2 (Chybicki 2017), both the excess in heterozygosity and the deficiency in M-ratio in each population were tested by the Wilcoxon signed-rank test which does not assume normality of data distribution and approximates the exact p-value after 1,000,000 permutations.
Population structure and gene flow
To identify genetically homogeneous populations of individuals in our samples, we applied two different approaches. Firstly, we examined this by a Principal Coordinates Analysis (PCoA) of the R package ‘adegenet’ 2.1.3 (Jombart 2008). This multivariate method considers the best approximation of a dissimilarity matrix of Euclidean and short distances while its principal components optimize the representation of the squared pairwise distances between individuals. Secondly, we used an individual-based clustering method implemented in the software Structure 2.3.4 (Pritchard et al. 2000; Hubisz et al. 2009) to infer optimal number of genetic clusters (K). We ran the admixture model with correlated allele frequencies without the prior population information and degree of admixture α = 1. For each value of K (range 1–6), we conducted 10 independent runs with uniform priors using a burn-in of 100,000 iterations followed by 100,000 Markov chain Monte Carlo iterations. The optimal number of clusters in the data set was inferred by the ΔK method (Evanno et al. 2005), which finds the breakpoint in the slope of the likelihood distribution for different K values, using the Structure Harvester Web 0.6.94 (Earl and vonHoldt 2012) and also through Q-matrix correlations, which determine the stable K solutions (identify anomalous runs) implemented in the R package ‘CorrSieve’ 1.6–8 (Campana et al. 2011).
We assumed that individual migration between two geographically distant population groups would extend far beyond the dispersal limits of an individual of the species under study and that linkage of groups would overestimate possible evidence of gene flow within groups. Therefore, to examine gene flow, we computed directional relative migration values between populations in urban and rural areas separately using the divMigrate function (Sundqvist et al. 2016) from the R package ‘diveRsity’ 1.9.89 (Keenan et al. 2013). Statistical significance of directional migration was calculated using 1,000 bootstraps. Furthermore, we detected possible immigrants from neighbouring populations using a machine-learning framework implemented in the R package ‘assignPOP’ 1.2.0 (Chen et al. 2021). A naïve Bayes classification method (a simple probabilistic classifier with strong independence assumptions between features) was used as the default setting of this package to build a predictive model that estimated individual membership probabilities for each study population using baseline data representing all microsatellite genotypes of three populations of the corresponding area (rural or urban).
Results
All 11 microsatellite loci were highly variable, with 12–22 alleles per locus, and showed relatively high observed (0.758–0.975) and expected heterozygosity (0.819–0.913). The inbreeding coefficient ranged from − 0.151 to 0.165 (Table 1). Genetic variation between populations differed significantly with respect to the number of alleles (χ2 = 27.5, p < 0.001) and heterozygosity (HO, χ2 = 20.9, p < 0.001; HE, χ2 = 23.5, p < 0.001). It was mainly decreased in two urban populations, uKVP and uZUZ. In contrast, uMSP urban population did not differ from rural populations (Fig. 2). Unique (private) alleles were found in all populations. The highest number of them was found in uMSP (14), and the lowest in uZUZ (3). Other populations displayed 5–7 private alleles. There were no significant differences in the FIS index of the populations. Genetic differentiation of urban populations was higher (mean ± SD; DPS = 0.608 ± 0.049, G′ST = 0.508 ± 0.095) compared to rural samples (DPS = 0.467 ± 0.019, G′ST = 0.247 ± 0.055), and this differentiation did not correlate with geographical distance between all sites (Mantel test with 999 replicates, p = 0.72). There were significant signatures of a demographic bottleneck (p < 0.05), which manifested as a deficiency in M-ratio in all populations, regardless of environment. However, we found no excess of heterozygosity (Table 2).

a Number of alleles, b observed heterozygosity, c expected heterozygosity, and d inbreeding coefficient in three rural (green) and three urban (blue) populations of Bufotes viridis calculated from 11 microsatellite loci. Different lowercase letters above box-plots denote groups that significantly differ (p < 0.05) from the others according to post-hoc Dunn tests
Multivariate PCoA analysis revealed significant variation in genotypes among individuals. The first two axes explained most of the variation (62.7%; Fig. 3a). Two urban populations (uKVP, uZUZ) differed significantly from the others, in contrast to uMSP, which did not differ from the rural sites. In the outputs of the admixture model simulated in the Structure analysis, both methods ΔK and Q-matrix correlations inferred stable genetic structure at K = 3 (Fig. 3b, Fig. S1 of Supplementary information). However, Q-matrix correlations using both the maximum average correlation and the rows-and-columns criteria (R = 0.99, p < 0.05) yielded a stable solution also at K = 5 (Fig. S2 of Supplementary information). This suggests that each urban site represented a single genetic population, while all rural sites likely belong to one homogeneous genetic cluster.

a Genetic distances between individuals in the studied populations using the Principal Coordinate Analysis according to the first and second principal components with 95% inertia ellipse. b Genetic structure of samples in the study area inferred by the Structure admixture analysis. Individuals in the stacked bar plots of different K are represented by vertical bars divided into parts proportional to their proposed ancestry in determined genetic clusters
Relative migration values between rural and urban populations also differed when using directional migration concept. Higher level of gene flow was calculated in the rural area, while lower level of gene flow between urban populations was significantly (p < 0.05) directional from the more isolated uKVP and uZUZ sites to the uMSP site (Fig. 4a). However, a machine-learning approach did not support this pattern in such resolution. Apart from two single immigrants between rural set of populations, all other individuals were assigned to the sites where they were sampled (Fig. 4b). Nevertheless, both analyses suggest very restricted gene flow in the urban area.

Gene flow between rural (left) and urban (right) set of populations. a Calculated relative migration values between rural and urban populations of Bufotes viridis. The larger of the two relative migration values indicates that it is likely to be a source population, and asterisk indicates a statistically significant (p < 0.05) directional migration. b Membership probability from the assignment test and the number of potential migrants (arrows). Individuals in the stacked bar plots are represented by vertical bars divided into parts proportional to their proposed membership to source population (coded in different colours)
Discussion
In this small-scale study, we found that urban populations of B. viridis exhibit lower genetic variability, higher genetic differentiation, and spatial structuring compared to sites in the adjacent rural landscape, although different bottleneck tests provided discrepant results for the dataset analysed (see Peery et al. 2012). Our results clearly indicate that gene flow was more restricted in the city of Košice agglomeration, although the geographic distances between sampled sites were much smaller (2–4 km versus 6–13 km). Genetic differentiation and isolation were the most pronounced in two populations that reproduced at sites with the least amount of suitable habitat (uKVP, uZUZ). Surprisingly, the third urban population in the large city park (uMSP) was genetically homogeneous with rural populations outside Košice. Thus, we found differential effects of the city environment on the population structure of the green toad, as distinct urban populations could be probably the result of a recent colonization event or fragmented remnants of a historical population that lived in the area before heavy urban development. Thus, despite emerging markers and high-throughput technologies in population genetics, we confirmed that microsatellites can still be a powerful tool to draw inferences about population patterns and processes (Putman and Carbone 2014). To compensate for potential weaknesses of these markers, we have successfully integrated several complementary computational methods to answer our questions.
Urban populations are often founded by the immigration of individuals from the surrounding rural areas, while amphibians are generally considered species with weak dispersal abilities, often migrating only up to a few kilometres (e.g., Smith and Green 2005; Vences et al. 2019). In natural populations, they show a typical pattern of isolation-by-distance (Palo et al. 2003; Spear et al. 2005; Arioli et al. 2010) because B. viridis is a poor disperser (Indermaur et al. 2009; Sinsch and Leskovar 2011). However, greater geographic distance between populations does not necessarily correlate with greater genetic differentiation between them (Degani et al. 2013). This is likely due to small and isolated populations in urban environments, which should be more likely to exhibit genetic drift resulting from founder effect (demographic bottleneck).
Urbanization reduces the size of local populations and gene flow between them through habitat loss, degradation, or fragmentation (Johnson and Munshi-South 2017). In our urban populations, this effect was observed in lower genetic diversity and higher genetic differentiation compared to rural populations. Such a pattern was also found in populations of Pelophylax ridibundus by Mikulíček and Pišút (2012). In this case, very close (0.4 km) urban populations were genetically more differentiated (FST = 0.070) than more distant populations (8 km) connected by a migration corridor of a water channel (FST = 0.010), while neighbouring populations isolated by a four-lane highway were significantly more differentiated (FST = 0.062–0.089). Similar results were demonstrated in Rana temporaria, although no physical barriers prevented the movement of individuals. Significant genetic differentiation (FST = 0.388) was found between urban populations separated by 2 km compared to populations at less disturbed rural sites (FST = 0.145) with an average distance of 41 km (Hitchings and Beebee 1997). In addition to the founder effect, strong selection pressure at breeding sites with unpredictable conditions (e.g., regular desiccation of pools due to temperature rise in the city) can produce populations with specific genotypes of adapted individuals, which can increase genetic differentiation among sites (Degani et al. 2013). Thus, the imbalance between migration and genetic drift may be a result of colonization of new sites by a limited number of founders or the creation of artificial barriers to gene flow (Mikulíček and Pišút 2012).
Both the Bayesian approach and multivariate analysis suggested genetic isolation of two urban populations at sites with less suitable habitat for green toads. In contrast, admixture of the population inhabiting the large city park with all rural populations and the highest number of private alleles is likely the result of intense historical gene flow (Smith and Green 2005). This site is located in the former floodplain of the Hornád River and its tributaries (Fig. 1), which could serve as a vector for migration of individuals from the surrounding area (e.g., Mikulíček and Pišút 2012; Vences et al. 2019). Thus, the city park was established on a natural wetland approximately 200 years ago (Municipality of Košice, personal communication) and still resembles the natural habitat of the species due to the high proportion of diverse vegetation, various roosting opportunities, and safe breeding pools in an extensive fountain system. In addition, the limited human presence (the park is closed to visitors at night) may accommodate toads in their nocturnal terrestrial activities (e.g., Rodríguez-Prieto and Fernández-Juricic 2005).
In contrast, the dense development around two other urban fountains in settlements on a terrace about 100 m above the level of the Hornád River, built in the 1960–1990s, is probably a strong barrier to gene flow. The lack of suitable wetlands prior to urban development suggests that these sites were colonized by a limited number of founders only after the construction of the fountains (strong founder effect). Based on the approximate generation time of the green toad (Sinsch et al. 2007), the effect of the demographic bottleneck there could already last 10–20 generations. If there is some gene flow between urban sites, it is more likely to be an occasional migration from small and regularly drained fountains towards the population in the city park below. To further examine this possibility, one should investigate the possible role of the city drainage system in the passive transport of individuals (cf. Semlitsch 2008). Another explanation for possible long-distance migration and the occurrence of private alleles in the city park could be anecdotal evidence of voluntary rescue transfers of some toads from other sites or even transport of goods, as it is located close to the main railway station.
Although the green toad successfully inhabits urban areas throughout Europe, its life history traits might differ due to specific local environmental conditions in cities, including intensity of development, pond water regime, predation, anthropogenic stressors, or chemical pollution (e.g., Ensabella et al. 2003; Kaczmarski et al. 2019; Mazgajska and Mazgajski 2020). Nevertheless, the low levels of the FIS index in all populations in our study suggest that the species is not yet suffering from increased inbreeding even in the most isolated urban populations. Also, examination of the level of fluctuating asymmetry, which could serve as an indicator of urban-induced environmental stress, revealed no significant differences between rural and urban populations (Vargová et al. 2022). However, considering the planned reconstructions of the fountains and their surroundings, the survival of the green toad may be at risk (Municipality of Košice, personal communication).
Future conservation plans for the studied populations should therefore include habitat protection and management (Semlitsch and Bodie 2003; Wei et al. 2021), especially maintaining adequate hydroperiod length in breeding ponds (Guderyahn et al. 2016), suitable roosting and wintering sites within sufficient distance from the breeding pond, and connectivity between sites (Furman et al. 2016). Green spaces, such as the large urban park in our study, typically function as species refugia in urban landscapes (Niemeier et al. 2020) and could be core habitats that provide sources for other populations. However, we believe that within the urban area of Košice, it is almost impossible to establish habitat connectivity that could maintain gene flow between small populations. Therefore, at least in the case of fountain reconstruction or temporary draining of the pool, maintaining green areas around the breeding site could benefit toad populations by providing roosting opportunities throughout the season (Semlitsch and Bodie 2003; Guderyahn et al. 2016; Wei et al. 2021). In terms of species adaptation to new and changing urban environments, long-term monitoring of local genetic variation and structure is therefore important for the favourable status of species in Central Europe (Sinsch et al. 2007).
Data availability
The microsatellite data presented in this study are available on request from the first author.
References
Adamack AT, Gruber B (2014) PopGenReport: simplifying basic population genetic analyses in R. Methods Ecol Evol 5:384–387. https://doi.org/10.1111/2041-210X.12158
Arioli M, Jakob C, Reyer HU (2010) Genetic diversity in water frog hybrids (Pelophylax esculentus) varies with population structure and geographic location. Mol Ecol 19:1814–1828. https://doi.org/10.1111/j.1365-294x.2010.04603.x
Becker CG, Fonseca CR, Haddad CFB, Batista RF, Prado PI (2007) Habitat split and the global decline of amphibians. Science 318:1775–1777. https://doi.org/10.1126/science.1149374
Bowcock AM, Ruiz-Linares A, Tomfohrde J, Minch E, Kidd JR, Cavalli-Sforza LL (1994) High resolution of human evolutionary trees with polymorphic microsatellites. Nature 368:455–457. https://doi.org/10.1038/368455a0
Brede EG, Beebee TJC (2004) Contrasting population structures in two sympatric anurans: implications for species conservation. Heredity 92:110–117. https://doi.org/10.1038/sj.hdy.6800391
Brookfield JFY (1996) A simple new method for estimating null allele frequency from heterozygote deficiency. Mol Ecol 5:453–455. https://doi.org/10.1046/j.1365-294X.1996.00098.x
Budzik KA, Budzik KM, Żuwała K (2013) Amphibian situation in urban environment: history of the common toad Bufo bufo in Kraków (Poland). Ecol Quest 18:73–77. https://doi.org/10.2478/ecoq-2013-0009
Campana MG, Hunt HV, Jones H, White J (2011) CorrSieve: software for summarising and evaluating STRUCTURE output. Mol Ecol Resour 11:349–352. https://doi.org/10.1111/j.1755-0998.2010.02917.x
Cayuela H, Valenzuela-Sánchez A, Teulier L, Martínez-Solano Í, Léna JP, Merilä J, Muths E, Shine R, Quay L, Denoël M, Clobert J, Schmidt BR (2020) Determinants and consequences of dispersal in vertebrates with complex life cycles: a review of pond-breeding amphibians. Q Rev Biol 95:1–36. https://doi.org/10.1086/707862
Chakraborty R, Zhong Y, Jin L, Budowle B (1994) Nondetectability of restriction fragments and independence of DNA fragment sizes within and between loci in RFLP typing of DNA. Am J Hum Genet 55:391–401
Chen K-Y, Marschall EA, Sovic MG, Fries AC, Gibbs HL, Ludsin SA (2021) assignPOP: population assignment using genetic, non-genetic or integrated data in a machine learning framework. R package version 1.2.4. https://cran.r-project.org/package=assignPOP
Chybicki IJ (2017) INEST 2.2. http://www.ukw.edu.pl/pracownicy/strona/igor_chybicki/software_ukw/
Cox K, Maes J, Van Calster H, Mergeay J (2017) Effect of the landscape matrix on gene flow in a coastal amphibian metapopulation. Conserv Genet 18:1359–1375. https://doi.org/10.1007/s10592-017-0985-z
Degani G, Goldberg T, Gasith A, Elron E, Nevo E (2013) DNA variations of the green toad Pseudepidalea viridis (syn. Bufo viridis) from various habitats. Zool Stud 52:18. https://doi.org/10.1186/1810-522X-52-18
Earl DA, vonHoldt BM (2012) Structure harvester: a website and program for visualizing Structure output and implementing the Evanno method. Conserv Genet Resour 4:359–361. https://doi.org/10.1007/s12686-011-9548-7
Elmqvist T, Fragkias M, Goodness J, Güneralp B, Marcotullio PJ, Mcdonald RI, Parnell S, Schewenius M, Sendstad M, Seto KC, Wilkinson C (2013) Urbanization, biodiversity and ecosystem services: challenges and opportunities. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-7088-1
Ensabella F, Loriga S, Formichetti P, Isotti R, Sorace A (2003) Breeding site selection of Bufo viridis in the city of Rome (Italy). Amphibia-Reptilia 24:396–400. https://doi.org/10.1163/156853803322440853
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol 14:2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Faeth SH, Bang C, Saari S (2011) Urban biodiversity: patterns and mechanisms. Ann N Y Acad Sci 1223:69–81. https://doi.org/10.1111/j.1749-6632.2010.05925.x
Fahrig L, Pedlar JH, Pope SE, Taylor PD, Wegner JF (1995) Effect of road traffic on amphibian density. Biol Cons 73:177–182. https://doi.org/10.1016/0006-3207(94)00102-V
Furman BL, Scheffers BR, Taylor M, Davis C, Paszkowski CA (2016) Limited genetic structure in a wood frog (Lithobates sylvaticus) population in an urban landscape inhabiting natural and constructed wetlands. Conserv Genet 17:19–30. https://doi.org/10.1007/s10592-015-0757-6
Garcia VOS, Ivy C, Fu J (2017) Syntopic frogs reveal different patterns of interaction with the landscape: a comparative landscape genetic study of Pelophylax nigromaculatus and Fejervarya limnocharis from central China. Ecol Evol 7:9294–9306. https://doi.org/10.1002/ece3.3459
Garza JC, Williamson EG (2001) Detection of reduction in population size using data from microsatellite loci. Mol Ecol 10:305–318. https://doi.org/10.1046/j.1365-294x.2001.01190.x
Goldberg CS, Waits LP (2010) Comparative landscape genetics of two pond-breeding amphibian species in a highly modified agricultural landscape. Mol Ecol 19:3650–3663. https://doi.org/10.1111/j.1365-294x.2010.04673.x
Guderyahn LB, Smithers AP, Mims MC (2016) Assessing habitat requirements of pond-breeding amphibians in a highly urbanized landscape: implications for management. Urban Ecosyst 19:1801–1821. https://doi.org/10.1007/s11252-016-0569-6
Hamer AJ, McDonnell MJ (2008) Amphibian ecology and conservation in the urbanising world: a review. Biol Cons 141:2432–2449. https://doi.org/10.1016/j.biocon.2008.07.020
Harper EB, Rittenhouse TA, Semlitsch RD (2008) Demographic consequences of terrestrial habitat loss for pool-breeding amphibians: predicting extinction risks associated with inadequate size of buffer zones. Conserv Biol 22:1205–1215. https://doi.org/10.1111/j.1523-1739.2008.01015.x
Hedrick PW (2005) A standardized genetic differentiation measure. Evolution 59:1633–1638. https://doi.org/10.1111/j.0014-3820.2005.tb01814.x
Hitchings SP, Beebee TJ (1997) Genetic substructuring as a result of barriers to gene flow in urban Rana temporaria (common frog) populations: implications for biodiversity conservation. Heredity 79:117–127. https://doi.org/10.1038/hdy.1997.134
Hitchings SP, Beebee TJC (1998) Loss of genetic diversity and fitness in common toad (Bufo bufo) populations isolated by inimical habitat. J Evol Biol 11:269–283. https://doi.org/10.1046/j.1420-9101.1998.11030269.x
Holderegger R, Di Giulio M (2010) The genetic effects of roads: a review of empirical evidence. Basic Appl Ecol 11:522–531. https://doi.org/10.1016/j.baae.2010.06.006
Homola JJ, Loftin CS, Kinnison MT (2019) Landscape genetics reveals unique and shared effects of urbanization for two sympatric pool-breeding amphibians. Ecol Evol 9:11799–11823. https://doi.org/10.1002/ece3.5685
Hubisz MJ, Falush D, Stephens M, Pritchard JK (2009) Inferring weak population structure with the assistance of sample group information. Mol Ecol Resour 9:1322–1332. https://doi.org/10.1111/j.1755-0998.2009.02591.x
Indermaur L, Gehring M, Wehrle W, Tockner K, Naef-Daenzer B (2009) Behavior-based scale definitions for determining individual space use: requirements of two amphibians. Am Nat 173:60–71. https://doi.org/10.1086/593355
Johnson MT, Munshi-South J (2017) Evolution of life in urban environments. Science 358:eaam8327. https://doi.org/10.1126/science.aam8327
Jombart T (2008) adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405. https://doi.org/10.1093/bioinformatics/btn129
Kaczmarski M, Szala K, Kloskowski J (2019) Early onset of breeding season in the green toad Bufotes viridis in Western Poland. Herpetozoa 32:109–112. https://doi.org/10.3897/herpetozoa.32.e35825
Keenan K, McGinnity P, Cross TF, Crozier WW, Prodöhl PA (2013) diversity: an r package for the estimation and exploration of population genetics parameters and their associated errors. Methods Ecol Evol 4:782–788. https://doi.org/10.1111/2041-210X.12067
Lourenço A, Álvarez D, Wang IJ, Velo-Antón G (2017) Trapped within the city: integrating demography, time since isolation and population-specific traits to assess the genetic effects of urbanization. Mol Ecol 26:1498–1514. https://doi.org/10.1111/mec.14019
Mazgajska J, Mazgajski TD (2020) Two amphibian species in the urban environment: changes in the occurrence, spawning phenology and adult condition of common and green toads. Eur Zool J 87:170–179. https://doi.org/10.1080/24750263.2020.1744743
Mikulíček P, Pišút P (2012) Genetic structure of the marsh frog (Pelophylax ridibundus) populations in urban landscape. Eur J Wildl Res 58:833–845. https://doi.org/10.1007/s10344-012-0631-5
Miles LS, Rivkin LR, Johnson MT, Munshi-South J, Verrelli BC (2019) Gene flow and genetic drift in urban environments. Mol Ecol 28:4138–4151. https://doi.org/10.1111/mec.15221
Niemeier S, Müller J, Struck U, Rödel MO (2020) Superfrogs in the city: 150 year impact of urbanization and agriculture on the European common frog. Glob Change Biol 26:6729–6741. https://doi.org/10.1111/gcb.15337
Ogle DH, Wheeler P, Dinno A (2020) FSA: fisheries stock analysis. R package version 0.8.30. https://github.com/droglenc/FSA
Palo JU, O’Hara RB, Lauge AT, Laurila A, Primmer CR, Merilä J (2003) Latitudinal divergence of common frog (Rana temporaria) life history traits by natural selection: evidence from a comparison of molecular and quantitative genetic data. Mol Ecol 12:1963–1978. https://doi.org/10.1046/j.1365-294x.2003.01865.x
Peery MZ, Kirby R, Reid BN, Stoelting R, Doucet-Bëer E, Robinson S, Vásquez-Carrillo C, Pauli JN, Palsbøll PJ (2012) Reliability of genetic bottleneck tests for detecting recent population declines. Mol Ecol 21:3403–3418. https://doi.org/10.1111/j.1365-294X.2012.05635.x
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959. https://doi.org/10.1093/genetics/155.2.945
Putman AI, Carbone I (2014) Challenges in analysis and interpretation of microsatellite data for population genetic studies. Ecol Evol 4:4399–4428. https://doi.org/10.1002/ece3.1305
R Core Team (2021) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna. https://www.r-project.org/
Richardson JL (2012) Divergent landscape effects on population connectivity in two co-occurring amphibian species. Mol Ecol 21:4437–4451. https://doi.org/10.1111/j.1365-294X.2012.05708.x
Rodríguez-Prieto I, Fernández-Juricic E (2005) Effects of direct human disturbance on the endemic Iberian frog Rana iberica at individual and population levels. Biol Cons 123:1–9. https://doi.org/10.1016/j.biocon.2004.10.003
Ronce O (2007) How does it feel to be like a rolling stone? Ten questions about dispersal evolution. Annu Rev Ecol Evol Syst 38:231–253. https://doi.org/10.1146/annurev.ecolsys.38.091206.095611
Safner T, Miaud C, Gaggiotti O, Decout S, Rioux D, Zundel S, Manel S (2011) Combining demography and genetic analysis to assess the population structure of an amphibian in a human-dominated landscape. Conserv Genet 12:161–173. https://doi.org/10.1007/s10592-010-0129-1
Savary P, Foltête J, Moal H, Vuidel G, Garnier S (2020) graph4lg: a package for constructing and analysing graphs for landscape genetics in R. Methods Ecol Evol 12:539–547. https://doi.org/10.1111/2041-210X.13530
Schmidt C, Garroway CJ (2021) The population genetics of urban and rural amphibians in North America. Mol Ecol 30:3918–3929. https://doi.org/10.1111/mec.16005
Semlitsch RD (2008) Differentiating migration and dispersal processes for pond-breeding amphibians. J Wildl Manag 72:260–267. https://doi.org/10.2193/2007-082
Semlitsch RD, Bodie JR (2003) Biological criteria for buffer zones around wetlands and riparian habitats for amphibians and reptiles. Conserv Biol 17:1219–1228. https://doi.org/10.1046/j.1523-1739.2003.02177.x
Sinsch U, Leskovar C (2011) Does thermoregulatory behaviour of green toads (Bufo viridis) constrain geographical range in the west? A comparison with the performance of syntopic natterjacks (Bufo calamita). J Therm Biol 36:346–354. https://doi.org/10.1016/j.jtherbio.2011.06.012
Sinsch U, Leskovar C, Drobig A, König A, Grosse WR (2007) Life-history traits in green toad (Bufo viridis) populations: indicators of habitat quality. Can J Zool 85:665–673. https://doi.org/10.1139/Z07-046
Sinsch U, Oromi N, Miaud C, Denton J, Sanuy D (2012) Connectivity of local amphibian populations: modelling the migratory capacity of radio-tracked natterjack toads. Anim Conserv 15:388–396. https://doi.org/10.1111/j.1469-1795.2012.00527.x
Smith MA, Green DM (2005) Dispersal and the metapopulation paradigm in amphibian ecology and conservation: are all amphibian populations metapopulations? Ecography 28:110–128. https://doi.org/10.1111/j.0906-7590.2005.04042.x
Spear SF, Peterson CR, Matocq MD, Storfer A (2005) Landscape genetics of the blotched tiger salamander (Ambystoma tigrinum melanostictum). Mol Ecol 14:2553–2564. https://doi.org/10.1111/j.1365-294X.2005.02573.x
Stuart SN, Chanson JS, Cox NA, Young BE, Rodrigues AS, Fischman DL, Waller RW (2004) Status and trends of amphibian declines and extinctions worldwide. Science 306:1783–1786. https://doi.org/10.1126/science.1103538
Sundqvist L, Keenan K, Zackrisson M, Prodöhl P, Kleinhans D (2016) Directional genetic differentiation and relative migration. Ecol Evol 6:3461–3475. https://doi.org/10.1002/ece3.2096
Trenham PC, Shaffer HB (2005) Amphibian upland habitat use and its consequences for population viability. Ecol Appl 15:1158–1168. https://doi.org/10.1890/04-1150
Vargová V, Balogová M, Figurová M, Bočkay A, Pipová N, Kaňuch P, Uhrin M (2022) Skeletal morphology and fluctuating asymmetry of the European green toad, Bufotes viridis, in contrasting habitats. Amphibia-Reptilia 43:449–455. https://doi.org/10.1163/15685381-bja10111
Vences M, Perl RGB, Giesen K, Schluckebier R, Klaus S, Schmidt E, Steinfartz S, Ziegler T (2019) Development of new microsatellite markers for the green toad, Bufotes viridis, to assess population structure at its northwestern range boundary in Germany. Salamandra 55:191–199
Wei X, Huang M, Yue Q, Ma S, Li B, Mu Z, Peng C, Gao W, Liu W, Zheng J, Weng X, Sun X, Zuo Q, Bo S, Yuan X, Zhang W, Yang G, Ding Y, Wang X, Wang T, Hua P, Wang Z (2021) Long-term urbanization impacts the eastern golden frog (Pelophylax plancyi) in Shanghai City: demographic history, genetic structure, and implications for amphibian conservation in intensively urbanizing environments. Evol Appl 14:117–135. https://doi.org/10.1111/eva.13156
Williamson-Natesan EG (2005) Comparison of methods for detecting bottlenecks from microsatellite loci. Conserv Genet 6:551–562. https://doi.org/10.1007/s10592-005-9009-5
Acknowledgements
This research was funded by the Scientific Grant Agency VEGA (1/0298/19) and VVGS-2021-1988.
Funding
Open access funding provided by The Ministry of Education, Science, Research and Sport of the Slovak Republic in cooperation with Centre for Scientific and Technical Information of the Slovak Republic.
Author information
Authors and Affiliations
Contributions
MU and MB conceived the idea, and PK designed the methodology; VV, MB, and NP collected samples; DG and VV performed laboratory work and genotyping; PK analysed the data and prepared figures; VV and PK drafted the manuscript; MU supervised the project. All authors discussed the results and edited the manuscript.
Corresponding author
Ethics declarations
Compliance with ethical standards
All applicable institutional and/or national guidelines for the care and use of animals were followed.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Vargová, V., Gužiová, D., Balogová, M. et al. Urban environment determines population genetics in the green toad, Bufotes viridis. Eur J Wildl Res 69, 86 (2023). https://doi.org/10.1007/s10344-023-01716-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10344-023-01716-9