
Abstract
Understanding populational genetic diversity is crucial for making proper decisions about conservation and sustainable species management. In this work, we overviewed the conservation genetics of economically exploited tree species that are vulnerable to extinction in the Atlantic Forest of Brazil. For this, data on genetic parameters from research on 10 species of evergreen trees dwelling in that environment were compiled. Genetic variability differences between young and adults were assessed when data were available for both stages. Very low values observed for the probability of identity (PI) suggested that the SSR markers used had sufficient statistical power to consistently evaluate genetic variability of the populations. An innovative analytical approach using linear mixed-effect models revealed an integrated influence of the number of individuals sampled per population and the number of SSR markers on the output of the genetic estimators assessed. A large number of different alleles (NA) were observed in four out of the 10 species, indicating these populations may still hold unique and rare alleles. HO was smaller than HE for all but one of the studied species, suggesting higher numbers of homozygotes than expected. Comparison of data between ontogenic stages suggested that the time frame of habitat loss and fragmentation was not yet sufficient to cause significant loss of genetic diversity and differentiate populations. Our findings are discussed considering that intensity and duration of selective lodging and economic exploitation appear to be crucial for the underlying ecological patterns and for the definition of proper diversity conservation strategies.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Among all the existing biodiversity on earth, Brazil is characterized as one of the countries with the greatest diversity of species (Tabarelli et al. 2010). The Atlantic Forest of southeastern Bahia is the most representative of this biome in the northeast region of the country, being one of the main areas of endemism and considered a biodiversity hotspot on the planet (Myers et. al.2000; Martini et al. 2007; Bosa et al. 2015). However, the forest remnants in this region are immersed in anthropic-driven landscapes, in a mosaic pattern of matrix consisting of “cabrucas” (shaded-cacao plantations), “capoeiras” (newly recovering forest areas) in different stages of regeneration, and pasture areas scattered among them (Cassano et al. 2009; Piasentin and Gois 2016; Rolim et al. 2016). With the increasing anthropic actions in recent decades in the Atlantic Forest biome, resulting in habitat loss and forest fragmentation, those remnants have become progressively small and isolated (Santo-Silva et al. 2016; Arroyo-Rodriguez et al. 2020). As a consequence, gene flow among remnant populations is severely affected, potentially causing inbreeding and reducing the genetic diversity of the surviving tree species (Lowe et al. 2005; Lander et al. 2011; Haddad et al. 2015).
In this context of a degraded environment, the structural modifications in habitats, as well as the general characteristics of the forest fragments can cause long-term changes in the genetic constitution of tree species with long life-cycles (Sarmento and Villela 2010). Thus, the levels of genetic variability of trees subjected to these unfavorable conditions are informative indicators of the actual and current situation of tropical forests under anthropic influence (Diniz-Filho et al. 2009). Understanding these conditions is useful for the development of strategies for the diversity conservation (Spear et al. 2010; Sommer et al. 2013), aiming at a long-term maintenance of tree species with ecological and commercial values.
Habitat loss and forest fragmentation are of particular concern for tropical regions, such as the Brazilian Atlantic Forest, where only 28% of the original area is comprised by forest remnants (Rezende et al. 2018). Such processes tend to trigger a reduction in population sizes, which leads to genetic effects that can affect both the evolutionary processes and the definition of conservation strategies (Browne and Karubian, 2018). The persistence of populations can be negatively impacted by reductions in population density and by frequent processes of genetic structuring (Collevatti and Hay 2011; Ortego et al. 2012). The Atlantic Forest remnants display different levels of vegetation cover and fragmentation caused by a variety of anthropogenic disturbances, including selective logging to obtain wood, cleaning of areas for establishment of other crops (often by fire), extraction of fruits and seeds for use in food and health, etc. (Gerstner et al. 2014; Albuquerque et al. 2018). Moreover, species with different ecological requirements are expected to respond in different ways to these anthropogenic pressures (Kunz et al. 2014).
By means of molecular markers, it is possible to obtain refined information on the genetic diversity of forest species in natural populations. A frequently used tool in conservation genetics studies is microsatellite or SSRs (simple sequence repeats) markers (Rafalski 1996; Ouinsavi et al. 2009). These short DNA stretches consist of tandemly repeated di-, tri-, tetra- or penta-nucleotide motifs, which have been found in all eukaryotic species that have been scrutinized (Tautz and Renz 1984); different genotypes frequently show length polymorphisms in the SSR amplicons due to variation of the number of repeated motifs, which allow these markers to appear as co-dominant alleles, a very useful and convenient attribute for description of natural populations (Kumar et al. 2009). Through SSRs, it has been possible to address and quantify a series of impacts on populations of tree species associated with human exploitation (Nair 2010; Heuertz et al. 2020; Coelho et al. 2020; Borges et al. 2020). This includes the understanding of the genetic diversity within and among populations, the quantification of pollen and seed dispersal distances (Atsushi et al. 2021), and the detection of asexual reproduction in natural populations (Zucchi et al. 2005). These studies aimed to understand the drivers genetic in fragmented forests (Poorter et al. 2015), which is critical for development of biodiversity conservation strategies (Bambach 2006; Haddad et al. 2015). Special interest has been devoted to species that present some form of economic exploitation, such as wood, fibers, fruits, and seeds (Sampaio and Salazar 2007).
In recent years, several studies using microsatellite markers have investigated the effects of different anthropogenic disturbances on gene flow, diversity, and genetic structure in Atlantic Forest tree species (Buzatti et al. 2012; Martins et al. 2016; Carvalho et al. 2015, 2017; Santos et al. 2015, 2016, 2019; Soares et al. 2019; Borges et al. 2020; Waqar et al. 2021). Overall, these studies revealed that the species still have high genetic variability, which is being eroded by selective logging (Soares et al. 2019), deforestation (Borges et al. 2020) and defaunation (Carvalho et al. 2017). Furthermore, deforestation has been reported to reduce contemporary gene flow (Santos et al. 2016), increase genetic structuring, and reduce genetic variability between generations (Martins et al. 2016; Waqar et al. 2021). In this study, we used the SSR data from these studies conducted in northeast Atlantic forests and from studies conducted in southern regions of this biome. We provide an overview of the remaining genetic diversity of economically exploited tree species in the Atlantic Forest, aiming to furnish an updated picture of general conditions for their conservation. To achieve this goal, this research was guided by the following biological question: what is known about the remaining genetic diversity of economically exploited tree species in the Atlantic Forest? To answer it, we employed a set of genotyping data generated with microsatellite markers in populations of 10 different species in the Atlantic Forest in northeastern and southeastern Brazil. Compatible and innovative genetic and statistical analyses were presented.
Methods
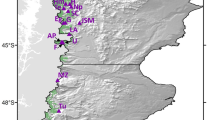
To offer an integrated view of the genetic variability for economically exploited evergreen tree species in the Brazilian Atlantic Forest, we consolidated microsatellite genotype data from previous studies of our group (dissertations and scientific articles, S1). In total, raw microsatellite data from natural populations for 10 species were obtained. The sampling area encompassed six Brazilian states (Bahia, Espírito Santo, Minas Gerais, São Paulo, Paraná and Mato Grosso do Sul), covering a large portion of the country's Atlantic coast, in a range of latitude between 10º 25′ 52'' and 26º 27′ 29'' S, although the majority were located in the southeastern Bahia State (Fig. 1). Out of these 10 species, four displayed data for the ontogenetic stages of adults and juveniles, whereas six had data only for adults (Table 1). We consider juveniles to be individuals who are already well established in the population, according to the classification of primary studies (for more details, see table S1). We then compile the sample size per population, number of sampled populations, and the number of SSR loci for the datasets (Table 1). Subsequently, we carried out bibliographic research on the reproductive system, pollination and dispersion mechanism of the seeds, type of economic exploitation, number of sampled populations inserted in conservation units, threat status and geographic distribution of the 10 tree species evaluated here (Table S1).

Map of Brazil with the remnants of the Atlantic Forest, highlighting the states and the spots where the populations of the tree species in this study were collected
From these data, possible genotyping errors and null alleles were verified using the Micro-Checker 2.2.3 (Van Oosterhout et al. 2004), and the statistical power of each set of SSR loci was verified by the probability of identity (PI) parameter, using GenAlex 6.5 (Peakall and Smouse 2012). PI is the chance of two individuals in a given population having the same genotype in an offspring. GenAlex was also used to estimate the average number of different alleles (NA), average number of effective alleles (NE), expected heterozygosity (HE) and observed heterozygosity (HO) for the 10 species. Estimating NE becomes especially relevant to compare populations/species where the number and distribution of alleles differ drastically (Weir 1990). As NE reflects the number of equally frequent alleles that would be required to achieve a given level of gene diversity, for any number of alleles, expected heterozygosity is highest when all allele frequencies are equal (Weir 1990). The inbreeding coefficient (f) and the 95% confidence interval, obtained with 10,000 bootstraps, were estimated with the divBasic function of the diveRsity library (Keenan et al. 2013) in the ‘R’ program (R Core Team). Finally, the analysis of molecular variance (AMOVA) was performed in GenAlex to estimate the partition of genetic variability within individuals (genotypes), among individuals within populations, and among populations. It is important to emphasize that we evaluated the number of alleles, because this estimator represents a global average of all the populations for each species under assessment, making it suitable to describe the global pattern of genetic diversity in each species, which is the central objective of our study. Finally, considering that each species has distinct biological, ecological and evolutionary history, which are characteristics that certainly influence genetic estimators, we did not make comparisons between species. To the four species with adult and juvenile data, t-tests were performed in R to compare average estimates for NA, NE, f, HE and HO. In addition, we estimated the magnitude of the differences between adult and juveniles, by calculating the percent decrease or increase in diversity between the two stages.
To assess the influence of the number of sampled individuals per population and the number of SSR loci per species on genetic estimators, we used linear mixed-effect models of the 'lme4' package in the R program (Bates et al. 2015). As this type of model requires compliance with the assumptions of normality and homoscedasticity of the residuals, a log scale transformation was performed. An independent model was created for each genetic estimator, as follows:
Genetic estimator ~ number of microsatellite loci + (1 | species).
Genetic estimator ~ number of individuals per population + (1 | species).
In these models, the genetic estimator is the response variable; the number of microsatellite loci or the number of individuals per population is the fixed effect; the terms in parentheses are the random effect variables, with the number “1” indicating that the intercept is random between observations of each species.
The models were tested comparatively to verify the most plausible ones through the Akaike Information Criterion corrected for small samples (AICC) and the Akaike Weights (Anderson 2008). The model that presented the smallest AICC with at least two units of difference to other models and with the largest Akaike Weights, was considered the most plausible. Using the ‘ggplot2’ package in R (Wickham 2016), only the models that met the established criteria for plausibility were graphically expressed, except when the most plausible was the null model.
Results
In this study, we analyzed microsatellite data from 10 tree species sampled in forest remnants widely distributed across the Atlantic Forest range (Fig. 1). Sample sizes varied across species, from 2 to 9 adult populations for 10 species and from 2 to 18 juvenile populations for four species (Table 1). Total sample sizes varied from 64 to 619 individuals and the number of microsatellite loci per species varied from 7 to 17 (Table 1).
Genetic diversity of adults and juveniles
The probability of identity (PI) was quite low for all the species, ranging from 2.4 × 10–5 for adults of Myroxylon peruiferum to 3.1 × 10–18 in Manilkara maxima (Table 1), indicating the genetic estimates shown in this study are statistically robust, as the chance of observing repeated genotypes is negligible. The average number of different alleles (NA) was 8.0 for adults, varying from 20.55 for Copaifera langsdorffii to 4.0 for Centrolobium tomentosum; the global average of number of effective alleles (NE) was 4.33, with these same species presented the highest estimates of NE (10.64) and HE (0.90) and the lowest estimates of NE (2.44) and HE (0.49) for adults, respectively. As for the observed heterozygosity (HO) for the adult stage, the values ranged between 0.37 (Plathymenia reticulata) and 0.73 (Manilkara multifida). The inbreeding coefficient (f) for adults was overall moderate (mean = 0.16), ranging from –0.27 (Melanoxylon brauna) to 0.34 (P. reticulata) (Table 1). For the juveniles, the overall PI was also quite low, ranging from 1.6 × 10–8 for Cariniana legalis to 4.5 × 10–19 for M. maxima (Table 1). For the four species with data for juveniles, the global mean of NA was 8.66, ranging from 8.0 alleles for M. brauna to 9.0 alleles for M. maxima; on the other hand, the number of effective alleles had an average of 4.69, with these same species presented the lowest (4.51) and the highest estimates of NE (4.84), respectively. The HE values varied between 0.70 (M. brauna) and 0.74 (M. maxima) with an average of 0.71; the HO ranged between 0.51 (M. multifida) and 0.67 (M. maxima) with a mean of 0.59; and the f coefficient ranged from 0.09 for M. maxima to 0.28 for M. multifida, with an average of 0.18 for the four species (Table 1).
The analysis molecular of variance (AMOVA) has demonstrated that the largest portion of genetic diversity occurs within individuals (genotypes), for both adults and juveniles, with the next relevant proportion of genetic diversity being found among individuals (Fig. 2). For adults, C. echinata showed the smallest proportion of variability within individuals (39%), while C. langsdorffii had the highest proportion (75%). For juveniles, the smallest partition of genetic diversity within individuals was observed for C. legalis (54%) and the largest for M. maxima (78%) (Fig. 2). With regards to the partition of diversity among individuals and among populations, two species displayed a distinct pattern than the others, i.e., adults of C. echinata and both stages of M. brauna showed higher genetic variation among populations than among individuals (Fig. 2).

Partition of genetic diversity components after analysis of molecular variance (AMOVA) for adult and juvenile stages of the tree species under study
Comparative genetics between the adults and the juveniles
Comparisons of genetic diversity patterns between ontogenic stages are useful to provide insights on historical events and possible courses of the genetic variability of a tree species through time (Santos et al. 2016; Caballero et al. 2019). Considering the four species with genetic data for adult and juvenile trees, pairwise tests of means indicated significant increases in NA, NE, f and HE for M. brauna and reductions in HO and HE for M. multifida for juveniles in relation to adults. The other genetic estimators did not significantly differ between the two stages (Table 1).
Despite the absence of statistical significance for all parameters, the assessment of the magnitudes of differences between the ontogenic stages (generations) can provide a view toward possible trends in genetic variation occurring in the species. It was observed a decrease in the average number of individuals per population (Ind/Pop) for M. multifida and an increase for C. legalis and M. brauna species (Table2). The average NA and NE showed a reduction only for the M. multifida, with increases for the other species, with NA ranging between 8 and 45% and NE between 4.11 to 37.61% (Table 2). The observed heterozygosity (Ho) showed a greater reduction for the M. multifida species and a less marked increase for C. legalis; the expected heterozygosity (HE) was reduced for M. multifida and sharply increased for M. brauna. Finally, the f coefficient values showed a reduction for the C. legalis and M. maxima and a large increase for the M. brauna and M. multifida (Table 2). In general, we observed that the variations in the increases of the genetic parameters values from adults to juvenile individuals were of greater magnitudes than the reductions (Table 2); the greatest positive variation (increase) between the parameters occurred for the f coefficient for the M. brauna species (119.3%) and the greatest negative variation (decrease) occurred for Ho, for the M. multifida species (29.85%).
Descriptive models for the genetic parameters based on the sampling characteristics
In general, finding model(s) to allow one to describe how certain variables/parameters interact is always useful for predictive purposes. By means of employing the sampling characteristics of numbers of SSR markers or numbers of individuals per population as fixed effects, a series of linear mixed-effects models were generated, using the genetic estimators (Table 1) as the response variables (see methods). The results showed that a model with the number of individuals per population was the most plausible to explain the NA estimator for the adult and juvenile stages (Fig. 3; Table S2 – supplementary material). This model evidenced a positive relationship between the variables, i.e., when the number of individuals increases, so does the number of different alleles (Fig. 3). It is also important to highlight that the models in which the number of markers interacted with the number of individuals per population was also more plausible to explain the number of alleles than the null model, presenting lower AICc values and higher Akaike Weights values for adult and juvenile individuals; for the other genetic estimators (NE, He, Ho and f), the null model appeared as the most plausible.

Effect of the number of individuals sampled in the population on the number of alleles for the ontogenetic stage of adults (a) and juvenile (b) of the tree species under analysis. Black lines correspond to the best-fit model (see Table S2)
Discussion
With the reduction in native forests mostly caused by anthropic activity (which includes logging and fire), the survival and maintenance of tree species depend on adequate sustainable management strategies to avoid higher risks of extinction. The Atlantic Forest is a relevant area of endemism and a biodiversity hotspot (Myers et al.2000; Bosa et al. 2015), which has faced forest loss and fragmentation, leading to conditions of small and isolated remnants (Nascimento and Laurence 2006; Rezende et al. 2018). Under these degrading conditions, gene flow between tree populations can be prevented, which can affect genetic constitution and diversity of surviving species (Sarmento and Villela 2010; Haddad et al. 2015; de Azevedo 2017). Hence, understanding the levels of genetic variability of trees under these conditions can help developing more consistent strategies for their conservation (Spear et al. 2010; Sommer et al. 2013), and so, a long-term maintenance of tree species with commercial and ecological values. From the compilation of information on the genetic diversity of 10 economically exploited species in the Brazilian Atlantic Forest, we verified that six of those species are endemic and classified as threatened by extinction. It was observed from the genetic parameters assessed that populations have maintained an overall high genetic variability, even with a history of recent exploration. Given the longevity of these trees, generations considered to be parental still have showed genes with potential to boost the levels of observed heterozygosity in this work (Caballero et al. 2019; Carvalho et al. 2010). Our results suggest that the presence of individuals of these species has preceded the processes of fragmentation and loss of habitat in the regions they are located (Newbold et al. 2015; Miraldo et al. 2016).
As it has been shown for other species (e.g., Paetkau et al. 1995; Ganzhorn 2015; Leal et al. 2014), our results of very low probabilities of identity (PI) found for the two ontogenetic stages evaluated (Table 1) clearly indicated that finding two identical genotypes by descent, in all assessed tree populations, was a practically null possibility. In addition, despite a large variation among the studied species in terms of the number of SSR used (Table 1), these markers allowed a proper discrimination of the individuals, and so, an adequate evaluation of the genetic variability of these species for adults, and for the juvenile stages when available (Collevati et al. 1999; Almeida-Rocha et al. 2020). Coupling these results with the analysis of other genetic parameters, a surprisingly high genetic diversity appears to be yet existing in the arboreal individuals of these species from the Atlantic Forest remnants of Brazil. As discussed below, these findings bring hope for the development of appropriate conservation strategies for these economically and environmentally relevant resources.
The number of different alleles (NA) identified for the used markers is another variable that helps describing the levels of diversity in populations, so that assessing this genetic parameter is considered as being essential to understand how species respond to environmental changes; high numbers of alleles in plant populations tend to promote a greater adaptive potential for the corresponding species (Guidugli 2011). In this regard, the 10 species showed a great variation in NA for the adult stage, with some species such as C. langsdorffii showing more than 20 different alleles (Table 1). This suggests even further that populations of this tree species do display high levels of genetic variability and adaptive potential (Sebbenn et al. 2011). As seen below, its type of commercial exploration offers a plausible explanation for this feature, thereby linking its survival directly to a specific kind anthropic activity/management. On the other hand, it was observed that some species such as C. tomentosum presented low NA, suggesting possible restrictions to their adaptive potential. In terms of conservation, the number of different alleles has been considered a very informative parameter for hypervariable makers (e.g., SSR; Hollingsworth et al. 2005), since it is linked to effective size of populations; this parameter is associated with demographic changes in the past that might have affected not only neutral markers, but also genes related to adaptive traits (Petit et al. 1998). The evaluated species presented NE approximately half of NA, indicating an imbalance in allele frequencies, since alleles with frequencies far from the average contribute very little to the effective number of alleles (Weir 1990). This implies the fact that about half of the alleles may have a low frequency and be lost with the intensification of deforestation and illegal exploitation of these species. Alternatively, the frequency of certain alleles for given loci may be very high, and in extreme cases, may be fixed in the population, leading to the observed imbalance in allele frequencies. An example of this imbalance can be observed in the species Caesalpinia echinata, which has an NA of 7.14 and NE of 2.79, with several alleles with high or fixed frequencies, while other alleles are very rare or absent in populations (Fig.S1, Supplementary material). Thus, this calamitous reduction in the NE will probably lead to a reduction in gene diversity in the next generations (Weir 1990), which may compromise the adaptive capacity of species in a scenario of anthropic environmental changes.
In relation to the heterozygosity levels for adults, the results of HO being lower than HE for almost all studied species (except M. brauna), is pointing to a greater number of homozygotes in relation to those in the Hardy–Weinberg model (Allendorf and Luikart 2006). Even in a relatively short timeframe of exploration, a loss of heterozygosity is possible, which can not only influence in the adaptation of a given species’ population, but also affect other interacting populations that are geographically close (Almeida-Rocha et al. 2020). The C. langsdorffii species, which is economically explored as an oil source for cosmetics industry (Stupp et al. 2008), showed the greatest genetic diversity for the adult stage considering both the highest value of HE and the second largest value of HO (Table 1). A likely explanation for this result would be the fact this species already presents a great conservation value, since the oil is extracted from the main trunk of adult individuals, which therefore helps maintaining the specimens (Duminil et al. 2013). At the other end of the heterozygosity rank, the P. reticulata species showed the lowest HO and HE values, coupled with the highest inbreeding coefficient (f) among the adult species (Table 1). This suggests this species had likely suffered the most from the effects of anthropic activities among those under assessment in this study. Another hypothesis to explain those low values of HE and HO is linked to the reproductive system of P. reticulata, in which its hermaphrodite flowers that undergo self-fertilization can favor the occurrence of inbreeding (Goulart et al. 2005). Thus, understanding aspects associated with the biology, ecology, and evolution of species can also be crucial to determine population genetic characteristics (Duminil et al. 2013). According to Tarazi et al. (2009), in a tree population isolated by the effect of fragmentation, the probability of crosses between related individuals (inbreeding) increases, thereby reducing the proportion of heterozygotes in the population. Nevertheless, although this could have theoretically happened, it is noteworthy that the 10 tree species of the Brazilian Atlantic Forest under study seem to have not been more intensively affected by such mechanism, as a considerable genetic diversity in their populations could be observed (Table 1).
We believe that the maintenance of such genetic variability, even with a recent history of high logging and forest fragmentation, appears to be related to the partitioning of this variability. As seen from the AMOVA, despite the differences among the 10 species (Fig. 2), the greatest proportion of genetic diversity showed to occur within individuals for all species evaluated. This highlights the importance of the factor ‘genotypes’ for the populational variability, which is in clear agreement with the overall levels of heterozygosity observed. Therefore, under these conditions, fewer individuals could maintain a large proportion of genetic diversity for these tree species, in a range that varied from as little as 39% for C. echinata (one of the species most explored since the begin of the country’s colonization time in 1500 a.d.; Cardoso et al. 2005; Melo et al. 2007) to 75% for C. langsdorffii (Fig. 2). The results so far are suggesting that these 10 species, particularly, have been able to maintain sufficient levels of open-pollination through time to assure heterozygosity, and so, variation at genotypic level (Paschoa et al. 2018). This is probably since these species are pollinated by animals, such as bees, which favors crossing and maintaining a certain level of genetic variability (see table S1). The species with the highest proportion of genotypic diversity also showed a generally greater genetic variability overall (Fig. 2; Table 1). Due to this observed pattern, it is suggested that the few adult individuals still inhabiting small fragments of the Atlantic Forest store a considerable genetic diversity for the species, and so, are very likely to be relevant sources of variability. This finding has a great relevance for the design of proper strategies for genetic conservation, mainly considering that most of the 10 species are currently threatened by extinction (Godoy et al. 2018). The results also brought forward a warning sign in the sense that the persistence of only a few individuals in a population, in a given area over time can lead to the crossing between related individuals, which is known to reduce genetic diversity and adaptive potential (Fietz et al. 2014).
Considering the four species with data for adults and juveniles’ trees, our descriptive population genetics demonstrated that effects associated with habitat loss, fragmentation and logging are noticeable, given the differences found between HE and HO (Tables 1 and 2). However, the fact that tree species currently in a vulnerable conservation state have yet shown similar levels of high genetic diversity might be explained by the notion that the increased rates of forest loss and fragmentation are more recent events if compared to their life cycles (Collevatti et al. 2001; Souza et al. 2012). Particularly for the M. maxima species, the small f coefficient estimated, coupled with the values of HE and HO, suggest that for this species, gene flow is occurring and enabling the exchange of important alleles for the maintenance of a high genetic diversity (Guidugli 2011). On the other hand, for the species M. multifida, the significantly lower HO value for the juveniles compared to adults (Tables 1 and 2), and the highest value of f among the juveniles’ populations of the four species point to a reduction in genetic variability between generations. It is possible that particular anthropic processes have led to specific patterns of M. multifida specimens’ loss within the fragments, thereby affecting the distribution of the remaining populations within and among the fragments, and so, the partitioning of diversity (Fig. 2). This has likely accentuated the genetic differentiation observed from adults to juveniles’ individuals with regards to variability among individuals and populations.
The way in which genetic variability is distributed within and among populations is usually related (i) to the pollen and/or seed dispersal syndrome, (ii) to the type of reproduction the species present, and (iii) to how the individuals are distributed within the considered populations (Sebbenn et al. 2011). The way in which populations of a species reproduce has a direct influence on the levels of inbreeding and relatedness of individuals in future generations (Guidugli 2011). Most species of long-lived woody trees with seeds dispersed by animals often maintain greater genetic variation within habitat patches than between patches (Hamrick et al.1992; Silva et al. 2013; De Kort et al. 2021). Species such as M. maxima, which showed the highest values of HO for both adults and juveniles (although with a noticeable decrease between these stages as in M. multifida – Table 1) was one with most of the genetic diversity identified being found within individuals (genotypes); similarly, adults from M. multifida, C. tomentosum, C. langsdorffii, and M. peruiferum also showed the largest partition of variability in the genotypic portion (Fig. 2), independently from the corresponding levels of diversity (Table 1). This corroborates the view that understanding the underlying characteristics of both reproductive and dispersal features of tree species is critical for a proper design of conservation strategies (Prata and Freitas, 2008).
Some particular results are worth discussing in more details. The M. multifida species, for instance, showed a decrease in the number of different alleles and effective alleles, with a significant reduction in both types of heterozygosity from adults to juvenile individuals, followed by a relevant increase in the inbreeding coefficient (f) (Tables 1 and 2). Such changes observed in these parameters between developmental stages suggest that this species has been suffering reduction in genetic variability, most likely due to anthropic actions that led to the loss of specimens in the forest fragments. Since this species is considered to be long-lived, it can be inferred that the parental generations (adults) likely bear a given set of genes that defines the high levels of heterozygosity displayed. Thus, it is plausible to assume that these mature trees have preceded the intense process of habitat loss and fragmentation that have shaped the current Atlantic Forest remnants (Moreira et al. 2021). Under such circumstances, juvenile individuals have likely inherited alleles from a reduced set of remaining trees, thereby increasing the probability of union of alleles by descent (Azevedo et al. 2005), and so, explaining the patterns observed for the genetic parameters (Table 1). Moreover, decreased levels of heterozygosity and number of distinct alleles are expected to also reduce the contribution of genotypes for the partitioning of populational diversity (Fig. 2). Regarding the M. brauna, the very low f value observed for adults (-0.27), coupled with the high number of heterozygotes are likely to be also reflecting genetic diversity prior to the anthropically-driven Atlantic Forest loss and fragmentation. A high level of heterozygosity leads to lower average fixation indexes that describe populational structuring, which is, thus, suggestive that there were no deviations from panmixia due to the reproductive system for most populations assessed (Reis et al. 2015).
Despite the overall levels of diversity found for the 10 species in this study, we could notice, not unexpectedly, that different mechanisms have shaped the specific characteristics of that diversity across generations (Table 2). For instance, considering the four species with data for adults and juveniles, M. multifida and M. brauna pointed to decreases in diversity with time (although with distinct patterns), whereas M. maxima and C. legalis displayed an opposite tendency. The trends observed for their f values among generations pointed to differences in the crossing conditions among individuals that led to increased or reduced populational relatedness. The results suggest that the recent habitat loss and fragmentation events in the Atlantic Forest have differentially affected these species in terms of specificity and magnitude, at least in the timeframe involved and at the detection capacity of this study. For example, while populations of M. multifida and M. brauna have likely suffered direct effects of selective cutting and/or fragmentation of forest patches (Allendorf and Luikart 2006), these effects seem to have not affected M. maxima and C. legalis (Tables 1 and 2; Fig. 2). Alternatively, the different types of management/exploitation may also contribute to those specific differences in diversity outcomes (Reichmann et al. 2017). Interestingly, M. brauna have shown the juvenile populations with a significant increase in the number of different alleles from adults (Tables 1 and 2), which was a tendency that would be in contrast to what might be expected from the overall trends of diversity decrease and inbreeding discussed above, based on its HO and f values. This result is probably due to the extreme logging of adult individuals of M. brauna, leaving only a very small number or, in most cases, the total loss of individuals of this stage in the populations (see Table 1). Thus, the remaining adult individuals represent a subsampling of the genetic variability of this stage, while the greater variability in the juveniles is probably due to the reproduction of adults before being removed from populations (Borges et al. 2020).
The process of exploitation of timber species in Brazil reached alarming levels from the nineteenth century onwards, with greater intensity in the exploitation of the 80 s (Dean 1996). In the southern region of Bahia, for example, where most of these species evaluated here were sampled, the process of clearing forests and, consequently, fragmentation, began in the mid-1980s, and intensified in the 1990s, during the crisis cocoa, the main economic product at the time (Rocha 2006). On a larger time scale, however, with an ever increasing forest loss and fragmentation processes, as well as with a reduction in pollinators and seed dispersal, an increase in the deleterious damages can eventually occur, and consequently increase the probability of local species extinction (Ganzhorn et al. 2015). Based on such sustained and deleterious anthropic actions, which have been leading to a continuous loss of specimens within the forest fragments that affect the distribution of populations in those remnants, it is plausible to foresee a relevant reduction in genetic variability for future generations of the tree species under study (Vranckx et al. 2012; Carvalho et al. 2015). This concern becomes especially relevant since most of the populations evaluated here are outside conservation units (see table S1) and, therefore, susceptible to increasing deforestation in the Atlantic Forest and logging (Borges et al. 2020). It is worth noting that the genetic analysis of adults allows a look at the variability in the past as a whole, when several factors were mixed as evolutionary driving forces of diversity (Sujii et al. 2017). In this sense, adult individuals reflect changes that occurred in the past, which may or not be directly related to anthropic processes (Harguindegury 2013). Thus, comparing ontogenetic stages of species (which reflect current coexistence of generations) is an important strategy to understand the fate of genetic diversity in given populations (Caballero et al. 2019).
Our approach of descriptive models’ selection revealed that the number of sampled adults or juvenile individuals had a positive correlation with the number of different alleles (NA) identified for the 10 tree species evaluated (Fig. 3). The results suggest that NA was the parameter showing a significantly greater sensitivity in relation to the number of individuals sampled. Since this independent variable is the first to suffer the impacts of fragmentation and selective cutting, we can realize that both the power of the genetic analyses endeavored and the real variability existing in an area are compromised by these impacts. At the beginning of a process of genetic erosion, it is remarkable that the first phenomenon to occur is the loss of alleles (Hartl and Clark 2010), so that it is expected that any genetic estimator based on the NA would be the first affected by those undesirable conditions (Santos et al. 2011; Barbosa et al. 2015). However, considering that NA is highly dependent on sample size, analytical biases can be introduced when comparing populations with different samplings (Allendorf and Luikart, 2006). Nonetheless, it is important to note that we evaluated the number of alleles, as this estimator represents a global average of all populations of each species in question, and we did not perform cross-population comparisons. In this way, this genetic estimator is suitable for describing the global pattern of genetic diversity, thereby reflecting the real conservation status (the central objective of our study). This is especially so for those species for which the population census was carried out, as is the case of M. brauna. Although models with only the number of individuals were the most plausible to explain NA in adult and juveniles’ trees, models with the number of markers interacting with the number of individuals per population were more plausible to explain the number of alleles than the null model (Fig. 3). Thus, by demonstrating that the association/link between the number of individuals sampled and the number of markers used can remarkably influence the estimates of genetic variability, we suggest that further research in conservation genetics should take into account these two independent variables (number of individuals and markers) in an integrated manner. We claim that not only the methodological strategy here employed, but also the constitution and possible directions of genetic diversity identified in this study can serve as valuable tools for the definition of more sound and sustainable conservation proposals, for these and other tree species in threatened forest biomes.
Data availability
The files containing sampling sites and genotyping data with microsatellite markers will be deposited in the Dryad database.
References
Borges DB, Mariano-Neto E, Caribé DS, Corrêa RX, Gaiotto FA (2020) Changes in fine-scale spatial genetic structure related to protection status in Atlantic rain forest fragment. J Nat Conserv 53:125784
Dean W (1996) A ferro e fogo: a história e a devastação da mata atlântica Brasileira. Cia. das Letras, São Paulo
Haddad NM, Brudvig LA, Clobert J, Davies KF, Gonzalez A, Holt RD, Townshend JR (2015) Habitat fragmentation and its lasting impact on Earth’s ecosystems. Sci Adv 1(2):e1500052
De Kort JG, Runier S, Ducatez O, Honnay M, Baguette VM, Stevens S, Blanchet, (2021) Life history, climate and biogeography interactively affect worldwide genetic diversity of plant and animal populations. Nat Commun 12:516
Miraldo A, Li S, Borregaard MK, Flórez-Rodríguez A, Gopalakrishnan S, Rizvanovic M (2016) An anthropocene map of genetic diversity. Science 353:1532–1535
Petit RJ, Mousadik EL, A, Pons O, (1998) Identifying populations for conservation on the basis of genetic markers. Conserv Biol 12:844–855
Piasentin FB, Gois SLL (2016) Conservação de remanescentes florestais no Brasil: considerações sobre os principais instrumentos de gestão ambiental. Desenv Meio Amb 36:115–134
Santos ER, Barros HB, Ferraz EC, Cella AJS, Capone A, Santos AF, Fidelis RR (2011) Divergence between soybean genotypes grown in irrigated lowland Brazil. Rev Ceres 58:755–764
Sebbenn AM, Carvalho ACM, Freitas MLM, Moraes SMB, Gaino APSC, Silva JM, Jolivet C, Moraes MLT (2011) Low levels of realized seed and pollen gene flow and strong spatial genetic structure in a small, isolated and fragmented population of the tropical tree Copaifera langsdorffii Desf. Heredity 106:134–145
Silva DSBS, Barboza BG, Anuska CFS, Oliveira B, Estevam CSA, Neto V, Santos ALLM, Dias AS, Scher R, Pantaleao SM (2013) Investigation of protective effects of Erythrina velutina extract against MMS induced damages in the root meristem cells of Allium cepa. Rev Bras Farmacogn 23:273–278
Waqar Z, Moraes RCS, Benchimol M, Morante-Filho JC, Mariano-Neto E, Gaiotto FA (2021) Gene flow and genetic structure reveal reduced diversity between generations of a tropical tree Manilkara multifida penn in atlantic forest fragments. Genes 12(12):2025
Albuquerque UP, Gonçalves P, Ferreira J, Washington S, Chaves L (2018) Humans as niche constructors: revisiting the concept of chronic anthropogenic disturbances in ecology. Perspect Ecol Conserv 16:1–11
Allendorf F, Luikart G (2006) Conservation and genetics of populations. Blackwell Publishing
Almeida-Rocha JM, Soares L, De Andrade ER, Gaiotto FA, Cazetta E (2020) The impact of anthropogenic disturbances on the genetic diversity of terrestrial species: a global meta-analysis. Mol Ecol. https://doi.org/10.1111/mec.15688
Anderson DR (2008) Model based inference in the life sciences: a primer on evidence. Springer, New York
Arroyo-Rodríguez V, Fahrig L, Tabarelli M, Watling JI, Tischendorf L, Benchimol M, Cazetta E, Faria D, Leal IR, Melo FPL, Morante’Filho JC, Santos BA, Arasa-Gisbert R, Arce-Pea N, Cervantes-López MJ, Cudney-Valenzuela S, Galán-Acedo C, San José M, Vieira ICG, Slik JW, Ferry NAJ, Tscharntke T (2020) Designing optimal human-modified landscapes for forest biodiversity conservation. Ecol Lett 23(9):1404–1420
Atsushi N, Susumu G, Chikako S, Yuji I (2021) Directional seed and pollen dispersal and their separate effects on anisotropy of fine-scale spatial genetic structure among seedlings in a dioecious, wind-pollinated, and wind-dispersed tree species, Cercidiphyllum japonicum. Ecol Evol 11:7754–7767
Azevedo VCR, Vinson CC, Ciampi AY (2005) Twelve microsatellite loci in Manilkara huberi (Ducke) standl (Sapotaceae), an amazonian timber species. Mol Ecol Notes 5:13–15
Bambach RK (2006) Phanerozoic biodiversity mass extinctions. Annu Rev Earth Planet Sci 34:127–155
Barbosa ACOF, Collevatti RG, Chaves LJ, Guedes LBS, Diniz-Filho JAF, Telles MPC (2015) Range-wide genetic differentiation of Eugenia dysenterica (Myrtaceae) populations in Brazilian Cerrado. Biochem Syst Ecol 59:288–296
Bates D, Mächler M, Bolker B, Walker S (2015) Fitting linear mixedeffects models using lme4. J Stat Softw 67:1–48
Bosa N, Calvete EO, Klein VA, Susin M (2015) Growth of gypsophila seedlings on different substrates. Hortic Bras 21:514–519
Browne L, Karubian J (2018) Habitat loss and fragmentation reduce effective gene flow by disrupting seed dispersal in a neotropical palm. Mol Ecol 27:3055–3069
Buzatti RSDO, Ribeiro RA, Lemos Filho JPD, Lovato MB (2012) Fine-scale spatial genetic structure of Dalbergia nigra (Fabaceae), a threatened and endemic tree of the Brazilian atlantic forest. Genet Mol Biol 35:838–846
Caballero S, Dove V, Jackson-Ricketts J, Junchompoo C, Cohen S, Hines E (2019) Mitochondrial DNA diversity and population structure in the irrawaddy dolphin (Orcaella brevirostris) from the gulf of Thailand and the mekong river. Mar Mamm Sci 35:300–310
Cardoso SRS, Provani J, Lira CF, Pereira LOR, Ferreira PCG, Cardoso MA (2005) High levels of genetic structuring as a result of population fragmentation in the tropical tree species Caesalpinia echinata lam. Biodivers Conserv 14:1047–1057
Carvalho ACM, Freitas MLM, Moraes SMB, Stranghetti V, Alzate-Marin AL, Sebbenn AM (2010) Genetic diversity, inbreeding and gene flow in a small fragmented population of Copaifera langsdorffii. Rev Bras Bot 33:599–606
Carvalho CS, Ribeiro MC, Cortes MC, Galetti M, Collevatti RG (2015) Contemporary and historic factors influence differently genetic differentiation and diversity in a tropical palm. Heredity 115:216–224
Carvalho CS, Ballesteros-Mejia L, Ribeiro MC, Côrtes MC, Santos AS, Collevatti RG (2017) Climatic stability and contemporary human impacts affect the genetic diversity and conservation status of a tropical palm in the atlantic forest of Brazil. Conserv Genet 18(2):467–478. https://doi.org/10.1007/s10592-016-0921-7
Cassano CR, Schroth G, Faria D (2009) Landscape and farm scale management to enhance biodiversity conservation in the cocoa producing region of southern bahia, Brazil. Biodivers Conserv 18:577–603
Coelho GM, Santos AS, Menezes IPP, Tarazi R, Sousa FMO, Silva MGCPC, Gaiotto FA (2020) Genetic structure among morphotypes of the endangered Brazilian palm Euterpe edulis Mart (Arecaceae). Ecol Evol 10:6039–6048
Collevatti RG, Hay JD (2011) Kin structure and genotype-dependent mortality: a study using the neotropical tree Caryocar brasiliense. J Ecol 99:757–763
Collevatti RG, Grattapaglia D, Hay JD (2001) Population genetic structure of the endangered tropical tree species Caryocar brasiliense, based on variability at microsatellite loci. Mol Ecol 10:349–356
Diniz-Filho JAF, Nabout JC, Telles MPC, Soares TN, Rangel TFLVB (2009) A review of techniques for spatial modeling in geographical, conservation and landscape genetics. Genet Mol Biol 32:203–211
Dumini J, Brown RP, Ewédjè E-EBK, Mardulyn P, Doucet J-L, Hardy OJ (2013) Large-scale pattern of genetic differentiation within African rainforest trees: insights on the roles of ecological gradients and past climate changes on the evolution of Erythrophleum spp (Fabaceae). BMC Evol Biol 13:195
Fietz J, Tomiuk J, Loeschcke V, Weis-Dootz T, Segelbacher G (2014) Genetic consequences of forest fragmentation for a highly specialized arboreal mammal - the edible dormouse. PLoS ONE 9:e88092
Ganzhorn S, Perez-Sweeney B, Thomas W, Gaiotto F, Lewis J (2015) Effects of fragmentation on density and population genetics of a threatened tree species in a biodiversity hotspot. Endang Species Res 26:189–199
Gerstner K, Dormann CF, Stein A (2014) Effects of land use on plant diversity – a global meta-analysis. J Appl Ecol 51:1690–1700
Godoy FMDR, Lenzi M, Ferreira BHDS, Silva LVD, Zanella CM, Paggi GM (2018) High genetic diversity and moderate genetic structure in the self-incompatible, clonal Bromelia hieronymi (Bromeliaceae). Bot J Linn Soc 187:672–688
Goulart MF, Ribeiro SP, Lovato MB (2005) Genetic, morphological and spatial characterization of two populations of Mabea fistulifera Mart. (Euphorbiaceae), in different successional stages. Braz Arch Biol Technol 48:275–284
Guidugli MC (2011) Genetic studies of the forest species Cariniana estrellensis (Raddi) kuntez: diversity, Crossing system and contemporary gene flow. Doctoral Dissertation. University of Sao Paulo.
Hamrick JL, Godt MJW, Sherman-Broyles S (1992) Factors influencing levels of genetic diversity in wood plant species. New for (dordr) 6:95–124
Harguindeguy I (2013) Physiological responses to hypoxia and manganese in eucalyptus clones with differential drought tolerance of pointers from Vale do Rio Doce. 2013. Masters Thesis. Federal University of Viçosa
Heuertz M, Caron H, Scotti-Saintagne C, Pétronelli P, Engel J, Tysklind N, Miloudi S, Gaiotto FA, Chave J, Molino JF, Sabatier D, Loureiro J, Budde KB (2020) The hyperdominant tropical tree Eschweilera coriacea (Lecythidaceae) shows higher genetic heterogeneity than sympatric Eschweilera species in French guiana. Plant Ecol Evol 153:67–81
Hollingsworth PM, Dawson IK, Goodall-Copestake WP, Richardson JE, Weber JC, Sotelo Montes C, Pennington RT (2005) Do farmers reduce genetic diversity when they domesticate tropical trees? A case study from Amazonia. Mol Ecol 14:497–501
Keenan K, Mcginnity P, Cross TF, Crozier WW, Prodohl PA (2013) diveRsity: An R package for the estimation and exploration of population genetics parameters and their associated errors. Method Ecol Evol 4:782–788
Kumar S, Kumaria S, Sharma SK, Rao SR, Tandon P (2009) Genetic diversity assessment of Jatropha curcas L. germplasm from northeast India. Biomass Bioenerg 35:3063–3070
Kunz N, Reiner G, Gold S (2014) Investing in disaster management capabilities versus pre-positioning inventory: a new approach to disaster preparedness. Int J Prod Econ 157:261–272
Lander TA, Bebber D, Choy TL, Harris SA, Boshier DH (2011) The circe principle explains how resource-rich land can waylay pollinators in fragmented landscapes. Curr Biol 21:1302–1307
Leal SRP, Gaiotto FA (2014) Effect of selective logging on genetic diversity and gene flow in Cariniana legalis sampled from a cacao agroforestry system. Genet Mol Res 13:626–635
Lowe AJ, Boshier D, Ward M, Bacles CFE, Navarro C (2005) Genetic resource loss following habitat fragmentation and degradation; reconciling predicted theory with empirical evidence. Heredity 95:255–273
Martini AMZ, Fiaschi P, Amorim AM, Paixão J (2007) A hot-point within a hotspot: a high diversity site in Brazil`s Atlantic forest. Biodivers Conserv 16:3111–3128
Martins K et al (2016) The role of very small fragments in conserving genetic diversity of a common tree in a hyper fragmented Brazilian Atlantic forest landscape. Conserv Genet 17(3):509–520
Melo SCO, Gaiotto FA, Cupertino FB (2007) Microsatellite markers for Caesalpinia echinata Lam. (Brazilwood), a tree that named a country. Conserv Genet 8:1269–1271
Moreira PAM, Frederico SN, Jorge AL (2021) Consequences of tropical dry forest conversion on diaspore fate of Enterolobium contortisiliquum (Fabaceae). Plant Ecol 222:525–535
Myers N, Mittermeier RA, Mittermeier CG, Fonseca GAB, Kent J (2000) Biodiversity hotspots for conservation priorities. Nature 403:853–858
Nair R, Varghese SH, Nair BG, Maekawa T, Yoshida Y, Kumar DS (2010) Nanoparticulate material delivery to plants. Plant Sci 179:154–163
Nascimento HEM, Laurance WF (2006) Area and edge effects on forest structure in upland forest fragments after 13–17 years of isolation. Acta Amaz 36:183–192
Newbold T, Hudson LN, Hill SLL, Contu S, Lysenko I, Senior RA (2015) Global effects of land use on local terrestrial biodiversity. Nature 520:45–50
Van Oosterhout OC, Utchinson WF, Wills DPM, Shipley P (2004) MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538
Ouinsavi C, Sokpon N, DE Khasa P (2009) Genetic diversity and population structure of a threatened african tree species, Milicia excelsa, using nuclear microsatellites DNA markers. Int J for Res 2009:1–8
Paetkau D, Calvert W, Stirling I, Strobeck C (1995) Microsatellite analysis of population structure in Canadian polar bears. Mol Ecol 4:347–354
Paschoa RPDA, Christ JA, Valente CS, Ferreira MFDAS, Miranda FDDE, Garbin ML, Carrijo TT (2018) Genetic diversity of populations of the dioecious Myrsine coriacea (Primulaceae) in the Atlantic Forest. Acta Bot Brasilica 32:376–385
Peakall R, Smouse PF (2012) GenAlEx 6.5: genetic analysis in Excel. population genetic software for teaching and research - an update. Bioinform 28:2537–2539
Poorter H, Jagodzinski AM, Ruiz-Peinado R, Kuyah S, Luo Y, Oleksyn J, Sack L (2015) How does biomass distribution change with size and differ among species? An analysis for 1200 plant species from five continents. New Phytol 208:736–749
Rafalski JA, Morganti M, Powell W, Vogel JM, Tingey SV (1996) Generating and using DNA markers in plants. In: Bierren B, Lai E (eds) Non-mammalian Genomic Analysis: a practical guide. Springer, New York
Reichmann MC, Zanella CÂ, Valério Júnior C, Borges ACP, Sausen TL, Paroul N, Cansian RL (2017) Genetic diversity in populations of Maytenus dasyclada (Celastraceae) in forest reserves and unprotected Araucaria forest remnants. Acta Bot Brasilica 31:93–101
Reis MVM, Damasceno Júnior PC, Campos TO, Diegues IP, Feitas SC (2015) Genetic variability and association between characteristics in germplasm of the physic nut (Jatropha curcas L.) germplasm. Rev Ciênc Agron 46:412–420
Rezende CL, Scarano FR, Assad ED, Joly CA, Metzger JP, Strassburg BBN, Mittermeier RA (2018) From hotspot to hopespot: an opportunity for the Brazilian Atlantic forest. Perspect Ecol Conserv 16:208–214
Rocha LB (2006) A região cacaueira da Bahia: uma abordagem fenomenológica. Universidade Federal de Sergipe, Thesis
Rolim SG, Sambuichi RH, Schroth G, Nascimento MT, Gomes JML (2016) Recovery of forest and phylogenetic structure in abandoned cocoa agroforestry in the Atlantic forest of Brazil. Environ Manage 59:410–418
Santos AS, Cazetta E, Morante Filho JC, Baumgarten J, Faria D, Gaiotto FA (2015) Lessons from a palm: genetic diversity and structure in anthropogenic landscapes from Atlantic Forest. Brazil Conserv Genet 16(6):1295–1302
Santos AS, Cazetta E, Dodonov P, Faria D, Gaiotto FA (2016) Landscape-scale deforestation decreases gene flow distance of a keystone tropical palm, mart (Arecaceae). Ecol Evol 6:6586–6598
Santos AS, Borges DB, Vivas CV, Berg CVD, Rodrigues PS, Tarazi R, Gaiotto FA (2019) Gene pool sharing and genetic bottleneck effects in subpopulations of eschweilera ovata (Cambess.) mart. ex miers (Lecythidaceae) in the Atlantic forest of Southern Bahia. Brazil Genetic Mol Biol 42:655–665
Santo-Silva E, Almeida W, Tabarelli M, Peres C (2016) Habitat fragmentation and the future structure of tree assemblages in a fragmented Atlantic forest landscape. Plant Ecol (dordr) 217:1129–1140
Sarmento MB, Villela FA (2010) Sementes de espécies florestais nativas do sul do Brasil. Inform Abrates. 20:39–44
Silva FKG, Lopes SF, Lopez LCS, Melo JIM, Trovão DMBM (2014) Patterns of species richness and conservation in the Caatinga along elevational gradients in a semiarid ecosystem. J Arid Environ 110:47–52
Silva-Junior JA, Franca DS, Moraes RCS, Gaiotto FA (2016) Development of microsatellite markers for Manilkara maxima T.D. Penn. (Sapotaceae) and their use in conservation genetics. Mol Biol Rep 44:1–5
Soares LASS, Cazetta E, Santos LR, França DDES, Gaiotto FA (2019) Anthropogenic disturbances eroding the genetic diversity of a threatened palm tree: a multiscale approach. Front Genet 10:1090
Sommer AB, Freitas MLM, Zannato AS, Tambarussi EV, Moraes MLT, Ambrosano MN, Pereira LCSM, Gandara FB, Kageyama PY, Alexandre M et al (2013) The effects of pollen dispersal and mating pattern on inbreeding depression and hybrid vigor in Balfourodendron riedelianum (Engl.) Engl. (Rutaceae). Conserv Genet 21:305–317
Souza PB, Souza AL, Silva Costa W, Del Peloso RV, Delana JM (2012) Floristic and diversity of tree species and shrub under a stand of Anadenanthera peregrina (L.) Speg. Cerne 18:413–421
Spear SF, Balkenhol N, Fortin MJ, Mcrae BH, Scribner K (2010) Use of resistance surfaces for landscape genetics studies: considerations for parameterization and analysis. Mol Ecol 19:3576–3591
Stupp T, de Freitas RA, Sierakowski MR, Deschamps FC, Wisniewski A, Biavatti MW (2008) Characterization and potential uses of Copaifera langsdorfii seeds and seed oil. Bioresour Technol 99:2659–2663
Sujii P, Schwarcz K, Grando C, De Aguiar S, Ellida M, Gustavo M, Brancalion PHS, Zucchi M (2017) Recovery of genetic diversity levels of a neotropical tree in Atlantic Forest restoration plantations. Biol Conserv 211:110–116
Tabarelli M, Aguiar AV, Ribeiro MC, Metzger JP, Peres CA (2010) Prospects for biodiversity conservation in the Atlantic Forest: lessons from aging human-modified landscapes. Biol Conserv 143:2328–2340
Tarazi R, Mantovani A, Reis MS (2009) Fine-scale spatial genetic structure and allozymic diversity in natural populations of Ocotea catharinensis Mez. (Lauraceae). Conserv Genet 11:965–976
Tautz D, Renz M (1984) Simple sequences are ubiquitous repetitive components of eukaryotic genomes. Nucl Acid Res 12:4127–4138
Vranckx G, Jacquemyn H, Muys B, Honnay O (2012) Meta-analysis of susceptibility of woody plants to loss of genetic diversity through habitat fragmentation. Conserv Biol 26:228–237
Weir BS (1990) Genetic Data Analysis. Sunderland, Massachusetts: Sinauer Associates, Inc
Wickham H (2016) Package ‘ggplot2’: elegant graphics for data analysis. Springer-Verlag, New York, 10, 978-0
Zucchi MI, Pinheiro JB, Chaves LJ, Coelho ASG, Couto MA, Morais LKD et al (2005) Genetic structure and gene flow of Eugenia dysenterica natural populations. Pesq Agropec Bras 40:975–980
Acknowledgements
Thanks to the Coordenação de Aperfeiçoamento de Pessoal de nível Superior (CAPES), who provided postdoctoral researcher fellowship to ASS, and doctoral studies scholarship to NSS (Finance Code 001). The research was also supported by grants from the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) provided to FAG. (CNPq Proj. #306160/2017-0). The authors would like to thank the colleagues in the Conservation Genetics group at UESC by scientific discussions with regards to this manuscript. We also thank the Universidade Estadual de Santa Cruz (UESC) for the laboratory facilities and infrastructure provided for the research.
Funding
This work was supported by Coordenação de Aperfeiçoamento de Pessoal de nível Superior – CAPES (Grant numbers: Code 001) and Conselho Nacional de Desenvolvimento Científico e Tecnológico – CNPq (Grant numbers: 306160/2017–0).
Author information
Authors and Affiliations
Contributions
The authors ASS, FAG and LLL contributed to the study conception and design. Material preparation and analysis were performed by ASS. All authors contributed to the data collection. The first draft of the manuscript was written by NSS, ASS and LLL, and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript. The authors NSS, ASS and LLL have contributed equally to this work and shared first authorship.
Corresponding author
Ethics declarations
Conflict of interests
The authors have no relevant financial or non-financial interests to disclose.
Additional information
Communicated by Oliver Gailing.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
de Santana, N.S., Santos, A.S., Borges, D.B. et al. Genetic resilience of Atlantic forest trees to impacts of biome loss and fragmentation. Eur J Forest Res 142, 161–174 (2023). https://doi.org/10.1007/s10342-022-01516-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10342-022-01516-x