
Abstract
Polypeptide antibiotics abuse can lead to antibiotic residues in food products and have unwanted effects on human health. A selective, accurate, and sensitive analytical method for the simultaneous determination of four polypeptide antibiotics (gramicidin S, bacitracin, polymyxin B, and polymyxin E) in infant formula powder was developed using solid-phase extraction (SPE) combined with high performance liquid chromatography–tandem mass spectrometry (HPLC–MS/MS). The samples were extracted with acidified methanol, deproteinized with acetonitrile, and degreased with n-hexane before SPE. After cleanup with 0.1% formic acid and methanol (3:1, v/v) in Oasis HLB cartridges, the extracts were analyzed by HPLC–MS/MS with electrospray ionization (ESI) source and time-scheduled multiple reaction monitoring (MRM). Linearity was assessed by using matrix-matched standard calibration and good determination coefficients (r2 > 0.995) were obtained. The average recoveries for blank sample at three spiked concentration levels were in the range of 82.8–101.2%. The limits of detection (LODs) and limits of quantitation (LOQs) of all analytes were in the range of 5–15 μg kg−1 and 20–50 μg kg−1, respectively. The intra-day and inter-day precisions were lower than 10%. The results of method validation demonstrated that the developed method is accurate and reliable, and it can be applied for screening and quantitation of target polypeptide antibiotics in food.
Graphic Abstract

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Polypeptide antibiotics are members of a huge family of antibiotics containing non-protein polypeptide chains. Compared with traditional antibiotics, polypeptide antibiotics have different target sites and duration of action, and they have a long history of use in human medicine for anti-infective and antitumor purposes. The polypeptide antibiotics gramicidin S, bacitracin, polymyxin B, and polymyxin E are metabolites of Bacillus brevis, Bacillus subtilis, Bacillus polymyxa, and Bacillus licheniformis [1]. Gramicidin S and bacitracin have strong therapeutic activity against gram-positive cocci and bacilli [2, 3]. Polymyxin B and polymyxin E have been used to treat severe infections of Pseudomonas aeruginosa or other gram-negative bacilli. Since these drugs have serious side effects, including fever, hemolysis, nephrotoxicity, neurotoxicity, and cytotoxicity [4,5,6], they have been replaced by other lower toxicity and better-tolerated antibiotics. Hence, they are no longer used as a drug for systemic treatment, only as a variety of topical therapeutic administration, such as the treatment of the external auditory canal, cornea, or skin infections [7].
However, these four polypeptide antibiotics are extensively used as veterinary drugs for infectious disease in modern animal husbandry and may also be added into animal feed to improve feed conversion efficiency and growth rate. Polypeptide antibiotics abuse can lead to antibiotic residues in food products and have unwanted effects on human health [6]. Therefore, the application of polypeptide antibiotics in veterinary practice is regulated by several regulatory agencies. For example, Japan has established maximum residue limits (MRLs) for bacitracin (400 μg kg−1) and polymyxin E (10 μg kg−1) in milk. The European Union (EU) has forbidden the use of bacitracin as an animal feed additives since 1999 and set MRLs for bacitracin (100 μg kg−1) and polymyxin E (50 μg kg−1) in milk [8, 9]. Taking the side effects of these drug residue into account, an accurate and high-throughput analytical method for determining polypeptide antibiotic residues in food is critical and necessary.
Several techniques have been developed for the analysis of polypeptide antibiotics, including immunoassay [10, 11], fluorescence [12, 13], capillary electrophoresis [14,15,16,17], and high performance liquid chromatography (HPLC) with various detection systems [9, 18,19,20]. However, the processing procedure of immunoassay methods for polypeptide antibiotics is cumbersome and capillary electrophoresis has the drawback of poor reproducibility. HPLC methods need time-consuming sample pretreatment including protein precipitation, solid-phase extraction (SPE), and derivatization. Besides, these methods have the common disadvantage of low detection sensitivity. As a highly selective and sensitive analytical method, high performance liquid chromatography–tandem mass spectrometry (HPLC–MS/MS) has been reported for the determination of polymyxin B and polymyxin E in food samples and biological matrices [21,22,23,24,25], as well as bacitracin in rabbit serum and food matrices [26, 27]. However, no sensitive method for the simultaneous quantitative analysis of gramicidin S, bacitracin, polymyxin B, and polymyxin E in food products has been reported.
The aim of this study was to develop a new, highly sensitive HPLC–MS/MS method for simultaneous determination of four polypeptide antibiotics (gramicidin S, bacitracin, polymyxin B, and polymyxin E) in infant formula milk powder samples. The HPLC–MS/MS conditions and cleanup procedure were optimized. The method was validated to demonstrate the matrix effect, linearity, LODs, LOQs, recovery, and precision. This method was also applied for the analysis of commercial samples.
Materials and Methods
Reagents, Materials, and Standards
The standard of gramicidin S (purity 95%) was obtained from BOC Sciences (NY, USA). Bacitracin (purity 78%), polymyxin B (polymyxin B1 purity 86%), and polymyxin E (polymyxin E1 purity 87%) were purchased from Dr. Ehrenstorfer GmbH (Augsburg, Germany). Methanol (MeOH), acetonitrile (ACN), and n-hexane were obtained from Sigma-Aldrich (St. Quentin Fallavier, France). Formic acid (FA) was purchased from Baillingwei Company (Beijing, China). Ultrapure water (resistivity 18.2 MΩ) was prepared using the Milli-Q water purification system (Millipore, Brussels, Belgium). All solvents and reagents were of HPLC and analytical grade. Formula infant milk powder was purchased from local supermarkets.
Individual stock solution of gramicidin S was prepared at a concentration of 1000 mg L−1 in MeOH. Individual stock standard solutions of bacitracin, polymyxin B, and polymyxin E were prepared at a concentration of 1000 mg L−1 in 0.1% FA. Then a mixed working standard solution of the four analytes was prepared at different concentrations (1000, 500, 200, and 100 μg L−1) by combining suitable aliquots of stock solutions of each individual standard and diluting them with appropriate volumes of 0.1% FA in MeOH (4:1, v/v). All of them were prepared weekly and stored in screw-capped glass tubes at − 20 °C in the dark.
HPLC–MS/MS Conditions
A 1260 HPLC (Agilent, CA, USA) integrated system coupled to an AB 5500 (Applied Biosystems, USA) triple quadrupole mass spectrometer equipped with ESI source was used. Data was acquired using the Analyst 1.6.1 software. Precursor and production ion selection and optimization of collision energies were performed manually by flow injection of analytical standards. The analysis was carried out using a Poroshell 120 SB-C18 analytical column (2.7 μm, 4.6 × 150 mm, Agilent, CA, USA) maintained at 30 °C. A binary gradient of mobile phase A (0.1% FA in water) and B (0.1% FA in MeOH) was delivered at a constant flow rate of 0.5 mL min−1. A 5-μL sample was injected. The following gradient elution program was used: 0–2 min (20% B), 6–16 min (95% B), 19–20 min (20% B).
The mass spectrometer was operated in positive electrospray ionization mode (ESI+). Targets ions were examined using multiple reaction monitoring (MRM) mode. The general source settings in the positive ionization modes were as follows: ion source gas 1 (GS1) 50 psi; ion source gas 2 (GS2) 45 psi; ion spray voltage (IS) 5000 V; ion source temperature 450 °C; and curtain gas 0.2 MPa. The precursor ions, product ions, decluster potential (DP), collision energy (CE), and collision cell exit potential (CXP) values of all analytes and other parameters are shown in Table 1.
Sample Preparation
All formula milk powder samples were collected from supermarkets in Beijing, China. Typically, the milk powder samples were dissolved in water at room temperature for each experiment. For recovery and validation studies, the homogenized blank sample was spiked with known variable amounts of four mixed standard solutions.
Formula milk powder (1.00 g) was precisely weighed and dissolved in Milli-Q water (10 mL). Then each sample was transferred to 50-mL polypropylene centrifuge tubes. For method development and validation, blank samples were spiked with mixed standard solutions. Afterwards, the mixture was vortexed for 1 min and left to stand at room temperature for 10 min for equilibration. Then, 20 mL 0.1% FA/MeOH (3:1, v/v) with 2 mL ACN was added into the mixture and the tube was shaken and centrifuged for 10 min at 4 °C.
Next, the supernatant was transferred to another tube and the residue was treated by following the extraction procedure described above. After the supernatant of the second extraction was combined with the first extraction, 20 mL n-hexane was added and the tube was vortexed for 1 min and then centrifuged for 5 min at 4 °C. The supernatant was transferred to another tube. Then, 20 mL n-hexane was added into the residue and the operation described above was repeated. After centrifugation, the supernatant was collected again in the same tube.
Solid-Phase Extraction
Initially, the Oasis HLB SPE cartridge (6 ml/500 mg) was conditioned by passing 6 mL of MeOH followed by 6 mL of Milli-Q water. Then the obtained sample extract was loaded. The cartridge was washed with 15 mL Milli-Q water and the analytes were eluted by adding 6 mL MeOH. Finally, the eluate was evaporated to nearly dryness under a gentle stream of nitrogen in a 40 °C water bath, and then reconstituted with 2 mL 0.1% FA/MeOH (4:1, v/v). The sample extract was vortexed for 30 s, and then filtered through a 0.22-μm nylon membrane for HPLC–MS/MS analysis.
Results and Discussion
Optimization of MS Conditions
The optimum MS/MS parameters of the four analytes were obtained by analyzing single standard solutions at a concentration of 100 ng mL−1. Precursor ions were selected in both positive and negative mode. In agreement with a previous report [28], the positive ion mode produced more abundant m/z values in this work. This can be explained as follows: the analytes contain peptide bonds and the nitrogen atoms of amino groups in peptide bonds add hydrogen ions very easily; therefore, the positive ion mode was selected as the ionization mode.
Full-scan mass spectral acquisitions were performed to identify the precursors. In contrast to the formation of triply charged ions in previous reports [25, 29, 30], it was noted that the doubly charged peak of bacitracin ([M + 2H]2+ at m/z 712.2) has the highest response (see Electronic Supplementary Material Fig. S1a), which is in line with another related report [30]. The possible reason for the phenomenon is that the charged status of bacitracin was likely to be affected by the type of mass analyzer, MS interface, ion source design, or other objective reasons [31]. Similarly, the doubly charged peak [M + 2H]2+ (m/z 602.7) of polymyxin B, [M + 2H]2+ (m/z 585.6) of polymyxin E, and [M + 2H]2+ (m/z 571.6) of gramicidin S (see Electronic Supplementary Material Fig. S1) were observed to have the highest response value in the full-scan mass spectra. The ring structure of gramicidin S is centrosymmetrical, which may easily produce two identical structures and was attributed to the doubly charged peak. Thus, the doubly charged ions of gramicidin S (m/z 571.6), bacitracin (m/z 712.2), polymyxin B (m/z 602.7), and polymyxin E (m/z 585.6) were selected as precursor ions.
The suitable fragment ions of the precursor ion were selected in the product ion scan mode. It is better to select two ions that are sensitive in MRM mode for qualitative and quantitative determination. Product ions of m/z 169.2, m/z 199.2, m/z 233.3, and m/z 101.1, which are relatively high in kurtosis, were used as the quantitative ions for gramicidin S, bacitracin, polymyxin B, and polymyxin E, respectively. Collision energy (CE) was a critical parameter which affected the sensitivity toward the target compounds. The values were set from 0 to 180 eV during the optimization process of CE in MRM mode. As shown in Table 1, CE value of the highest abundance was selected for each target. The optimized CE values of the four polypeptide antibiotics were in the range of 32–50 eV. Moreover, in order to eliminate the solvent ionic cluster effectively and enhance the signal abundance, DP and CXP were optimized to obtain more abundant m/z values.
Optimization of Chromatographic Conditions
In a preliminary study, the separation performance of three columns, Agilent Poroshell 120 SB-C18 (150 mm × 4.6 mm, 2.7 μm), Agilent Extend-C18 (150 mm × 4.6 mm, 5 μm), and Waters Atlantis T3 (150 mm × 4.6 mm, 5 μm), was evaluated. The best separation effect and the highest response for total analytes were observed with the Poroshell 120 SB-C18 column, which was therefore chosen as the HPLC column.
In order to optimize the separation for the four target analytes, various types of mobile phases such as MeOH/water, 0.1% FA in water, and 0.1% FA in MeOH were tested. When MeOH/water was used as the mobile phase, good peak shape and response value were not obtained because the polypeptide antibiotics were unstable under neutral conditions [32]. So, the volatile organic acid formic acid was added as an acidity regulator in this study to improve the separation effect. When FA was added to the mobile phase (0.1% FA in water/MeOH, 0.1% FA in water, or 0.1% FA in MeOH), good response and peak shape were obtained in the chromatographic traces for the test compound polymyxin E. This is because FA not only provided an acidic environment but also supplied the essential proton source to improve the ionization efficiency. Compared with 0.1% FA in water/MeOH, the mixture of 0.1% FA in water and 0.1% FA in MeOH as mobile phases could produce better peak shape and reduce the tailing factor from 1.6 to 1.1 (see Electronic Supplementary Material Fig. S2). Therefore, 0.1% FA in water (A) and 0.1% FA in MeOH (B) were chosen as the mobile phases.
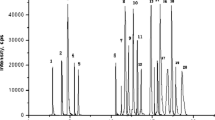
Furthermore, several gradient profiles were optimized to obtain better target separation and less analysis time. Parameters such as flow rate, injection volume, and temperature were also optimized to a achieve better separation effect. As shown in Fig. 1, the target compounds could be well separated in less than 13 min, except polymyxin B and polymyxin E, which have nearly the same retention time (10.7 and 10.8 min, respectively) owing to their similar chemical structure and properties. However, they can be distinguished well under MRM scan mode in mass spectrometry.

Optimized chromatograms of extracted ion current for sample spiked at 100 μg kg−1 of four polypeptide antibiotics
Optimization of Sample Preparation
When using ESI–MS in the analysis of samples, the ion suppression caused by matrix effects may reduce the signal and increase detection limits. In this study, SPE was used to purify the sample matrix. Three different SPE columns (C18, Oasis MAX, and Oasis HLB) were investigated. Compared with the average recoveries of the four analytes purified by C18 columns (73%) and Oasis MAX column (69%), the Oasis HLB columns (6 ml/500 mg) provided the best purification effect and had the highest average recoveries (88%). The packing of Oasis HLB (hydrophilic-lipophilic balance) is a reversed-phase adsorbent which is more suitable for retaining the polar compounds. So, the Oasis HLB was chosen as the SPE column for sample purification.
In this work, water, MeOH, water/MeOH (1:1, v/v), and different concentrations of formic acid (0.1% FA and 1% FA in water, v/v) in MeOH (1:1, v/v) were investigated in order to get the best solvent extraction efficiency of a spiked blank sample (all of the extraction solvents are in excess). As shown in Fig. 2, when using water or MeOH alone as extraction solvent, water showed a relatively better extraction effect than MeOH for all the targets except gramicidin S, because bacitracin, polymyxin B, and polymyxin E were readily soluble in water whereas gramicidin S was readily soluble in MeOH. Since the average recoveries were below 75%, mixed extraction solvents with/without acidified reagent (FA) were also investigated. Results showed that the addition of FA could provide recoveries higher than 80%. Besides, the mixture of 0.1% FA/MeOH (1:1, v/v) showed better extraction effect (average recoveries 96%) than 1% FA/MeOH (1:1, v/v). A possible explanation was that polypeptide antibiotic needs an appropriate pH environment; the lower pH value of the extraction solvent was likely to cause degradation of bacitracin and polymyxin E, which has been reported elsewhere [21, 30].

Effect of different extraction solvents on the recoveries of target analytes
The effect of different volume ratios of 0.1% FA/MeOH (1:1, 2:1, 3:1, 4:1, v/v) on the extraction was studied. Figure 3 shows that when the volume ratio of 0.1% FA/MeOH increased from 1:1 to 3:1, the recovery of analytes was higher. Whereas when the volume ratio of 0.1% FA/MeOH increased to 4:1, the recovery of polymyxin B was lower than 70%. In order to ensure the best recoveries, 0.1% FA/MeOH (3:1, v/v) was chosen as the extraction solvent.

Effect of different volume ratios of extraction solvent on the recoveries of target analytes
Additionally, the effect of volume of solvent on the recoveries of target compounds was also studied. The recoveries of target compounds were higher with increasing solvent volume (Fig. 4). When the volume of extraction solvent increased to 15 mL, the recovery values began to stabilize. In order to ensure the stability of target compound recoveries, 20 mL of extraction solvent was chosen as the optimal volume of extraction solvent.

Effect of different volumes of extraction solvent on the recoveries of target analytes
Methods Validation
Matrix Effect
Suppression or enhancement of the target signal usually occurs in MS/MS determination with electrospray ionization as a result of the matrix effect (ME), especially in complex matrices. Generally, it will produced a strong suppression effect when the positive ion mode is used. In this study, the matrix effect of the developed method was evaluated by comparing the slopes of standard solution calibration plots to the matrix-matched calibration plots. For this purpose, the following equation was used [33]:
where Sm is the slope of calibration plot in matrix and Ss is the slope of calibration plot in solvent. The values of ME for gramicidin S, bacitracin, polymyxin B, and polymyxin E were 9.4%, 46.9%, 88.4%, and 22.2%, respectively. Soft matrix effects (ME > − 20%, ME < 20%) are negligible. However, the matrix effects in milk powder samples for bacitracin and polymyxin E were medium (−50% < ME < − 20%, 20% < ME < 50%) and that for polymyxin B was significant (ME < − 50%, ME > 50%) which resulted in signal suppression in MS. Therefore, it is necessary to overcome the influence of the matrix on the analytes. In this study it can be solved by employing a matrix-matched calibration standard. Thus, the blank matrix solution was used to dilute the standard stock solution step by step and to establish a calibration standard curve and external standard method for accurate quantification of all the compounds.
Linearity, LODs, and LOQs
To evaluate the analytical features of the developed method, the calibration curves of gramicidin S, bacitracin, polymyxin B, and polymyxin E were evaluated using matrix-matched samples spiked at three different concentration levels in the range of 20–2000 μg kg−1. Three replicates for each concentration level were used. For all analytes in milk powder matrices, the detector response was linear with a coefficient of determination (r2) equal to or higher than 0.99. However, the linear range was different: for gramicidin S, polymyxin B, and polymyxin E, the linear range started at a concentration of 20 or 30 μg kg−1 and the detector response was linear up to 1000 μg kg−1 (Table 2). Signals for bacitracin were linear from 50 up to 2000 μg kg−1.
Limits of detection (LODs) of the instrumental method were calculated by the injection of a series of diluted standard solutions until a signal-to-noise (S/N) ratio equal to or slightly higher than 3 was reached. Limits of quantification (LOQs) were determined by injecting a series of spiked samples until an S/N ratio equal to or higher than 10 was reached. Under the optimum conditions of determination, the LODs were 5 μg kg−1 for gramicidin S, 15 μg kg−1 for bacitracin, 10 μg kg−1 for polymyxin B, and 6.5 μg kg−1 for polymyxin E, which were significantly lower than those obtained with capillary chromatography [15] and LC-fluorimetric detection [18] and also lower than the MRLs regulated in Japan and EU [8]. The LOQs were 20 μg kg−1 for gramicidin S, 50 μg kg−1 for bacitracin, 30 μg kg−1 for polymyxin B, and 20 μg kg−1 for polymyxin E. The results indicated that good sensitivity was obtained.
Recovery
The recovery studies were implemented by using blank samples to assess the effectiveness of the extraction. The blank infant formula milk powder samples were spiked with working standard solutions at three concentration levels (1 × LOQ, 2 × LOQ, and 4 × LOQ) for each target compound. Six replicates were analyzed at each concentration level. The recoveries of the four analytes at various spiked levels were in the range of 82.8–101.2% (Table 2).
Precision
The precision was evaluated in terms of intra-day precision and inter-day precision, expressed as relative standard deviation (RSD). Intra-day precision was measured by spiking blank sample with the low (1 × LOQ), intermediate (2 × LOQ), and high (4 × LOQ) levels of standards, using six replicates for each concentration level in 1 day. To evaluate inter-day precision, the same concentration levels were analyzed over 5 consecutive days. The RSD values of intra-day precision ranged from 2.2% to 8.8% and the RSD values of inter-day precision ranged from 2.7% to 9.8%, indicating the stability of the developed method.
Application of the Developed and Validated Methods
In order to estimate the reliability and practicality of the developed HPLC–MS/MS method, 30 samples (including stage 1, stage 2, stage 3, and stage 4) of formula infant milk powder purchased from local supermarkets were analyzed in this study. The sample matrix was extracted and analyzed using the developed processing procedure. The spiked samples were also analyzed in the same process. The results of the detection demonstrated that gramicidin S, bacitracin, polymyxin B, and polymyxin E were detected at concentrations below LODs in the real samples and have a recovery higher than 80% in the spiked samples.
Conclusions
In this work, a multi-residue method for simultaneous determination of gramicidin S, bacitracin, polymyxin B, and polymyxin E in milk powder was developed. The proposed detection method achieved superior sensitivity, selectivity, and accuracy through using the MRM mode. Matrix-matched standard calibration was used to eliminate matrix effects. The LOQs of the four target analytes were 20 μg kg−1, 50 μg kg−1, 30 μg kg−1, and 20 μg kg−1, respectively. The method was successfully applied to real samples from local markets. The developed method is suitable for monitoring the contamination of polypeptide antibiotics in infant formula milk powder and is expected to be applied to the detection of polypeptide antibiotics in other animal-derived foods.
References
Choi SK, Park SY, Kim R, Kim SB, Lee CH, Kim JF, Park SH (2009) Identification of a polymyxin synthetase gene cluster of Paenibacillus polymyxa and heterologous expression of the gene in Bacillus subtilis. J Bacteriol 191(10):3350–3358
Swierstra J, Kapoerchan V, Knijnenburg A, Van BA, Overhand M (2016) Structure, toxicity and antibiotic activity of gramicidin S and derivatives. Eur J Clin Microbiol 35(5):763–769
Ming LJ, Epperson JD (2002) Metal binding and structure-activity relationship of the metalloantibiotic peptide bacitracin. J Inorg Biochem 91(1):46
Falagas ME, Rizos M, Bliziotis IA, Rellos K, Kasiakou SK, Michalopoulos A (2005) Toxicity after prolonged (more than four weeks) administration of intravenous colistin. BMC Infect Dis 5(1):1
Marr AK, Gooderham WJ, Hancock RE (2006) Antibacterial peptides for therapeutic use: obstacles and realistic outlook. Curr Opin Pharmacol 6(5):468–472
Mogi T, Kita K (2009) Gramicidin S and polymyxins: the revival of cationic cyclic peptide antibiotics. Cell Mol Life Sci 66(23):3821–3826
Sınırtaş M, Akalın H, Gedikoğlu S (2009) Investigation of colistin sensitivity via three different methods in Acinetobacter baumannii isolates with multiple antibiotic resistance. Int J Infect Dis 13(5):e217
Liu JJ, Jin F, She YX, Liu HB, Shi XM, Wang M, Wang J, Xu SY (2011) Simultaneous determination of 5 peptide antibiotics in bovine milk samples by liquid chromatography–tandem mass spectrometry. Chin J Anal Chem 39(5):652–657
Capitán-Vallvey LF, Navas N, Titos A, Checa R (2001) Determination of the antibiotic zinc bacitracin in animal food by high-performance liquid chromatography with ultraviolet detection. Chromatographia 54(1–2):15–20
Matsumoto M, Tsunematsu K, Tsuji A, Kido Y (1997) Enzyme immunoassay using peroxidase as a label and a dip-strip test for monitoring residual bacitracin in chicken plasma. Anal Chim Acta 346(2):207–213
Suhren G, Knappstein K (2005) Detection of colistin in spiked and incurred milk samples by LC-and ELISA-technique. Anal Chim Acta 529(1–2):97–101
Lantz AE, Jørgensen P, Poulsen E, Lindemann C, Olsson L (2006) Determination of cell mass and polymyxin using multi-wavelength fluorescence. J Biotechnol 121(4):544–554
Caudron E, Baghriche S, Prognon P, Pradeau D (2013) Simultaneous quantification of gentamicin and colistin sulfate in pharmaceuticals using ion-pairing and polarity gradient chromatography with low-UV detection. Chromatographia 76(13–14):747–755
Kang JW, De Reymaeker G, Van Schepdael A, Roets E, Hoogmartens J (2001) Analysis of bacitracin by micellar electrokinetic capillary chromatography with mixed micelle in acidic solution. Electrophoresis 22(7):1356
Chaisuwan P, Moonta T, Sangcakul A, Nacapricha D, Wilairat P, Uraisin K (2015) Simple in-house flow-injection capillary electrophoresis with capacitively coupled contactless conductivity method for the determination of colistin. J Sep Sci 38(6):1035–1041
Srisom P, Liawruangrath B, Liawruangrath S, Slater JM, Wangkarn S (2007) Simultaneous determination of neomycin sulfate and polymyxin B sulfate by capillary electrophoresis with indirect UV detection. J Pharm Biomed Anal 43(3):1013–1018
Injac R, Mlinaric A, Djorjevic-Milic V, Karljikovic-Rajic K, Strukelj B (2008) Optimal conditions for determination of zinc bacitracin, polymyxin B, oxytetracycline and sulfacetamide in animal feed by micellar electrokinetic capillary chromatography. Food Addit Contam 25(4):424–431
Morales-Muñoz S, de Castro MD (2005) Dynamic ultrasound-assisted extraction of colistin from feeds with on-line pre-column derivatization and liquid chromatography-fluorimetric detection. J Chromatogr A 1066(1–2):1
Gmur DJ, Bredl CR, Steele SJ, Cai S, VanDevanter DR, Nardella PA (2003) Determination of polymyxin E1 in rat plasma by high-performance liquid chromatography. J Chromatogr B 789(2):365–372
Thomas TA, Broun EC, Abildskov KM, Kubin CJ, Horan J, Yin MT, Cremers S (2012) High performance liquid chromatography-mass spectrometry (LC-MS) assay for polymyxin B1 and B2 in human plasma. Ther Drug Monit 34(4):398
Dotsikas Y, Markopoulou CK, Koundourellis JE, Loukas YL (2011) Validation of a novel LC-MS/MS method for the quantitation of colistin A and B in human plasma. J Sep Sci 34(1):37–45
Jansson B, Karvanen M, Cars O, Plachouras D, Friberg LE (2009) Quantitative analysis of colistin A and colistin B in plasma and culture medium using a simple precipitation step followed by LC/MS/MS. J Pharm Biomed Anal 49(3):760
Cheng C, Liu S, Xiao D, Hollembaek J, Yao L, Lin J, Hansel S (2010) LC–MS/MS method development and validation for the determination of polymyxins and vancomycin in rat plasma. J Chromatogr B 878(28):2831–2838
Soon-Ee C, Bulitta JB, Jian L, Nation RL (2014) Development and validation of a liquid chromatography-mass spectrometry assay for polymyxin B in bacterial growth media. J Pharm Biomed Anal 92:177–182
Boison JO, Lee S, Matus J (2015) A multi-residue method for the determination of seven polypeptide drug residues in chicken muscle tissues by LC-MS/MS. Anal Bioanal Chem 407(14):4065
Mascher DG, Unger CP, Mascher HJ (2007) Determination of neomycin and bacitracin in human or rabbit serum by HPLC–MS/MS. J Pharm Biomed Anal 43(2):691–700
Kaufmann A, Widmer M (2013) Quantitative analysis of polypeptide antibiotic residues in a variety of food matrices by liquid chromatography coupled to tandem mass spectrometry. Anal Chim Acta 797(40):81
Wan CH, Ho C, Sin WM, Wong YC (2006) Detection of residual bacitracin A, colistin A, and colistin B in milk and animal tissues by liquid chromatography tandem mass spectrometry. Anal Bioanal Chem 385(1):181–188
Lee SC, Matus JL, Gedir RG, Boison JO (2011) A valid LC-MS method for the determination of bacitracin drug residues in edible pork tissue with confirmation by LC-tandem mass spectrometry. J Liq Chromatogr Relat Technol 34(20):2699–2722
Zhang D, Park JA, Kim DS, Kim NH, Kim SK, Cho KS, Jeong D, Shim JH, Abd El-Aty AM, Shin HC (2015) Simultaneous detection of bacitracin and polymyxin B in livestock products using liquid chromatography with tandem mass spectrometry. J Sep Sci 38(14):2371
Sin WM, Ho C, Wong YC, Ho SK, Ip CB (2005) Analysis of major components of residual bacitracin and colistin in food samples by liquid chromatography tandem mass spectrometry. Anal Chim Acta 535(1–2):23–31
Kang J, Vankeirsbilck T, Van SA, Orwa J, Roets E, Hoogmartens J (2000) Analysis of colistin sulfate by capillary zone electrophoresis with cyclodextrins as additive. Electrophoresis 21(15):3199–3204
Rajski Ł, Lozano A, Uclés A, Ferrer C, Fernándezalba AR (2013) Determination of pesticide residues in high oil vegetal commodities by using various multi-residue methods and clean-ups followed by liquid chromatography tandem mass spectrometry. J Chromatogr A 1304(16):109–120
Acknowledgements
The authors wish to acknowledge the financial support given by the project of National Key Research and Development Program of China and Public Welfare Fund of Inspection and Quarantine of China.
Funding
This study was funded by the project of National Key Research and Development Program of China and Public Welfare Fund of Inspection and Quarantine of China (Grant Numbers 2018YFC1603606, 201510038, 2016YFD0401103, and 2016YFF0203903).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Liu, T., Zhang, C., Zhang, F. et al. Sensitive Determination of Four Polypeptide Antibiotic Residues in Milk Powder by High Performance Liquid Chromatography–Electrospray Tandem Mass Spectrometry. Chromatographia 82, 1479–1487 (2019). https://doi.org/10.1007/s10337-019-03777-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10337-019-03777-y