
Abstract
Urbanization is a pervasive process causing habitat fragmentation, spatial isolation of populations, and reduction of biological diversity. In this study, we applied 11 microsatellite loci and Bayesian analyses to investigate genetic diversity and population structure in marsh frogs (Pelophylax ridibundus) living in two types of environment—highly fragmented urban landscapes, and landscapes characterized by the presence of a river and artificial canals. Our results show reduced genetic diversity, lower effective population sizes, and higher genetic differentiation for spatially isolated urban populations in comparison with populations outside intensely urbanized areas. Reduction of allelic diversity in urban localities isolated for 13–37 generations is more conspicuous than reduction of expected heterozygosity. Populations living close to the River Danube, its branches, and artificial canals are genetically more homogenous. Our results also suggest that the Danube in Bratislava is not a natural barrier to gene flow. In contrast, it acts as a natural corridor for water frog dispersal. Population structure of P. ridibundus also shows higher genetic connectivity within water paths than between them, suggesting limited overland dispersal, and reflects the historical landscape structure associated with the distribution of the lost river branches.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Urbanization is an important cause of landscape change, having strong impact on biological diversity. Frequent outcome of urbanization is loss of habitats and subsequent fragmentation of the remaining habitat matrix, thereby influencing species composition and population dynamics (Pillsbury and Miller 2008; Sutherland et al. 2010). The effects of fragmentation vary with many factors including the size, quality and age of habitat patches, the vagility and life history of the species, and the characteristics of the landscape matrix between the patches (Keyghobadi et al. 2005; Pavlacky et al. 2009; Delaney et al. 2010; Jarošík et al. 2011). From a population genetics point of view, if dispersal and gene flow between the patches are restricted or impeded, populations become genetically isolated with increasing risk of inbreeding, loss of genetic diversity, and even local extinction (Allendorf and Luikart 2007). Small and isolated populations are more prone to loss of genetic variation through drift than large and more connected populations.
The response of population genetic structure to habitat change and fragmentation is not immediate as landscapes typically change much faster than the genetic structure of organisms (Whitlock and McCauley 1999; Holzhauer et al. 2006; Orsini et al. 2008; Richmond et al. 2009). Populations that have been isolated only recently are not expected to be at equilibrium between migration and drift and will carry the genetic signature of historical landscape connectivity. The response of genetic changes to population subdivision depends on the rate of genetic drift and dispersal (Crow and Aoki 1984). As genetic drift drives allele frequencies within subpopulations to fixation, genetic variation is removed and heterozygosity within the subpopulations is reduced. Gene flow counteracts the effect of subdivision and maintains homogenized populations. While genetic divergence between subpopulations increases rapidly as a result of isolation, loss of genetic variation decreases slower (Keyghobadi et al. 2005). Of two components of within-population variation, allelic diversity and heterozygosity, it is the allelic diversity that is lost more readily. The extent of heterozygosity may remain relatively high even when the loss of allelic diversity is substantial (Allendorf and Luikart 2007).
In the present study, we investigated population genetic structure of the marsh frog, Pelophylax ridibundus (Pallas, 1771), former name Rana ridibunda Pallas, 1771, in urban landscape characterized by intensive changes throughout the last century. These changes included regulation of the Danube River, loss of the river tributaries, and intensification of urbanization, leading to a reduction of landscape permeability and spatial isolation of populations. On the other hand, man-made gravel pits connected by artificial canals may increase landscape permeability and allow marsh frogs to colonize new sites. The outcomes of landscape changes on population genetic structure are species specific and strongly depend on life histories, habitat preference, and dispersal abilities. P. ridibundus is a large water frog species of the family Ranidae widely distributed in lowlands of central Europe. It inhabits larger, sparsely vegetated water bodies near rivers such as oxbow lakes, backwaters with stagnant or slowly flowing water, gravel pits, and canals characterized by high levels of dissolved oxygen, low salinity, and near-to-neutral pH (Rybacki and Berger 1994; Plénet et al. 2000; Pagano et al. 2001a, b; Schmeller et al. 2007). Marsh frogs are bound to water bodies throughout the year, including both breeding and hibernation (Berger 1982). Adult dispersal between ponds is limited to several hundred meters in continuous terrestrial habitats: dispersal rate decreases with increasing pond-to-pond distance and degree of pond isolation (Holenweg Peter 2001). This limited terrestrial dispersal together with high site fidelity and strong homing behavior (Holenweg Peter et al. 2001) should result in reduced gene flow between spatially isolated ponds. On the other hand, the occurrence of small rivers, canals, and water bodies between breeding ponds should facilitate dispersal, making populations genetically more homogenous (Holenweg Peter 2001; Pagano et al. 2001a).
The main aim of the present study was to test three hypotheses: (1) If anthropogenic structures like buildings and busy roads are barriers to gene flow, then populations isolated with the urbanization of intervening habitat should have a reduced level of genetic diversity and their genetic differentiation should be larger due to genetic drift. (2) The Danube in the Bratislava city is a large river with strong current. If the Danube is a natural barrier to dispersal, this should result in genetic differentiation between the right and the left bank populations. (3) If river backwaters and artificial canals facilitate dispersal and gene flow, populations connected by these water corridors should be genetically more homogenous and genetically diverse than isolated urban populations.
Material and methods
Sampling
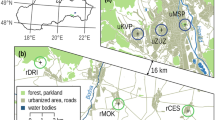
A total of 494 samples of marsh frogs were obtained from 17 sampling localities in the years 2006–2008 (Table 1). Localities are situated in an area of approximately 130 km2 and cover virtually all known breeding sites of this species in Bratislava and its vicinity. The shortest straight distance between two ponds was 420 m; the farthest ponds within the study area were 18.5 km apart. Most sampling sites were man-made gravel pits, oxbow lakes, and artificial canals with stagnant water situated on the right (DRA, CHOR), as well as the left (all other sites) bank of the river Danube (Fig. 1). Four breeding sites, referred here as urban, are ponds situated within the city (STR, ROH, ZLP, KAL). These ponds are surrounded by buildings and isolated by four-lane highways. Other breeding sites (suburban) are in close vicinity of the Danube (referred as the Danube localities) and artificial canals (referred as the “canal” localities). Besides P. ridibundus, one or two other water frog taxa, Pelophylax lessonae and/or Pelophylax esculentus, live in three localities (KOP, CHOR, DEV), but they are less abundant.

Population genetic structure of P. ridibundus in Bratislava and its vicinity. GENELAND assigned all individuals to three clusters (black, red, and yellow) associated with the river Danube, the lost Mill side channel, and the canals. BIS, VRA, and ZLP were assigned to separate clusters in BAPS. Arrows between localities indicate non-significant Fst values. Pairwise Fst comparisons between all other localities were statistically significant
Landscape structure
Landscape development in the city of Bratislava was influenced by three main processes during the last two centuries: urbanization; the regulation of the course of the Danube, connected with loss of the river braiding; and the construction of artificial canals and gravel pits. During the eighteenth and nineteenth centuries, more than 30 river braids formed a dynamic river system strongly influenced by regular floods (Pišút 2011). Most of these alternate river paths (braids) have been cut off the Danube. The modern Danube in Bratislava is now a channeled 300-m-wide river with a strong current and mean annual discharge of 2,024 m3/s (Pišút 2002). Two branches of the Danube are the most important ones for understanding the water frog distribution and their present-day population structure (Figs. 1 and 2). The Lesser Danube branch splits from the Danube in the south-eastern part of the city. The second branch is the Mill side channel (Mühlauer Donau Arm, Mlynské rameno) whose path in the eighteenth and nineteenth centuries split from the Danube flowing eastward to join the Lesser Danube. At the end of nineteenth century, the uppermost section of the Mill channel was buried, but the lower section remained connected to the Lesser Danube up to the beginning of 1970s, when the channel was lost completely to urbanization (Pišút 2002). In the 1960s, the gravel pits STR and ROH were built in close vicinity to the Mill channel; at sites where small ponds with high abundance of water frogs once occurred. Thus, marsh frog populations STR and ROH have been isolated from the Danube for about 113 years, i.e., approximately 37 frog generations (marsh frog generation time was taken as 3 years according to Zeisset and Beebee 2003; Socha and Ogielska 2010), and about 40 years (13 generations) from the Lesser Danube. The frogs at these sites appear to have been isolated from each other for about 40 years by construction of a four-lane highway and housing estates. The eastern part of the study area was not so strongly influenced by the Danube. The origins of two localities, fishery ponds SUR and SPR, date back to the seventeenth and eighteenth century, respectively. Other sampling sites in this part of the study area were formed mainly in the second half of the twentieth century after gravel mining. The sites ZLP, KAL, and VAJ have been isolated from each other by buildings and four-lane highways for about 40 years (13 generations). The gravel-pit localities are situated several meters to 4 km from the Šúrsky canal, which was built between 1941 and 1943 as a drainage canal flowing from SUR to the Lesser Danube. Several smaller drainage canals neighbor the Šúrsky canal in the northern part of the study area.

Historical map of Bratislava showing position of present-day localities in association with the Danube River, the Mill side channel, and the Lesser Danube. Map sheets of the Ist Military mapping of the Habsburg Empire, 1782–1785, original scale 1:28,800 (Arcanum 2004)
DNA extraction and genotyping
Tissue samples from 20 to 33 individuals per locality were obtained by toe clipping and stored in 96 % ethanol. Total genomic DNA was extracted using Wizard SV Genomic DNA Purification System (Promega, USA) following the manufacturer’s protocol.
Eleven microsatellite loci, originally developed for P. ridibundus and P. lessonae (Garner et al. 2000; Zeisset et al. 2000; Christiansen and Reyer 2009; Holsbeek et al. 2009), were applied in this study. These markers were amplified in two multiplex PCRs (multiplex 1—Res14, Res15, Res17, Res22, Rrid059A, Rrid082A, and Rrid171A; multiplex 2—Rrid169A, RLCA1b6, RLCA1b20, Ga1a19) using Qiagen multiplex PCR kit. PCR was performed in a total volume of 10 μL containing 1× Qiagen Master Mix, Q solution (only in multiplex 2), 2 μL of DNA and 0.3 μM (RLCA2a34), 0.2 μM (Res14), or 0.1 μM (rest loci) of each primer pair (forward primers were fluorescently labeled with FAM, VIC, NED, and PET). PCR amplification involved an initial cycle of denaturation at 95 °C for 15 min and 33 subsequent cycles of 94 °C for 30 s, 60 °C for 90 s, and 72 °C for 60 s, followed by a final extension step at 60 °C for 30 min. PCR products were run on an ABI 3130 genetic analyzer (Applied Biosystems) with a LIZ-500 size standard. Peaks were visualized using the software GeneMapper 3.7 (Applied Biosystems) and scored manually by a single observer.
Genetic diversity, bottleneck, and effective population size (Ne)
Genotypes were checked for stuttering, large allele dropout, and null alleles with Micro-Checker 2.2.3 (Van Oosterhout et al. 2004). A maximum-likelihood estimate of the frequency of null alleles for each locus and population was then calculated using individual inbreeding model (IIM) implemented in the program INEst (Chybicki and Burczyk 2009). Number of alleles (Na), their frequencies, and observed (Ho) and expected (He) heterozygosity were calculated using GenAlEx 6.1 (Peakall and Smouse 2006). Allelic richness (AR) and tests for Hardy–Weinberg (H–W) equilibrium and linkage disequilibria (LD) were calculated using FStat 2.9.3.2 (Goudet 2001). Coefficient of inbreeding (F IS), defined as a probability that the two alleles at a locus are identical by descent, was corrected for the presence of null alleles and estimated using the program INEst (Chybicki and Burczyk 2009). Differences among urban and suburban localities for Ho, He, and AR were carried out using two-sided permutation tests implemented in FStat 2.9.3.2 (Goudet 2001).
To test for bottlenecks, we applied the program Bottleneck (Cornuet and Luikart 1996). This program uses coalescent simulations to generate gene diversities for each population and locus that are expected from the observed number of alleles given the sample sizes and assuming mutation–drift equilibrium. The calculated average expected gene diversity is then compared to the observed gene diversity to assess whether there is gene diversity excess or deficit at each locus. Populations that have undergone recent bottlenecks show gene diversities that are excessive relative to that expected given the observed number of alleles because allele number declines more rapidly than heterozygosity during bottlenecks. The stepwise mutation model (SMM) and the two-phase model (TPM) of microsatellite evolution were employed in making calculations using Wilcoxon signed-rank tests (Luikart and Cornuet 1998) with 2,000 iterations. Under the TPM of mutation, 95 % single-step mutations (12 % variance of multi-step mutations) were used as suggested by Piry et al. (1999).
For estimation of effective population sizes (Ne), we applied two approaches—a linkage disequilibrium (LD) method implemented in the program ldne (Waples 2006) and approximate Bayesian computation (ABC) framework implemented in the program ONeSAMP (Tallmon et al. 2008). Because bias in Ne estimation using LD might be expected if allele frequencies are close to zero (Waples 2006), we excluded all alleles with intra-population frequencies less than 0.05.
Population genetic structure
Population differentiation was measured using Fst statistics following the method of Weir and Cockerham (1984) implemented in FStat 2.9.3.2 (Goudet 2001). A test of population differentiation was based on 10,000 permutations, standard Bonferroni correction and did not assume Hardy–Weinberg equilibrium within samples. Then, the unbiased Fst values using the ENA (excluding null alleles) correction for null alleles were calculated with the program FreeNA (Chapuis and Estoup 2007). Isolation-by-distance (IBD) was estimated using correlation between matrices of pairwise Fst and Euclidian log-transformed geographic distances (Mantel test), as implemented in the program SpaGeDi 1.3 (Hardy and Vekemans 2002).
To assess genetic structure among samples, two Bayesian-based methods implemented in the programs BAPS 5.2 (Corander et al. 2003, 2008a, b; Corander and Marttinen 2006) and GENELAND 3.1.4 (Guillot et al. 2005a, b) were applied. These programs assign individuals into the K clusters with minimized Hardy–Weinberg and linkage disequilibria. Both programs also include geographical information in the inference of population structure. In Geneland, first we ran analyses with K free to vary, to infer the optimal value of this parameter. Then the algorithm was run again with K fixed at the previously inferred value as recommended by Guillot et al. (2005a, b) in order to estimate the other parameters, mainly the assignment of individuals to the inferred populations. We ran the analysis 10 times to verify the consistency of the results, with the following parameters: 500,000 MCMC iterations, maximum rate of Poisson process fixed to 100, zero uncertainty of coordinates, minimum and maximum K fixed to 1 and 20, respectively, maximum number of nuclei in the Poisson–Voronoi tessellation fixed to 300, and null allele and uncorrelated allele models. All 10 replicates revealed the maximum a posteriori estimate of K = 3. Then we ran the MCMC 100 times with K fixed to 3 and similar parameter settings. The run with the highest log probability was chosen for post-process analyses. The posterior probability of population membership for each pixel of the spatial domain and for each individual was then computed with 420 pixels along the X axis and 330 pixels along the Y axis. The modal population of each individual, maps of population membership, and maps of probability of population membership were finally computed.
Using BAPS 5.2, a mixture model for spatial clustering of groups was chosen (Corander et al. 2008a), followed by an admixture analysis (Corander and Marttinen 2006; Corander et al. 2008b). Five replicates for every upper level of K (5, 10, 15, and 20) were run. When estimating individual ancestry coefficients via admixture analysis, only clusters (based on the mixture analysis) that had at least 20 individuals present within them were analyzed. The number of iterations that were used to estimate the admixture coefficients for the individuals, and the number of reference individuals from each population was 200. The number of iterations that were used to estimate the admixture for the reference individuals was set to 20. In addition to analyses with the number of clusters free to vary, BAPS 5.2 allows assignment of individuals to a fixed number of clusters. Another set of analyses was performed with a fixed number of clusters K = 3, as inferred by GENELAND. A mixture model for spatial clustering of groups was replicated 10 times and followed by admixture analyses.
Results
Genetic diversity, bottleneck, and effective population size (Ne)
The 11 microsatellite loci contained between four and 19 alleles, with a mean of 10.91 alleles per locus (Table 2). Estimated null allele frequencies per locus ranged from 0.007 to 0.131, with frequencies averaged over loci varying from 0.053 to 0.118 depending on the population (Table 3).
Urban localities ZLP, ROH, KAL, and STR showed significantly lower mean values of allelic richness (AR, mean = 3.927, P = 0.016) and expected (He, mean = 0.549, P = 0.027) but not observed (Ho, mean = 0.554, P = 0.351) heterozygosity in comparison with suburban localities (Table 3). The highest values of genetic diversity (Na, AR, He) were from localities near the Danube River (DEV, CHOR, KOP, DRA). Significant deviation from H–W equilibrium was observed in 10 out of 17 populations. Coefficient of inbreeding (F IS), estimated simultaneously with null allele frequencies, varied from 0.005 to 0.015 depending on the population (Table 3).
LD was found in all but not in two populations (CHOR, ZEL). The highest LD between pairs of loci was found in IVD, VRA, and VAK (Appendix 1). Pairwise LD values estimated over all samples were sufficiently low that we assume the studied microsatellite loci are physically unlinked or freely recombining.
We found no evidence that any of the populations have been subjected to a recent bottleneck under both the SMM and TPM mutation model (Table 4). All locality samples exhibited some loci with higher than expected heterozygosity given the observed number of alleles. The highest number of loci with a heterozygosity excess was found in two urban samples STR and ROH; however, none of the localities showed a significant (P < 0.05) deviation from equilibrium expectations.
Effective population sizes estimated from locality samples ranged from 2.6 to 201.5 (LD method) and from 15.0 to 72.7 (ABC method), respectively (Table 5). Ne values calculated by the ABC approach were usually lower than those generated from the LD method, with tighter 95 % confidence intervals. Correlation between ABC- and LD-based Ne values was highly significant (Spearman rank order correlation, r = 0.831, P < 0.001). Three localities with the highest LDs (IVD, VRA, VAK) had the smallest LD-based Ne, not confirmed using the ABC method. Ne estimates for urban locality samples STR, ROH, ZLP, and KAL were lower compared to many of the other localities.
Population genetic structure
Global Fst over all loci and localities revealed significant genetic differentiation (Fst = 0.056, P < 0.001, range 0.003–0.142). Out of 136 pairwise comparisons, only 7.35 % were not statistically significant (Appendix 2). Fst values corrected for the presence of null alleles were consistent with those calculated using standard approach. Relatively low genetic differentiation was observed between localities near the Danube River and in the vicinity of the canals. For instance, KOP and CHOR, lying on the opposite river banks, were not differentiated (Fst = 0.006). Similarly, no significant differentiation was found between sites SUR and ZEL (Fst = 0.010), almost 8 km distant, but connected by an artificial canal. In contrast, the two urban samples STR and ROH were significantly differentiated (Fst = 0.070, P < 0.001) in spite of their close geographic proximity (420 m). Similarly, geographically close ZLP and KAL (Fst = 0.089, P < 0.001) and ZLP and VAJ (Fst = 0.062, P < 0.001), isolated from each other by a four-lane highway and buildings, were genetically differentiated.
Mantel test revealed no correlation between genetic (Fst) and geographic distances (b-slope = −0.002, a-intercept = 0.059, r 2 = 0.002, P = 0.857), corroborating the fact that geographically close populations were often as genetically differentiated as distant populations.
GENELAND assigned all individuals to three clusters. Cluster 1 comprised localities near the Lesser Danube and canals (BIS, IVD, IVS, KAL, SPR, SUR, VAJ, VAK, ZEL, ZLP). Cluster 2 was composed of individuals from urban localities ROH and STR, plus VRA. Finally, all marsh frogs from localities situated near the Danube River (DEV, CHOR, KOP, DRA) were assigned to cluster 3 (Figs. 1 and 3).

Maps of posterior probability of population membership using GENELAND (above) and a bar plot of the proportional membership of individuals for each of the six inferred clusters using BAPS (below)
BAPS assigned all individuals to six clusters with a probability of 0.999. Clusters 1, 2, and 3 were mostly identical with those defined by GENELAND except that individuals from BIS, VRA, and ZLP were assigned to separate clusters 4, 5, and 6 (Figs. 1 and 3). When the BAPS analysis was performed with a fixed number of clusters, K = 3, assignment results were consistent with GENELAND except BIS that was assigned to the Danube cluster. Eight out of the 17 localities (BIS, VRA, ZLP, IVD, KAL, ROH, STR, DRA) showed uniform cluster assignment across individuals. Two individuals from DEV and one individual from IVS were assigned to clusters 4 (BIS) and 6 (ZLP), respectively, with a probability higher than 0.80. Only 3.64 % of individuals had unclear assignments to clusters.
Discussion
Our study suggests that landscape structure has a substantial effect on population genetic structure of the water frog species P. ridibundus. Spatially isolated urban breeding sites reveal lower genetic diversity, lower effective population sizes, and higher genetic differentiation in comparison with sites not exposed to intensive urbanization. Breeding sites in proximity to the Danube, its branches, and artificial canals are genetically more homogenous. Population structure reflects higher genetic connectivity within water paths than between them, indicating limits to overland dispersal. The present-day distribution of marsh frog genetic structure also reflects landscape changes that took place during the last century, resulting from urbanization, canalization of the Danube, and the creation of artificial ponds and canals.
Genetic diversity and effective population size
The lowest allelic diversity measured as an average number of alleles (Na) and allelic richness (AR) were observed in urban localities ROH, STR, ZLP, and KAL, but the loss of expected heterozygosity (He) in urban samples was not so considerable. The loss of allelic diversity in subdivided populations is expected to be faster than the loss of expected heterozygosity, which occurs mainly when populations are extremely small (Allendorf and Luikart 2007). When we compare the two urban locality samples ROH and STR with the Danube localities (DEV, DRA, KOP, CHOR), with which they were connected by the river branch about a century (37 frog generations) ago, we find that Na and AR are 35.9 % and 33.1 % lower, while He is only 10.9 % lower. Our findings are thus in agreement with theoretical expectations that the loss of allelic diversity in isolated populations is faster and earlier detectable than the loss of heterozygosity (Allendorf and Luikart 2007; Broquet et al. 2010). In contrast to urban sites, higher values of allelic diversity and heterozygosity were observed in localities associated with the Danube River and artificial canals most likely due to extensive current or past gene flow. Our results add to the weight of evidence for lower genetic diversity in amphibians living in human-dominated and fragmented landscape (Andersen et al. 2004; Arens et al. 2007; Noël and Lapointe 2010; Noël et al. 2007).
A significant deficit of heterozygotes was detected in 10 out of 17 localities and can indicate either inbreeding or the presence of null alleles (Chybicki and Burczyk 2009). We suppose that a heterozygote deficit in our sampling sites was more likely caused by null alleles in specific loci than by inbreeding. First, F IS values corrected for the presence of null alleles were low, indicating that null alleles rather than inbreeding caused deviations from Hardy–Weinberg equilibrium. Second, a significant deficit of heterozygotes was found mainly in suburban populations and not in isolated and small urban populations as we could expect theoretically. It would seem that the effective population sizes of populations living in the urban localities STR, ROH, ZLP, and KAL are sufficient to avoid inbreeding.
In three localities (CHOR, DEV, KOP), P. ridibundus lives together with other water frog taxa, P. lessonae and a hybridogenetic hybrid P. esculentus (for details of hybridogenetic reproduction of water frogs, see Graf and Polls Pelaz 1989; Plötner 2005). P. ridibundus shows the highest number of alleles and expected heterozygosities in these three ponds. Despite both P. lessonae and P. esculentus are less abundant in CHOR, DEV, and KOP, we can infer that high genetic diversity in these three localities might be influenced by introgression of “lessonae” genes into the genepool of P. ridibundus mediated by hybrids. P. esculentus is a hybridogenetic hybrid forming “ridibundus” gametes in most of its range, including western Slovakia (Mikulíček and Kotlík 2001). Its mating with syntopic P. ridibundus may result in P. ridibundus progeny. If a hybrid forming “ridibundus” gametes with introgressed “lessonae” genes mates with P. ridibundus, the resulting P. ridibundus progeny may share “lessonae”-specific genetic traits. However, interspecific gene flow between P. ridibundus and P. lessonae in western Slovakia was either not documented (allozymes; Mikulíček and Kotlík 2001) or was very low (AFLP markers; Mikulíček, unpublished data). The finding that introgression of “lessonae”-specific genes into the genepool of P. ridibundus is an infrequent process can be inferred also from the absence (CHOR) or low rate (DEV, KOP) of linkage disequilibria (comparable to other populations) found in the course of this study. If the rate of introgression would be high, the influx of “lessonae” genes into the genepool of P. ridibundus should generate increased linkage disequilibria (Szymura and Barton 1986; Goodman et al. 1999; Macholán et al. 2007). Thus, we argue that low rate of introgression would have a lower impact on genetic diversity of P. ridibundus in Bratislava, in contrast to limited landscape permeability decreasing genetic diversity and increasing genetic differentiation between urban localities.
Effective population size (Ne) is defined as the size of an ideal population that has the same rate of genetic drift as the observed population (Allendorf and Luikart 2007). In this study, we used two single-sample estimators of contemporary Ne—the linkage disequilibrium (LD) method (Hill 1981) and approximate Bayesian computation (ABC) based on summary statistics (Tallmon et al. 2008). In general, both methods provided similar and correlated values, although Ne calculated by the ABC approach was generally lower. Effective population size estimates in studied localities were similar to values found in P. ridibundus in Britain (Ne = 15.8–48.4 individuals, Zeisset and Beebee 2003). In our study, three sites (IVD, VRA, VAK) revealed high LD and consequently low LD-based Ne. The principle of the LD method is that as Ne decreases, genetic drift with few randomly mating individuals generates nonrandom associations among alleles at different loci, i.e., linkage disequilibrium. Estimation of Ne based on LD requires that the source of LD is derived from small Ne. However, in natural populations LD between unlinked loci can be generated by other processes, such as population substructure, immigration, extensive inbreeding, epistatic selection, and overlapping generations (Luikart et al. 2010). We suggest that high LD in some studied populations were likely influenced by factors other than low Ne and therefore the ABC method may be a better estimate of Ne in these populations.
Genetic differentiation and landscape permeability
Marsh frogs are highly aquatic species spending most of their life in or near water. They occupy permanent ponds and their movement though the terrestrial environment is limited. Taking into account habitat preferences, limited dispersal, strong site fidelity, and homing (Holenweg Peter 2001; Holenweg Peter et al. 2001), it is not surprising that genetic differentiation was detected between most of the breeding sites. Global genetic differentiation measured by Fst in this study was relatively low (Fst = 0.056), but statistically significant. Relatively low values of Fst reflect the limited differentiation of these populations at the small geographic scale and the high microsatellite diversity. Studies of Arioli et al. (2010) and Christiansen and Reyer (2011) found a positive correlation between geographic and genetic distances (an isolation by distance pattern), not shown at the scale of the current study. The localities we sampled in the Bratislava city and its vicinity are likely not in migration–drift equilibrium due to historical factors including the colonization of newly formed ponds (in the vicinity of the Šúrsky canal) and establishment of man-made barriers preventing gene flow.
Bayesian methods assigned each individual to one of three clusters, associated with the river Danube (CHOR, DEV, DRA, KOP, partially BIS), the lost Mill side channel (ROH, STR, VRA), and to the Šúrsky canal (the rest sites) built in the 1940s (Figs. 1 and 2). On the basis of these results, it could be assumed that gene flow among breeding sites within water paths is higher than gene flow between them and that the population genetic structure is influenced by historical landscape characterized by the presence of the river branches. Notable locality with genetics reflecting more historical than present-day landscape structure is the VRA, lying near the Lesser Danube, but genetically similar to urban sites STR and ROH (GENELAND). All these three sites were associated with the lost Mill channel flowing from the Danube to the Lesser Danube up to the end of nineteenth century. The lower section of the Mill channel was connected as a backwater of the Lesser Danube until the beginning of 1970s (Pišút 2002). This suggests the VRA sample retains genetic similarity to urban localities it has likely been separated from for 40 years (13 frog generations).
Marsh frogs probably disperse more readily through water (canals, rivers and their braids) than overland. Breeding ponds situated near the water corridors thus should not be necessarily equivalent to genetically distinct units and whereas ponds in close proximity isolated by man-made barriers may have a different genetic structure. Sites SUR and ZEL, about 8 km distant but connected by the artificial canal, are genetically not differentiated. Although marsh frogs have limited dispersal abilities in the terrestrial environment, they can disperse up to several hundred meters in a continuous habitat. The maximal distance recorded for the marsh frog during a field study of Holenweg Peter (2001) was 1,760 m (see also Smith and Green 2005). According to our results based on genetic differentiation of locality samples (Fst), dispersal distance taking into account the use of water corridors seems a better predictor for gene flow. Our results are in agreement with other studies, which showed that water corridors facilitate dispersal and gene flow between breeding sites of tree frogs and pool frogs (Ficetola and De Bernardi 2004), the salamanders of the families Ambystomatidae and Dicamptodontidae (Spear et al. 2005; Purrenhage et al. 2009; Mullen et al. 2010) and the Great plains toads (Jungels et al. 2010). On the other hand, urban areas and busy highways represent barriers to gene flow, allowing localities distant by only several hundred meters, to genetically differentiate: STR and ROH are only 420 m apart but genetically highly differentiated. In general, urban land cover and roads provide high landscape resistance and have been identified as important barriers to gene flow in many species of amphibians (Vos et al. 2001; Lesbarreres et al. 2006; Goldberg and Waits 2010; Safner et al. 2011).
The 300-m-wide Danube River with its strong current is definitely not a suitable habitat for active marsh frog dispersal, but passive transport during floods may explain the low genetic differentiation of breeding sites lying near the river (on the same as well as on opposite banks). Our results are in agreement with other population genetics studies, which showed that the rivers are not barriers for amphibians (Gascon et al. 1998; Spear et al. 2005; Jungels et al. 2010). In contrast, Marsh et al. (2007) found that even streams contributed to population genetic differentiation of the low dispersal salamander Plethodon cinereus. However, other studies either provided an ambiguous outcome that the rivers are barriers to gene flow between amphibian populations (Zhao et al. 2009) or found large rivers playing an important role in a biogeographical pattern of frogs leading to the allopatric distribution of phylogenetically closely related species (Zeisset and Beebee 2008). It could be concluded that the effect of rivers on genetic differentiation of amphibian populations is not uniform across studies and most likely depends on the characteristics of the rivers (their width, current, the distance from headwaters, flood occurrence) and species-specific traits (aquatic or terrestrial mode of life, dispersal abilities).
Urbanization represents a significant threat to biodiversity because it causes degradation and fragmentation of habitats, and changes species communities and structure of populations. This is particularly acute in areas where cities straddle rivers. Our study confirms that the river Danube shapes population structure of amphibians making their breeding sites genetically homogeneous. Historical and present-day changes associated with urbanization and the river regulations have a negative impact on genetic diversity of studied species. Water corridors between remnants of river braids seem to be necessary to keep populations interconnected.
References
Allendorf FW, Luikart G (2007) Conservation and the genetics of populations. Blackwell, Malden
Andersen LW, Fog K, Damgaard C (2004) Habitat fragmentation causes bottlenecks and inbreeding in the European tree frog (Hyla arborea). Proc R Soc B 271:1293–1302
arcanum (2004) The Ist Military mapping [Az első katonai felmérés]. DVD ROM. HM Hadtörténeti Intézet és Múzeum Térképtára and Arcanum Adatbázis Kft., Budapest
Arens P, van der Sluis T, van der Sluis WPC, Vosman B, Smulders MJM (2007) Genetic population differentiation and connectivity among fragmented Moor frog (Rana arvalis) populations in The Netherlands. Landsc Ecol 22:1489–1500
Arioli M, Jacob C, Reyer H-U (2010) Genetic diversity in water frog hybrids (Pelophylax esculentus) varies with population structure and geographic location. Mol Ecol 19:1814–1828
Berger L (1982) Hibernation of the European water frogs (Rana esculenta complex). Zool Pol 29:57–72
Broquet T, Angelone S, Jaquiery J, Joly P, Lena J-P, Lengagne T, Plenet S, Luquet E, Perrin N (2010) Genetic bottlenecks driven by population disconnection. Conserv Biol 24:1596–1605
Chapuis M-P, Estoup A (2007) Microsatellite null alleles and estimation of population differentiation. Mol Biol Evol 24:621–631
Christiansen D, Reyer H-U (2009) From clonal to sexual hybrids: genetic recombination via triploids in all-hybrid populations of water frogs. Evolution 63:1754–1768
Christiansen D, Reyer H-U (2011) Effects of geographic distance, sea barriers and habitat on the genetic structure and diversity of all-hybrid water frog populations. Heredity 106:25–36
Chybicki IJ, Burczyk J (2009) Simultaneous estimation of null alleles and inbreeding coefficients. J Hered 100:106–113
Corander J, Marttinen P (2006) Bayesian identification of admixture events using multi-locus molecular markers. Mol Ecol 15:2833–2843
Corander J, Waldmann P, Sillanpää MJ (2003) Bayesian analysis of genetic differentiation between populations. Genetics 163:367–374
Corander J, Sirén J, Arjas E (2008a) Bayesian spatial modelling of genetic population structure. Comput Stat 23:111–129
Corander J, Marttinen P, Sirén J, Tang J (2008b) Enhanced Bayesian modeling in BAPS software for learning genetic structures of populations. BMC Bioinforma 9:539
Cornuet JM, Luikart G (1996) Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144:2001–2014
Crow JF, Aoki K (1984) Group selection for a phylogenic behavioral trait: estimating the degree of population subdivision. Proc Natl Acad Sci USA 81:6073–6077
Delaney KS, Riley SPD, Fisher RN (2010) A rapid, strong, and convergent genetic response to urban habitat fragmentation in four divergent and widespread vertebrates. PLoS One 5:1–11
Ficetola GF, De Bernardi F (2004) Amphibians in a human-dominated landscape: the community structure is related to habitat features and isolation. Biol Cons 119:219–230
Garner TWJ, Gautschi B, Röthlisberger S, Reyer H-U (2000) A set of CA repeat microsatellite markers derived from the pool frog, Rana lessonae. Mol Ecol 9:2173–2175
Gascon C, Lougheed SC, Bogart JP (1998) Patterns of genetic population differentiation in four species of Amazonian frogs: a test of the riverine barrier hypothesis. Biotropica 30:104–119
Goldberg CS, Waits LP (2010) Comparative landscape genetics of two pond-breeding amphibian species in a highly modified agricultural landscape. Mol Ecol 19:3650–3663
Goodman SJ, Barton NH, Swanson G, Abernethy K, Pemberton JM (1999) Introgression through rare hybridization: a genetic study of a hybrid zone between Red and Sika Deer (genus Cervus) in Argyll, Scotland. Genetics 152:355–371
Goudet J (2001) FSTAT, a program to estimate and test gene diversities and fixation indices (version 2.9.3.2). http://www2.unil.ch/popgen/softwares/fstat.htm. Accessed 14 December 2011
Graf JD, Polls Pelaz M (1989) Evolutionary genetics of the Rana esculenta complex. In: Dawley RM, Bogart JP (eds) Evolution and ecology of unisexual vertebrates. New York State Museum Bulletin 466, pp 289–301
Guillot G, Mortier F, Estoup A (2005a) Geneland: a program for landscape genetics. Mol Ecol Notes 5:712–715
Guillot G, Estoup A, Mortier F, Cosson JF (2005b) A spatial statistical model for landscape genetics. Genetics 170:1261–1280
Hardy OJ, Vekemans X (2002) SPAGeDi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol Ecol Notes 2:618–620
Hill WG (1981) Estimation of effective population size from data on linkage disequilibrium. Genet Res 38:209–216
Holenweg Peter A-K (2001) Dispersal rates and distances in adult water frogs, Rana lessonae, Rana ridibunda, and their hybridogenetic associate Rana esculenta. Herpetologica 57:449–460
Holenweg Peter A-K, Reyer H-U, Tjetje GA (2001) Homing behavior of Rana lessonae, Rana ridibunda, and their hybridogenetic associate Rana esculenta after experimental displacement. Amphibia-Reptilia 22:475–480
Holsbeek G, Maes GE, De Meester L, Volckaert FAM (2009) Conservation of the introgressed European water frog complex using molecular tools. Mol Ecol 18:1071–1087
Holzhauer SIJ, Ekschmitt K, Sander A-C, Dauber J, Wolters V (2006) Effect of historic landscape change on the genetic structure of the bush-cricket Metrioptera roeseli. Landsc Ecol 21:891–899
Jarošík V, Konvička M, Pyšek P, Kadlec T, Beneš J (2011) Conservation in a city: do the same principles apply to different taxa? Biol Conserv 144:490–499
Jungels JM, Griffis-Kyle KL, Boeing WJ (2010) Low genetic differentiation among populations of the Great plains toad (Bufo cognatus) in Southern New Mexico. Copeia 2010(3):388–396
Keyghobadi N, Roland J, Matter SF, Strobeck C (2005) Among- and within-patch components of genetic diversity respond at different rates to habitat fragmentation: an empirical demonstration. Proc R Soc B 272:553–560
Lesbarreres D, Primmer CR, Lode T, Merilä J (2006) The effects of 20 years of highway presence on the genetic structure of Rana dalmatina populations. Ecoscience 13:531–538
Luikart G, Cornuet J-M (1998) Empirical evaluation of a test for identifying recently bottlenecked populations from allele frequency data. Conserv Biol 12:228–237
Luikart G, Ryman N, Tallmon DA, Schwartz MK, Allendorf FW (2010) Estimation of census and effective population sizes: the increasing usefulness of DNA-based approaches. Conserv Genet 11:355–373
Macholán M, Munclinger P, Šugerková M, Dufková P, Bímová B, Božíková E, Zima J, Piálek J (2007) Genetic analysis of autosomal and X-linked markers across a mouse hybrid zone. Evolution 61:746–771
Marsh DM, Pge RB, Hanlon TJ, Bareke H, Corritone R, Jetter N, Backman NG, Gardner K, Seifert DE, Cabe PR (2007) Ecological and genetic evidence that low-order streams inhibit dispersal by red-backed salamanders (Plethodon cinereus). Can J Zool 85:319–327
Mikulíček P, Kotlík P (2001) Two water frog populations from western Slovakia consisting of diploid females and diploid and triploid males of the hybridogenetic hybrid Rana esculenta (Anura, Ranidae). Mitt Mus Natkd Berl, Zool Reihe 77:59–64
Mullen LB, Woods HA, Schwartz MK, Sepulveda AJ, Lowe WH (2010) Scale-dependent genetic structure of the Idaho giant salamander (Dicamptodon aterrimus) in stream networks. Mol Ecol 19:898–909
Noël S, Lapointe F-J (2010) Urban conservation genetics: study of a terrestrial salamander in the city. Biol Cons 143:2823–2831
Noël S, Ouellet M, Galois P, Lapointe F-J (2007) Impact on urban fragmentation on the genetic structure of the eastern red-backed salamander. Conserv Genet 8:599–606
Orsini L, Corander J, Alasentie A, Hanski I (2008) Genetic spatial structure in a butterfly metapopulations correlates better with past than present demographic structure. Mol Ecol 17:2629–2642
Pagano A, Joly P, Plénet S, Lehman A, Grolet O (2001a) Breeding habitat partitioning in the Rana esculenta complex: the intermediate niche hypothesis supported. Ecoscience 8:294–300
Pagano A, Crochet PA, Graf JD, Joly P, Lodé T (2001b) Distribution and habitat use of water frog hybrid complexes in France. Global Ecol Biogeogr 10:433–441
Pavlacky DC Jr, Goldizen AW, Prentis PJ, Nicholls JA, Lowe AJ (2009) A landscape genetics approach for quantifying the relative influence of historic and contemporary habitat heterogeneity on the genetic connectivity of a rainforest bird. Mol Ecol 18:2945–2960
Peakall ROD, Smouse PE (2006) GenAlEx6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6:288–295
Pillsbury FC, Miller JR (2008) Habitat and landscape characteristics underlying anuran community structure along an urban-rural gradient. Ecol Appl 18:1107–1118
Piry S, Luikart G, Cornuet J-M (1999) BOTTLENECK: a computer program for detecting recent reductions in effective population size using allele frequency data. J Hered 90:502–503
Pišút P (2002) Channel evolution of the pre-channelized Danube River in Bratislava, Slovakia (1712–1886). Earth Surf Proc Land 27:369–390
Pišút P (2011) The 1787 flood of the River Danube in Bratislava. Geogr J 63:87–109
Plénet S, Pagano A, Joly P, Fouillet P (2000) Variation of plastic responses to oxygen availability within the hybridogenetic Rana esculenta complex. J Evol Biol 13:20–28
Plötner J (2005) Die westpaläarktischen Wasserfrösche. Von Märtyrern der Wissenschaft zur biologischen Sensation. Zeitschrift für Feldherpetologie, Beiheft 9, Laurenti, Bielefeld, Germany
Purrenhage JL, Niewiarowski PH, Moore FB-G (2009) Population structure of spotted salamanders (Ambystoma maculatum) in a fragmented landscape. Mol Ecol 18:235–247
Richmond JQ, Reid DT, Ashton KG, Zamudio KR (2009) Delayed genetic effects of habitat fragmentation on the ecologically specialized Florida sand skink (Plestiodon reynoldsi). Conserv Genet 10:1281–1297
Rybacki M, Berger L (1994) Distribution and ecology of water frogs in Poland. Zool Pol 39:293–303
Safner T, Miaud C, Gaggiotti O, Decout S, Rioux D, Zundel S, Manel S (2011) Combining demography and genetic analysis to assess the population structure of an amphibian in a human-dominated landscape. Conserv Genet 12:161–173
Schmeller DS, Pagano A, Plénet S, Veith M (2007) Introducing water frogs—is there a risk for indigenous species in France? C R Biologies 330:684–690
Smith MA, Green DM (2005) Dispersal and the metapopulations paradigm in amphibian ecology and conservation: are all amphibian populations metapopulations? Ecography 28:110–128
Socha M, Ogielska M (2010) Age structure, size and growth rate of water frogs from central European natural Pelophylax ridibundus–Pelophylax esculentus mixed populations estimated by skeletochronology. Amphibia-Reptilia 31:239–250
Spear SF, Peterson CR, Matocq MD, Storfer A (2005) Landscape genetics of the blotched tiger salamander (Ambystoma tigrinum melanostictum). Mol Ecol 14:2553–2564
Sutherland RW, Dunning PR, Baker WM (2010) Amphibian encounter rates on roads with different amounts of traffic and urbanization. Conserv Biol 24:1626–1635
Szymura JM, Barton NH (1986) Genetic analysis of a hybrid zone between the fire-bellied toads, Bombina bombina and B. variegata, near Cracow in southern Poland. Evolution 40:1141–1159
Tallmon DA, Koyuk A, Luikart G, Beaumont MA (2008) ONeSAMP: a program to estimate effective population size using approximate Bayesian computation. Mol Ecol Res 8:299–301
van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P (2004) Micro-Checker: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538
Vos CC, Antonise-De Jong AG, Goedhart PW, Smulders MJM (2001) Genetic similarity as a measure for connectivity between fragmented populations of the moor frog (Rana arvalis). Heredity 86:598–608
Waples RS (2006) A bias correction for estimates of effective population size based on linkage disequilibrium at unlinked gene loci. Conserv Genet 7:167–184
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Whitlock MC, McCauley DE (1999) Indirect measures of gene flow and migration: Fst ≠ 1/(4Nm + 1). Heredity 82:117–125
Zeisset I, Beebee TJC (2003) Population genetics of a successful invader: the marsh frog Rana ridibunda in Britain. Mol Ecol 12:639–646
Zeisset I, Beebee TJC (2008) Amphibian phylogeography: a model for understanding historical aspects of species distributions. Heredity 101:109–119
Zeisset I, Rowe G, Beebee TJC (2000) Polymerase chain reaction primer for microsatellite loci in the north European water frogs Rana ridibunda and R. lessonae. Mol Ecol 9:1173–1174
Zhao S, Dai Q, Fu J (2009) Do rivers function as a genetic barriers for the plateau wood frog at high elevations? J Zool 279:270–276
Acknowledgments
We are grateful to Heinz-Ulrich Reyer, Stuart Baird, David Jandzík, and Peter Puchala for their suggestions and valuable comments which improved the manuscript. We thank Michal Zaťovič and Jana Zajícová for laboratory assistance. This work was carried out under permit 1323/527/05-5.1 (Ministry of Environment of the Slovak Republic). The study was partially supported by the Grant Agency of the Slovak Republic VEGA (projects 1/0491/10 and 1/0362/09).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by C. Gortázar
Electronic supplementary material
Below is the link to the electronic supplementary material.
Appendix 1
(XLSX 20 kb)
Appendix 2
(XLSX 13 kb)
Rights and permissions
About this article
Cite this article
Mikulíček, P., Pišút, P. Genetic structure of the marsh frog (Pelophylax ridibundus) populations in urban landscape. Eur J Wildl Res 58, 833–845 (2012). https://doi.org/10.1007/s10344-012-0631-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10344-012-0631-5