
Abstract
Gramicidin S and polymyxins are small cationic cyclic peptides and act as potent antibiotics against Gram-negative and Gram-positive bacteria by perturbing integrity of the bacterial membranes. Screening of a natural antibiotics library with bacterial membrane vesicles identified gramicidin S as an inhibitor of cytochrome bd quinol oxidase and an alternative NADH dehydrogenase (NDH-2) and polymyxin B as an inhibitor of NDH-2 and malate: quinone oxidoreductase. Our studies showed that cationic cyclic peptide antibiotics have novel molecular targets in the membrane and interfere ligand binding on the hydrophobic surface of enzymes. Improvement of the toxicity and optimization of the structures and clinical uses are urgently needed for their effective application in combating drug-resistant bacteria.
Similar content being viewed by others
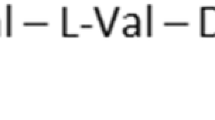

Avoid common mistakes on your manuscript.
Introduction
The emergence of drug-resistant strains of major pathogenic bacteria such as Staphylococcus aureus is an increasingly serious public health concern [1]. To evade bacterial drug resistance mechanisms, new effective chemotherapeutic agents, which have novel mechanisms of action as well as different cellular targets compared with conventional antibiotics, need to be developed [2]. The shortage of new antibiotics to combat multidrug-resistant (MDR) strains has led to a renewed interest in polymyxins [3–7]. Polymyxins are active against MDR Gram-negative bacteria such as Pseudomonas aeruginosa, Klebsiella pneumoniae, Acinetobacter baumannii, and Enterobacter species [3, 8]. Early reports described high incidences of nephrotoxocity and neurotoxicity during polymyxin therapy [9], and the use of polymyxins was replaced in the 1970s by antibiotics considered to be less toxic. However, recent studies showed that polymyxins have acceptable effectiveness and are considerably less toxic than originally reported [3].
Cationic amphiphilic peptide antibiotics as a defense system
Short cationic amphiphilic peptides form part of the biological defense system of a broad range of organisms [10]. Cationic amphiphilic peptides are used by microorganisms to suppress the growth of competitors in the same ecological niche, while similar peptides form part of the innate immune defenses of higher organisms. Membrane lipid bilayer, rather than cellular proteins, is considered the primary target of these peptides. Interestingly, in mammals, many host defense peptides have additional chemokine-like and immunomodulatory activities [10, 11]. Unlike conventional antibiotics, the acquisition of resistance against antimicrobial peptides is surprisingly rare. Naturally occurring cationic amphiphilic peptides have retained their antimicrobial activity for millions of years, and may hold promise as broad-spectrum antibiotics against the rapidly growing numbers of antibiotic-resistant microorganisms.
Gramicidins
The soil bacterium Aneurinibacillus migulanus (formerly known as Bacillus brevis) synthesizes nonribosomally and secretes the antibiotic tyrothricin, a mixture of linear pentadecapeptides (gramicidins A, B, and C) and backbone-cyclized cationic decapeptides [gramicidin S (GS) and tyrocidins [12, 13]] (Fig. 1). The primary structure of GS is [cyclo-(Val-Orn-Leu-D-Phe-Pro)2]. GS consists of an antiparallel β-sheet structure supported by two type II′ β-turns and is amphiphilic, with two charged Orn side chains and two d-Phe rings projecting from one side of the molecule and four hydrophobic Val and Leu side chains projecting from the other [12–14]. GS is an extremely powerful antibiotic drug against a broad spectrum of both Gram-negative and Gram-positive bacteria with the minimum inhibitory concentration of 3–11 µM [15, 16]. Regrettably, GS is very hemolytic, which presently restricts its use to topical applications [13, 14]. GS is also effective against several pathogenic fungi, and Otoguro et al. [17] reported nematocidal activities of GS [the 50% inhibitory concentration (IC50) = 0.08 µM] and polymyxins (IC50 = 0.8 µM) against the pine wood nematode Bursaphelenchus lignicolus. It was found later that GS was ineffective against the root-knot nematode Meloidogyne incognita even at 82 µM [18]. Effects and molecular targets of GS and polymyxins remain to be studied in lower eukaryotes.

Structures of gramicidins, tyrocidine A, and polymyxins. Tyrocidin B has Trp at position 6. l -DAB l-α-γ-diaminobutyric acid
Although the mechanism of antibacterial activity by GS is not completely understood, the primary mode of action is generally assumed to be the perturbation of lipid packing, resulting in destruction of the membrane integrity and enhancement of the permeability of the lipid bilayer of the bacterial cytoplasmic membrane [19–22]. Upon partitioning into lipid bilayers, GS displaces lipid molecules in the leaflet. Therefore, the accumulation of significant amounts of GS in a membrane is incompatible with the maintenance of a stable bilayer structure [23].
In contrast to GS, the structure of linear gramicidins like gramicidin A is an unconventional β-helix (6.3 amino acid residues per turn) with the alternating l- and d-amino acid composition except for position 2 (Gly). All side chains point outward, and linear gramicidins form N-to-N termini dimeric ion channels [24], which selectively transport alkaline metal cations and protons across the lipid bilayer [25].
Polymyxins
Polymyxins are an old class of cationic peptide antibiotics, and the emergence of MDR Gram-negative bacteria has led to the revival of polymyxins for salvage therapy [3, 4]. Polymyxins are pentabasic decapeptide antibiotics containing a cycloheptapeptide ring with a C9 or C10 fatty acid chain [6-methyl-octanoic acid (polymyxin B1, Polymyxin E1); 6-methyl-heptanoic acid (polymyxin B2, polymyxin E2)] through an α-amide linkage (Fig. 1) and nonribosomally synthesized in Bacillus polymyxa [26]. The target of antimicrobial activity is assumed to be the bacterial membrane. Cationic polypeptides bind to anionic lipopolysaccharide (LPS) molecules in the outer membrane of the Gram-negative bacteria, leading to a local disturbance of the membrane, which then causes an increase in the permeability [27, 28]. The polymyxin-mediated killing effect on the Gram-negative bacteria takes place prior to the increase in the membrane permeability, supporting a multi-hit hypothesis [29].
Screening of a natural antibiotics library
In order to identify new inhibitors for alternative respiratory enzymes (i.e., enzymes that are not present in human mitochondria), we screened the Kitasato Institute for Life Sciences Chemical Library [30] with the following enzymes: cytochrome bd quinol oxidase from Escherichia coli [31, 32], bacterial cyanide-insensitive oxidase (CIO, a variant of cytochrome bd) and a single-subunit NADH dehydrogenase (NDH-2) from the acetic acid bacterium Gluconobacter oxydans [33, 34], and NDH-2 and malate: quinone oxidoreductase (MQO) from Mycobacterium smegmatis [35] and Pseudomonas aeruginosa [36]. Cytochrome bd and CIO are widely distributed among bacteria and play an important role in microaerophilic respiration and protection against oxygen stress and in the survival and adaptation of pathogenic bacteria [37–39]. NDH-2s were shown to be crucial for the adaptation of M. tuberculosis [39] and malaria parasites [40, 41], but their specific inhibitors are rare [42]. Therefore, inhibitors of quinol oxidases [31–33, 43] and NDH-2 [34, 35, 40, 41, 44] are promising new chemotherapeutics.
Library screening identified GS as a mixed-type inhibitor of E. coli cytochrome bd, while gramicidin D (the naturally produced mixture of gramicidins A, B, and C of ~80% A, 5% B, and 15%C) [45] did not show inhibitory activity [31]. The IC50 of GS (3.5 µM) was comparable to the IC50 of the known quinol oxidation site inhibitors, 2-heptyl-4-hydroxyquinoline N-oxide (HQNO; 1 µM) and antimycin A (5 µM), and to the IC50 (9 µM) for the aerobic growth of E. coli cells. Cytochrome bo quinol oxidase and NDH-2 were tenfold less sensitive to GS than cytochrome bd oxidase, and succinate dehydrogenase (Complex II) was totally unaffected by GS. Notably, GS had a stimulatory effect at low concentrations by increasing the apparent V max value of cytochrome bd quinol oxidase [31, 43].
From screening with G. oxydans NDH-2, we identified GS and scopafungin as potent inhibitors and found the moderate inhibitory activity with polymyxin B [34]. GS serves as a noncompetitive inhibitor for NADH oxidation and a competitive inhibitor for quinone (Q) reduction. Furthermore, GS shows inhibitory activity towards NDH-2 of M. smegmatis, and the IC50 value of GS (2 µM) is significantly lower than that of trifluoroperazine (12 µM) for M. tuberculosis NDH-2 [44]. Recent screening of the natural antibiotics library revealed that that polymyxin B acts as a quinone reduction site inhibitor of M. smegmatis NDH-2 and MQO [35].
New mechanism for the action of cationic cyclic peptide antibiotics
GS and polymyxins are structurally unrelated cationic cyclic peptides, but their targets for antimicrobial activity are assumed to be the bacterial membranes. GS acts on the lipid bilayer of the cytoplasmic membrane and enhances the permeability by destroying membrane integrity [19–22]. Polymyxins bind to LPS in the outer membrane of the Gram-negative bacteria and lead to a local disturbance of the outer membrane which results in increased permeability of the cytoplasmic membrane [45–47]. Polymyxins can exhibit antibiotic activity towards mycobacterial species, which have the micolic acid-based cell wall in place of LPS [48, 49]. Thus, a universal mechanism of GS and polymyxins for their antibacterial activities is the direct or indirect destruction of the bacterial cytoplasmic membranes [19–22, 45–47].
Gramicidins have been shown to inhibit eukaryotic P-type ATPases. Kasamo [50] observed the inhibitory effect of GS (IC50 = 24 µM) and tyrocidine on plasma membrane Mg2+/K+-ATPase from tobacco leaves. Gramicidin D (linear gramicidins) was less effective than GS. Zhao and Dhalla [51] reported that GS inhibited rat heart plasma membrane Ca2+-ATPase uncompetitively (IC50 = 3 µM) and sarcoplasmic reticulum Ca2+-ATPase in a mixed-type manner (IC50 = 6 µM). Iglesias and Rega [52] found that GS inhibited Ca2+-ATPase of human red-cell membranes by lowering the maximum velocity of the high affinity component and the apparent affinity of the low-affinity component. Reversal of the inhibitory effect of GS by liposomes suggests that GS acts on the hydrophobic domain of Ca2+-ATPase. It was found recently that gramicidin A directly inhibits ATP hydrolysis of Na+/K+-ATPase (IC50 = 8 µM) from porcine cerebral cortex in the mixed-type manner while GS (IC50 = 41 µM) is less effective against Na+/K+-ATPase [53]. Notably, a mixture of linear gramicidins inhibits RNA synthesis by the purified A. migulanus RNA polymerase at high concentrations (50% inhibition at 54 µM), by interfering binding to DNA [54]. These observations indicate that GS and other peptide antibiotics interact with distinct targets other than the lipid bilayers.
By taking the advantages in structural variations of natural compounds and respiratory enzymes from different species, we carried out matrix screening of the natural antibiotics library with bacterial membranes. We demonstrated that the library is a potential source of species-specific novel inhibitors of respiratory enzymes [31–36, 43]. Cationic cyclic peptide antibiotics like GS and polymyxins would be accumulated in bacterial cytoplasmic membranes and then reach to the quinone-binding sites in the hydrophobic domains of peripheral or intrinsic membrane proteins (Fig. 2). So far, only membrane-bound respiratory enzymes [31–36, 43] and P-type ATPases [50–52] have been identified as molecular targets of GS and polymyxins. Penetration of peptide antibiotics across the bacterial membranes, and the identification of soluble molecular targets need to be examined in future studies. Considering their poor solubility in aqueous solutions, the upper limits of IC50s for chemotherapeutically important drug candidates are the µM concentration. Thus, our findings provide new insights for the molecular design and development of cationic cyclic peptide antibiotics targeting to bacterial membrane proteins like respiratory enzymes.

Interactions of gramicidin S (GS), gramicidin A, and polymyxin B in the cell surface of the Gram-negative bacteria. See details in text
Concluding remarks and perspectives
Recent studies have shown that structural analogs of GS [16, 55–57], tyrocidine [58], and polymyxin [59] can be designed with markedly reduced hemolytic activity and enhanced microbial activity, suggesting that cationic cyclic peptide antibiotics could be used as potent oral or injectable broad-spectrum antibiotics. In addition to the chemical synthesis approach, as shown for gramicidin S synthase [60], computational structure-based redesign of the nonribosomal peptide synthase is an alternative strategy to facilitate making new cyclic peptide antibiotics in a large quantity. Previous studies on polymyxins have likely been insufficient to abandon their clinical uses. Randomized controlled trials, the determination of the pharmacokinetic/pharmacodynamic properties, the development of improved formulations [61], dosage optimization, and the evaluation of toxicity of peptide antibiotics, are all urgently needed for treatments of infections with MDR bacterial strains. Continuing efforts to identify antibiotics with new targets and mechanisms of action, as well as careful examination of their clinical application, will lead to the development of new chemotherapeutics against drug-resistant bacteria. Further, tyrocidin A (IC50 = 0.6 nM) [62] and tryptophan-N-formylated gramicidin A (a nonhemolytic derivative with IC50 = 2.6 nM) [63, 64] have been shown to be potent antimalarial agents for the human malaria parasite Plasmodium falciparum. Application of peptide antibiotics to parasitic protists and other infectious disease needs to be examined.
References
Foster TJ (2004) The Staphylococcus aureus ‘superbug’. J Clin Invest 114:1693–1696
Brown ED, Wright GD (2005) New targets and screening approaches in antimicrobial drug discovery. Chem Rev 105:759–774
Falagas ME, Kasiakou SK (2005) Colistin: the revival of polymyxins for the management of multidrug-resistant Gram-negative bacterial infections. Clin Infect Dis 40:1333–1341
Evans ME, Feola DJ, Rapp RP (1999) Polymyxin B sulfate and colistin: old antibiotics for emerging multiresistant gram negative bacteria. Ann Pharmacother 33:960–967
Li J, Nation RL, Turnidge JD, Milne RW, Coulthard K, Rayner CR, Paterson DL (2006) Colistin: the re-emerging antibiotic for multidrug-resistant gram-negative bacterial infections. Lancet Infect Dis 6:589–601
Michalopoulos A, Falagas ME (2008) Colistin and polymyxin B in critical care. Crit Care Clin 24:377–391
Timurkaynak F, Can F, Azap ÖK, Demirbilek M, Arslan H, Karaman SÖ (2006) In vitro activities of non-traditional antimicrobials alone or in combination against multidrug-resistant strains of Pseudomonas aeruginosa and Acinetobacter baumannii isolated from intensive care units. Int J Antimicrob Agents 27:224–228
Arnold TA, Forrest G, Messmer KJ (2007) Polymyxin antibiotics for gram-negative infections. Am J Health Syst Pharm 64:819–826
Levin AS, Barone AA, Penço J, Santos MV, Marinho IS, Arruda EA, Manrique EI, Costa SF (1999) Intravenous colistin as therapy for nosocomial infections caused by multidrug-resistant Pseudomonas aeruginosa and Acinetobacter baumannii. Clin Infect Dis 28:1008–1011
Peschel A, Sahl H-G (2006) The co-evolution of host cationic antimicrobial peptides and microbial resistance. Nature Rev Microbiol 4:529–536
Bowdish DME, Davidson DJ, Hancock REW (2005) A re-evaluation of the role of host defense peptides in mammalian immunity. Curr Protein Pept Sci 6:35–51
Izumiya N, Kato T, Aoyaga H, Waki M, Kondo M (1979) Synthetic aspects of biologically active cyclic peptides: gramicidin S and tyrocidines. Halsted, New York
Waki M, Izumiya N (1990) Recent advances in the biotechnology of β-lactams and microbial bioactive peptides. In: Kleinhaug H, van Doren H (eds) Biochemistry of peptide antibiotics. de Gruyter, Berlin, pp 205–244
Xu Y, Sugár IP, Krishna NR (1995) A variable target intensity-restrained global optimization (VARTIGO) procedure for determining three-dimensional structures of polypeptides from NOESY data: application to gramicidin-S. J Biomol NMR 5:37–48
Ando S, Nishikawa H, Takiguchi H, Lee S, Sugihara G (1993) Antimicrobial specificity and hemolytic activity of cyclized basic amphiphilic β-structural model peptides and their interactions with phospholipid bilayers. Biochim Biophys Acta 1147:42–49
Kondejewski LH, Farmer SW, Wishart D, Kay CM, Hancock REW, Hodges RS (1996) Modulation of structure and antibacterial and hemolytic activity by ring size in cyclic gramicidin S analogs. J Biol Chem 271:25261–25268
Otoguro K, Liu ZX, Fukuda K, Li Y, Iwai Y, Tanaka H, Ōmura S (1988) Screening for new nematocidal substances of microbial origin by a new method using the pine wood nematode. J Antibiotics 41:573–575
Mayer A, Anke H, Sterner O (1997) Omphalotin, a new cyclic peptide with potent nematicidal activity from Omphalotus olearius I. Fermentation and biological activity. Nat Product Lett 10:25–32
Katsu T, Ninomiya C, Kuroko M, Kobayashi H, Hirota T, Fujita Y (1988) Action mechanism of amphipathic peptides gramicidin S and melittin on erythrocyte membrane. Biochim Biophys Acta 939:57–63
Prenner EJ, Lewis RNAH, Newman KC, Gruner SM, Kondejewski LH, Hodges RS, McElhaney RN (1997) Nonlamellar phases induced by the interaction of gramicidin S with lipid bilayers. A possible relationship to membrane disrupting activity. Biochemistry 36:7906–7916
Jelokhani-Niaraki M, Hodges RS, Meissner JE, Hassenstein UE, Wheaton L (2008) Interaction of gramicidin S and its aromatic amino-acid analog with phospholipid membranes. Biophysical J 95:3306–3321
Ashrafuzzaman Md, Andersen OS, McElhaney RN (2008) The antimicrobial peptide gramicidin S permeabilizes phospholipid bilayer membranes without forming discrete ion channels. Biochim Biophys Acta 1778:2814–2822
Salgado J, Grage SL, Kondejewski LH, Hodges RS, McElhaney RN, Ulrich AS (2001) Membrane-bound structure and alignment of the antimicrobial β-sheet peptide gramicidin S derived from angular and distance constraints by solid state 19F-NMR. J Biomol NMR 21:191–208
Wallace BA (1998) Recent advances in the high resolution structures of bacterial channels: gramicidin A. J Struct Biol 121:123–141
Kelkar DA, Chattopadhyay A (2007) The gramicidin ion channel: a model membrane protein. Biochim Biophys Acta 1768:2011–2025
Storm DR, Rosental KS, Swanson PE (1977) Polymyxin and related peptide antibiotics. Annu Rev Biochem 46:723–763
Scholar EM, Pratt WB (2000) Antibiotics that affect membrane permeability. Polymyxin B, colistin, Gramicidin A. In: Scholar EM, Pratt WB (eds) The antibacterial drugs. Oxford University Press, New York, pp 234–241
Schindler PRG, Teuber M (1975) Action of polymyxin B on bacterial membranes: morphological changes in the cytoplasm and in the outer membrane of Salmonella typhimurium and Escherichia coli B. Antimicrob Agents Chemother 8:95–104
Zhang L, Dhillon P, Yan H, Farmer S, Hancock REW (2000) Interactions of bacterial cationic peptide antibiotics with outer and cytoplasmic membranes of Pseudomonas aeruginosa. Antimicrobial Agents Chemother 44:3317–3321
Ui H, Ishiyama A, Sekiguchi H, Namatame M, Nishihara A, Takahashi A, Shiomi K, Otoguro K, Ōmura S (2007) Selective and potent in vitro antimalarial activities found in four microbial metabolites. J Antibiot 60:220–222
Mogi T, Ui H, Shiomi K, Ōmura S, Kita K (2008) Gramicidin S identified as a potent inhibitor for cytochrome bd-type quinol oxidase. FEBS Lett 582:2299–2302
Mogi T, Ui H, Shiomi K, Ōmura S, Miyoshi H, Kita K (2009) Antibiotics LL-Z1272 identified as novel inhibitors discriminating bacterial and mitochondrial quinol oxidases. Biochim Biophys Acta 1787:129–133
Mogi T, Ano Y, Nakatsuka T, Muroi A, Miyoshi H, Migita CT, Ui H, Shiomi K, Ōmura S, Kita K, Matsushita K (2009) Biochemical and spectroscopic properties of cyanide-insensitive quinol oxidase from Gluconobacter oxydans. J Biochem 146:263–271
Mogi T, Matsushita K, Murase Y, Kawahara K, Miyoshi H, Ui H, Shiomi K, Ōmura S, Kita K (2009) Identification of new inhibitors for alternative NADH dehydrogenase (NDH-II). FEMS Microbiol Lett 291:157–161
Mogi T, Murase Y, Mori M, Shiomi K, Ōmura S, Paranagama MP, Kita K (2009) Polymyxin B identified as an inhibitor of alternative NADH dehydrogenase and malate: quinone oxidoreductase from the Gram-positive bacterium Mycobacterium smegmatis. J Biochem 146. doi:10.1093/jb/mvp096
Mogi T, Kawakami T, Arai H, Igarashi Y, Matsushita K, Mori M, Shiomi K, Ōmura S, Kita K (2009) Siccanin rediscovered as a species-selective succinate dehydrogenase inhibitor. J Biochem 146. doi:10.1093/jb/mvp085
Way SS, Sallustio S, Magliozzo RS, Goldberg MB (1999) Impact of either elevated or decreased levels of cytochrome bd expression on Shigella flexneri virulence. J Bacteriol 181:1229–1237
Endley S, McMurray D, Ficht TA (2001) Interruption of the cydB locus in Brucella abortus attenuates intracellular survival and virulence in the mouse model of infection. J Bacteriol 183:2454–2462
Shi L, Sohaskey CD, Kana BD, Dawes S, North RJ, Mizrahi V, Gennaro ML (2005) Changes in energy metabolism of Mycobacterium tuberculosis in mouse lung and under in vitro conditions affecting aerobic respiration. Proc Natl Acad Sci USA 102:15629–15634
Fisher N, Bray PG, Ward SA, Biagini GA (2007) The malaria parasite type II NADH:quinone oxidoreductase: an alternative enzyme for an alternative lifestyle. Trends Parasitol 23:305–310
Kawahara K, Mogi T, Tanaka TQ, Hata M, Miyoshi H, Kita K (2009) Mitochondrial dehydrogenases in the aerobic respiratory chain of the rodent malaria parasite Plasmodium yoelii yoelii. J Biochem 145:229–237
Kerscher SJ (2000) Diversity and origin of alternative NADH:ubiquinone oxidoreductase. Biochim Biophys Acta 1459:274–283
Mogi T, Miyoshi H (2009) Characterization of cytochrome bd plasoquinol oxidase from the cyanobacterium Synechocystis sp. PCC 6803. J Biochem 145:395–401
Yano T, Li L-S, Weinstein E, The J-S, Rubin H (2006) Steady-state kinetics and inhibitory action of antitubercular phenothiazines on Mycobacterium tuberculosis type-II NADH-menaquinone oxidoreductase (NDH-2). J Biol Chem 281:11456–11463
Weinstein S, Wallace BA, Morrow JS, Veatch WR (1980) Conformation of the gramicidin A transmembrane channel: a 13C nuclear magnetic resonance study of 13C-enriched gramicidin in phosphatidylcholine vesicles. J Mol Biol 143:1–19
Fekety R (1990) Polymyxins. In: Mandell GL, Douglast RG Jr, Bennett JE (eds) Principles and practice of infectious diseases, 3rd edn. Churchill Livingstone, New York, pp 323–325
Vaara M, Viljanen P (1985) Binding of polymyxin B nonapeptide to gram-negative bacteria. Antimicrob Agents Chemother 27:548–554
David HL, Rastogi N (1985) Antibacterial action of colistin (polymyxin E) against Mycobacterium aurum. Antimicrob Agents Chemother 27:701–707
McGarvey JA, Bermudez LE (2001) Phenotypic and genomic analyses of the Mycobacterium avium complex reveal differences in gastrointestinal invasion and genomic composition. Infect Immun 69:7242–7249
Kasamo K (1982) Gramicidin S: a potent inhibitor of membrane-bound epidermal adenosine triphosphatase from Nicotiana tabacum L. leaves. Plant Cell Physiol 23:195–204
Zhao DY, Dhalla NS (1989) Influence of gramicidin S on cardiac membrane Ca2+/Mg2+ ATPase activities and contractile force development. Can J Physiol Pharmacol 67:546–552
Iglesias RO, Rega AF (1987) Gramicidin S inhibition of the Ca2+-ATPase of human red blood cells. Biochim Biophys Acta 905:383–389
Takada Y, Matsuo K, Kataoka T (2008) Gramicidin A directly inhibits mammalian Na+/K+-ATPase. Mol Cell Biochem 319:99–103
Sarkar N, Langrey D, Paulus H (1977) Biological function of gramicidin: selective inhibition of RNA polymerase. Proc Natl Acad Sci USA 74:1478–1482
Kondejewski LH, Jelokhani-Niaraki M, Farmer SW, Lix B, Kay CM, Sykes BD, Hancock REW, Hodges RS (1999) Dissociation of antimicrobial and hemolytic activities in cyclic peptide diastereomers by systematic alteration in amphipathicity. J Biol Chem 274:13181–13192
Prenner EJ, Kiricsi M, Jelokhani-Niaraki M, Lewis RNAH, Hodges RS, McElhane RN (2005) Structure-activity relationships of diastereomeric lysine ring size analogs of the antimicrobial peptide gramicidin S. Mechanism of action and discrimination between bacterial and animal cell membrane. J Biol Chem 280:2002–2011
Yamada K, Shinoda S, Oku H, Komagoe K, Katsu K, Katakai R (2006) Synthesis of low-hemolytic antimicrobial dehydropeptides based on gramicidin S. J Med Chem 49:7592–7595
Qin C, Zhong X, Bu X, Ng NLJ, Guo Z (2003) Dissociation of antimicrobial and hemeolytic activities of an amphipathic peptide antibiotics. J Med Chem 46:4830–4833
Vaara M, Fox J, Loidl G, Siikanen O, Apajalahti J, Hansen F, Frimodt-Møller N, Nagai J, Takano M, Vaara T (2008) Novel polymyxin derivatives carrying only three positive charges are effective antibacterial agents. Antimicrob Agents Chemother 52:3229–3236
Chen C, Georgiev I, Anderson AC, Donald BR (2009) Computational structure-based redesign of enzyme activity. Proc Natl Acad Sci USA 106:3764–3769
Pattani AS, Mandawgade SD, Patravale VB (2006) Development and comparative anti-microbial evaluation of lipid nanoparticles and nanoemulsion of polymyxin B. J Nanosci Nanotechnol 6:2986–2990
Rautenbach M, Vlok NM, Stander M, Hoppe HC (2007) Inhibition of malaria parasite blood stages by tyrocidines, membrane-active cyclic peptide antibiotics from Bacillus brevis. Biochim Biophys Acta 1768:1488–1497
Otten-Kuipers MA, Roelofsen B, Op den Kamp JAE (1995) Stage-dependent effects of analogs of gramicidin A on the growth of Plasmodium falciparum in vitro. Parasitol Res 81:26–31
Otten-Kuipers MA, Franssen FF, Nieuwenhuijs H, Overdulve JP, Roelofsen B, Op den Kamp JA (1997) Effect of tryptophan-N-formylated gramicidin on growth of Plasmodium berghei in mice. Antimicrob Agents Chemother 41:1778–1782
Acknowledgments
We would like to thank anonymous referees for their expert comments. This work was supported by grant-in-aid for Scientific Research (C) (20570124) and Creative Scientific Research (18GS0314) from the Japan Society for the Promotion of Science, and grant-in-aid for Scientific Research on Priority Areas (18073004) from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Mogi, T., Kita, K. Gramicidin S and polymyxins: the revival of cationic cyclic peptide antibiotics. Cell. Mol. Life Sci. 66, 3821–3826 (2009). https://doi.org/10.1007/s00018-009-0129-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-009-0129-9