
Abstract
A new multi-residue method for the determination of seven polypeptides, namely, polymixin B1, polymixin B2, polymixin E1 (colistin A), polymixin E2 (colistin B), enduracidin A (enramycin A), enduracidin B (enramycin B), and bacitracin A, in food of animal origin was developed and validated for chicken muscle tissue. Chicken muscle tissue was extracted with acidified methanol (1 % TFA). After homogenization, shaking, and centrifugation, the acidified methanol extract was decanted. A second extraction was performed with methanol (1 % TFA) and formic acid (1 %) 25:75, v/v. The pooled extract was cleaned up and concentrated on a solid-phase extraction cartridge. The retained analytes were eluted with methanol/acetonitrile. The extract was evaporated to dryness, reconstituted in mobile phase, filtered, and quantified by LC-MS/MS under ESI conditions. The method has a LOQ of 50.0 μg/kg for polymixin E2 (colistin B), 39.0 μg/kg for polymixin E1 (colistin A), 74.0 μg/kg for polymixin B1, 71.0 μg/kg for polymixin B2, 66.0 μg/kg for enduracidin A, 50.0 μg/kg for enduracidin B, and 30.0 μg/kg for bacitracin A in chicken muscle tissues. This is the first sensitive, suitable, multi-residue method reported for the seven polypeptide drug residues in chicken muscle tissue.

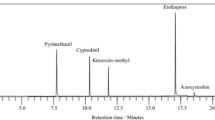
Effect of chicken muscle tissue on the ESI +ve detection signal for Colistin A showing signal suppression in the presence of matrix
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The intensive production of food of animal origin has led to the extensive use of antimicrobials for disease treatment, prophylaxis, and growth promotion. Sometimes they are added to animal feed at sub-therapeutic levels as antimicrobial growth promotants (AGPs) during the entire growing period to improve growth rate and feed conversion efficiency. The practice, however, has been suspected to have resulted in the possible transfer of resistance genes from animal enteric flora to human bacteria and pathogens and increased appearance of cross-resistance and thereby impaired the medical effectiveness of antibiotics reserved for human treatment. With increasing consumer concerns over the adverse effects of AGPs on human and animal health and the transfer of resistance between different bacteria and between humans and animals, the European Union (EU) passed a precautionary legislation in 1999 to ban/prohibit the use of seven specific AGPs, namely, bacitracin, avoparcin, spiramycin, olaquindox, carbadox, tylosin, and virginiamycin, in food animal production [1]. This ban is currently enforced within the EU under European Commission (EC) Directive 70/524/EEC [2].
Polymixin B (B1 and B2) and polymixin E (E1 and E2) are two other peptides (cyclic lipodecapeptides) used in veterinary practice for the treatment of food animals. Polymixin B is derived from the bacterium Bacillus polymyxa and used primarily for treating resistant Gram-negative infections. Listed in Table 1 are MRLs established by the Codex Alimentarius Commission and a few countries for the use of the polypeptides in poultry production. The chemical structures for the four polymixins are shown in Fig. 1.

Chemical structure of the polymixins B1, B2, E1, and E1
Bacitracin (Fig. 2) is a peptide antibiotic produced by certain strains of Bacillus subtilis and Bacillus licheniformis in which bacitracin A (57 %), bacitracin B (22 %), and bacitracin C (13 %) constitute the major components together with a small percentage of bacitracin F (2 %). The recommended dose in Canada for zinc bacitracin for the prevention of necrotic enteritis and bacterial enteritis in swine is 55 mg/kg in the complete feed and 110 mg/kg in the complete feed for the reduction of early mortality of chicks. In veterinary medicine, it is used in combination with tetracycline, neomycin, and prednisolone for intra-mammary treatment of mastitis in lactating cows.

Chemical structure of bacitracin A [C66H103N17O16S; molecular mass 1422.7]
Enduracidin (also called enramycin) is another polypeptide used for the treatment of food animals (Fig. 3). It is a 17 amino acid lipodepsipeptide produced by the bacterium Streptomyces fungicidicus ATCC 2103. In Japan and Korea, enduracidin is approved for use as a growth promotant in chickens (including broilers) with a MRL of 30 μg/kg for chicken muscle, fat, liver, kidney, and edible offal (Figs. 4 and 5).

Chemical structure of enduracidin A [C107H138N26Cl2O31; molecular mass 2355.3]; enduracidin B [C108H140N26Cl2O31; molecular mass 2369.4]

LC-Mass chromatogram of an extract from a drug-free chicken muscle tissue sample obtained using the described method

LC-Mass chromatogram of an extract from a drug-free chicken muscle tissue sample fortified with the seven polypeptides at the indicated concentrations obtained using the described method
In 2000, the EU audited Canada’s meat export program and observed that Canada permitted the use of the three polypeptides (avoparcin, virginiamycin, and bacitracin), the 2 quinoxaline N, N dioxides (olaquindox and carbadox), and the two macrolides (spiramycin and tylosin), all of which had been banned as feed additives in 1999 by the EU, in treating food animals. Additionally, they noted that Canada did not have adequate methods to test them. It was recommended that Canada must certify that its meat exports to the EU were free from these AGPs for those products to be acceptable in trade. Suitably validated methods had to be rapidly developed by Canada in order for the EU to not to use that as a basis for restricting Canadian exports from access to the EU market. At the time of that audit, most methods available for the determination of these compounds including those used in the EU were inhibition assays directed at animal feed at the milligram per kilogram concentrations and were, therefore, not considered suitable for the determination of these residues at the low microgram per kilogram concentrations likely to be found in animal tissues. Since then, Canada has stopped the market authorization of carbadox and Canadian scientists have developed suitable quantitative and confirmatory methods for olaquindox and carbadox [3], bacitracin [4], virginiamycin [5], tylosin, and spiramycin [6] for use in Canada’s residue monitoring program.
Very few methods had previously been published for the analysis of polypeptide residues in food of animal origin. A 1995 publication described thin layer chromatographic methods with UV and fluorescence (FL) detection for the analysis of bacitracin, colistin, virginiamycin, enduracidin, and thiopeptin residues in food of animal origin [7]. A LC/UV method with a LOD of 200 μg/kg for the analysis of enduracidin residues in beef, pork, and chicken muscle tissue had been published [8]. Nagase et al. [9] had reported a LC/FL method with a LOD of 100 μg/kg for virginiamycin M1 and 100 μg/kg for virginiamycin S. Moats [10] had published a LC/UV method with reported recoveries ranging from 83 to 98 % for virginiamycin M1 and Saito et al. [11] had reported another LC/UV method with a LOD of 50 μg/kg and recoveries ranging from 94 to 95 % for virginiamycin M1 in pork and chicken meat. A LC/FL method was described for the determination of colistin residues in bovine milk and tissues (liver, kidney, muscle, and fat) using trichloroacetic acid for protein precipitation, cleanup on a C18 SPE cartridge followed by precolumn derivatization with ortho-phthalaldehyde and 2-mercaptoethanol in borate buffer. The derivatized colistins in the extract were separated on a LiChrospher RP C18 column and quantified by fluorescence detection. LC-electrospray tandem mass spectrometry was used to characterize the identities of the derivatized colistins [12]. In 2006, Wan et al. [13] reported a sensitive method for the analysis of bacitracin A, polymixin E1 (colistin A), and polymixin E2 (colistin B) in animal tissue and milk with reported LOQs at two fifths the MRLs. The intra-day and inter-day method accuracy and precision evaluated at two fifths the concentration of the defined MRL for colistin A and colistin B were 91–104 % with a RSD <7 % and from 90 to 106 % with RSD <9 %, respectively. There were no determinative and confirmatory methods reported for the analysis of the seven polypeptide drug residues reported in this study. This paper describes a multi-residue LC-MS/MS method that was developed and validated as part of Canada’s effort to respond to the EU audit of Canada’s residue program in the year 2000, to increase the analytical sensitivity for the BAC method we had previously developed in our laboratory [4], and, even more importantly, to increase our laboratory’s capability and efficiency to analyze the additional polypeptide antibiotic residues, namely, polymixin B1, polymixin B2, polymixin E1, polymixin E2, enduracidin A, and enduracidin B in chicken muscle tissues to assure unimpeded access of Canadian chicken products to the EU market. The method was validated and demonstrated to be fit-for-use in both domestic and export regulatory enforcement programs. To the best of our knowledge, it is the first reported multi-residue method for the regulatory analysis of all the seven polypeptides in chicken muscle tissue. While this manuscript was in preparation for publication, Kaufmann and Widmer [14] reported in 2013 a method for the quantitative analysis of five of the seven polypeptides reported in our method in bovine muscle, kidney, liver and muscle tissues, bovine milk, and chicken eggs but not for chicken tissue. In their reported method, they had resorted to the addition of trifluoroacetic acid (TFA) as ion-pairing reagent in their mobile phase in order to obtain sharply resolved peaks especially for the polymixins. Also in that publication which did not include the enduracidins, only the elution profile of polymixins B1 and B2 out of the five analytes were shown with retention times of about 7 and 9 min in a 14-min run time. In our method, bacitracin and all the other polypeptides are eluted and analyzed by LC-MS/MS with a 15-min run time without the need to add TFA as ion-pairing reagent to the mobile phase.
Experimental
Chemicals
All water used in the method was purified by reverse osmosis followed by deionization, adsorption, and filtration. Acetonitrile, ethyl acetate, hexane, methanol, and all high HPLC grade reagents were obtained from Caledon (Georgetown, Canada). Formic acid (98.0 % purity) was obtained from EMD (Darmstadt, Germany). Trifluoroacetic acid (99 % purity) was obtained from Alfa Aesar (Ward Hill, MA, USA). Bacitracin A was obtained from Dr. Ehrenstorfer GmbH, Germany. Analytical reference standard materials for polymixin E1, polymixin E2, polymixin B1, and polymixin B2 were obtained as pure standards from the Katholieke Universiteit (Leuven, Belgium). Enduracidin hydrochloride was obtained from Sigma-Aldrich (Oakville, Canada) as a mixture of 46 % enduracidin A and 32 % enduracidin B. Mobile phase A was 0.1 % formic acid; mobile phase B was methanol.
Reagents and standard solutions
0.1 % Formic acid in water or methanol was prepared by transferring 1.0 mL formic acid to a 1-L volumetric flask half filled with water or methanol and bringing to volume with water or methanol. 0.1 M Hydrochloric acid was prepared by transferring 8.3 mL of concentrated hydrochloric acid to a 1-L volumetric flask half filled with water and bringing to volume with water. 1 % Trifluoroacetic acid in methanol [methanol (1 % TFA)] or water [water (1 % TFA)] was prepared by transferring 10 mL trifluoroacetic acid to a 1-L volumetric flask half filled with methanol or water and bringing to volume with methanol or water. The elution solution, methanol/acetonitrile (80:20, v/v), was also prepared. Mobile phase A (0.1 % formic acid in water) and mobile phase B (methanol) were prepared and filtered under vacuum through a 0.2-μm filter. An IRIS Polymeric SPE cartridge (C18/14 %; 100 mg/3 mL) was purchased from Canadian Life Sciences (Peterborough, Canada). A 0.2 μm PTFE (polytetrafluoroethylene) Acrodisc CR 13 mm syringe filter was obtained from Pall Canada Ltd. (Mississauga, Canada).
1000 mg/L of the standard solutions were prepared by accurately weighing 50 mg of standard material into a 50-mL volumetric flask and bringing to volume with methanol containing 0.1 % formic acid. A 10 mg/L working standard solution was prepared by pipetting 1 mL of the stock standard solution into a 100-mL volumetric flask and making it up to volume with methanol containing 0.1 % formic acid. Appropriate volumes of the 10 mg/L working standard solutions for each polypeptide were used for fortifying the chicken tissue samples for sample preparation.
Chicken tissue sample preparation
Control/blank (drug-free) chicken muscle tissues obtained from six different geographical areas of poultry production in Canada as well as test tissues were thawed and cut into very small pieces. 5.0 ± 0.05 g aliquots of test tissue were weighed into a 50-mL polypropylene centrifuge tube. Also, 5.0 ± 0.05 g control (drug-free) tissue were weighed into each of eight individual 50-mL polypropylene centrifuge tubes.
Preparation of calibration standard solutions
Chicken muscle tissue standard calibration curves were prepared by fortifying six of the eight weighed out control (drug-free) tissue samples with appropriate volumes of the 10 mg/L standard working solution to prepare tissue calibration standards ranging from 40 to 240 μg/kg for polymixin E1, 30–160 μg/kg for polymixin E2, 20–160 μg/kg for polymixin B2, 70–508 μg/kg for polymixin B1, 70–200 μg/kg for enduracidin A, 50–140 μg/kg for enduracidin B, and 30–160 μg/kg for bacitracin. The seventh sample was set aside and fortified by the Laboratory Quality Manager to serve as Quality Control (QC) sample. The eighth sample was not fortified and served as negative control sample for the analysis. [Note: The fortified samples and the test samples can be stored at 4 °C overnight before being analyzed the next morning.]
Muscle tissue sample extraction
To each fortified control tissue and all the other test samples was added 7 mL 1 % TFA in methanol. Each sample was homogenized for 30 s with a polytron. The polytron probe was rinsed with 2 × 1.5 mL portions of 1 % TFA in methanol. Samples were mechanically shaken at high speed for 10 min, centrifuged at 3900×g for 10 min, and the supernatant was transferred into a 50-mL polypropylene centrifuge tube. The residual pellet was extracted with 5 mL 1 % TFA in methanol and 15 mL 1 % TFA in water. The mixture was mechanically shaken at high speed for 15 min, centrifuged at 3900×g for 10 min, after which the supernatant was combined with the supernatant from the previous extraction. 15 mL 1 % TFA in water was added to the combined extract and mechanically shaken for 5 min and centrifuged at 3900×g for 10 min. The extract was filtered into a 125-mL filtering flask through a Buchner funnel fitted with a glass fiber filter paper. 15 mL 0.1 M HCl was added to the contents of the flask mixed thoroughly and readied for SPE cleanup.
Clean-up procedures
An IRIS Polymeric SPE cartridge was conditioned by passing 3 mL methanol, 3 mL water, and 3 mL 0.1 M HCl. About 200 μL of acid was left to sit in the cartridge. A 75-mL solvent reservoir was mounted onto the SPE cartridge, and the sample extract was loaded onto the cartridge. The cartridge was dried under full vacuum for at least 20 min making sure no water droplets remained sticking on the wall of the SPE cartridge. The solvent reservoir was removed and the cartridge was washed with 9 mL ethyl acetate, 15 mL 1 % formic acid in ethyl acetate, and 9 mL hexane. The cartridge was dried under full vacuum for 2 min or more after each solvent addition. The retained analytes were eluted with 2 × 2 mL methanol/acetonitrile (80:20) into a 10-mL glass centrifuge tube. [Note: The extract at this point can be stored at 4 °C where it has been determined to be stable for a couple of days before being evaporated to dryness.] The organic extract was evaporated to dryness using the N-evap at 40 °C and each reconstituted with 300 μL 0.1 % formic acid in methanol. Each reconstituted sample was vortex mixed briefly and sonicated for 5 min after which 700 μL 0.1 % formic acid in water was added. The sample was vortex mixed briefly, sonicated for 5 min, and filtered through a 0.2-μm PTFE Acrodisc CR 13 mm syringe into an LC sample vial for LC-MS/MS analysis. With a 30-place vacuum manifold, it is possible for an analyst to analyze 30 samples in a day. [Note: It is important not to use PVDF (polyvinylidene fluoride) filters for filtration of these samples].
Instrumentation
UPLC-MS/MS analysis
The UPLC-MS/MS system consisted of a Waters Acquity UPLC interfaced to a Waters Micromass triple quadrupole Premier mass spectrometer equipped with an ESI source and controlled by MassLynx 4.1 software. Chromatographic separation was conducted on a Poroshell 120, 100 × 2.1 mm id, 2.7 μm (Agilent Technologies, Mississauga, Canada), at a mobile phase flow rate of 0.40 mL/min using the following gradient conditions: 0.00–1.50 min with 90 % A, 1.50–1.60 min with 75 % A, 1.60–9.00 min with 50 % A, 9.01–11.00 min with 2 % A, and 11.20–15.00 with 90 % A. The extracted samples were held in a 10 °C temperature-controlled chamber and 4-μL aliquots of the samples were injected into the analytical system.
The ion source parameters were optimized as follows for each analyte by monitoring the MS/MS spectra of the analytes: Data acquisition was conducted in positive ESI mode using nitrogen as nebulizer and desolvation gas at flow rates of 50 and 1100 L/h, respectively. The capillary voltage was kept at 0.8 kV. The ion source temperature was held at 110 °C and the desolvation temperature at 450 °C. The low mass and high mass lenses were set for unit mass resolving power and the electron multiplier were held at 700 V. For MS/MS experiments, the flow of argon gas used as collision gas was set at 0.25 mL/min which results in a pressure of 4.56 × 10−3 mbar. Selected reaction monitoring (SRM) experiments were conducted acquiring two to four product transition ions for every precursor ion of interest. The precursor and product transition ions that were monitored for the quantitative and confirmatory analysis of each of the polypeptides in the matrices evaluated in this study are shown in Table 2.
Confirmation of the identity of a polypeptide was achieved if the retention time of an identified peak in the mass extracted SRM chromatogram matched the retention time of the known polypeptide in a similar matrix, the precursor ion and its associated transition product ions are present, and the calculated ion ratios of the analyte in the identified matrix match those of the analyte in a similar matrix.
Calculations
Before each day’s run, a wet prime of the injector was performed, the syringe was purged, and the needle wash of the LC system was primed. The sample cone was cleaned before the mass spectrometer was turned on. The UPLC system was allowed to equilibrate for 15 min. Using a QC sample from a previous run, four injections or more were made into the LC-MS/MS system to condition the column and the sample cone. The MS detector response (peak area) for each analyte measured at the specified retention time from the TIC chromatograms were plotted against the corresponding concentration X k of the analyte. After examining the data to see if they meet the criteria for linear regression analysis, a regression equation was derived for each of the polypeptides included in the study as follows:
where
- m :
-
Slope of the regression line
- b :
-
Intercept on the response axis
- Y k :
-
Predicted/calculated response
- X k :
-
Concentration of analyte added to control tissue
- s e :
-
Standard estimate of the error in the slope or intercept
The concentration of a polypeptide in a “test sample” was obtained by interpolating its mass detector response (peak area under the triple-charge product/transition ion) at the appropriate retention parameter using the regression Eq. 1.
Method validation
The method was validated to determine whether it is fit for use in a regulatory control program for the analysis of residues of the seven polypeptides included in the method. This included the evaluation of linearity, within run and between run accuracy and precision, limit of detection (LOD), limit of quantification (LOQ), limit of confirmation (LOC), selectivity, applicability of the method, stability in matrix during storage, stability in stock and working solutions during storage, stability of analytes in extract during sample analysis, and during freeze-thaw cycles of tissues during storage.
Selectivity
Chicken muscle tissue samples without any known previous treatment with any of the polypeptides under study were obtained from six different geographical regions of poultry production in Canada and tested for the seven polypeptides.
Interference experiments
To determine whether other veterinary drugs likely to be used in poultry production would impact the detector responses for these polypeptides and, therefore, impact the ability to accurately quantify the amounts of polypeptides in chicken muscle samples, five cocktail mixes (I–V) were prepared as follows: Cocktail mix I contained six tranquilizers, Cocktail mix II included ten non-steroidal anti-inflammatory drugs, Cocktail mix III contained 11 corticosteroids, and Cocktail mix IV contained eight steroids. These cocktail mixtures were selected because the laboratory had methods for all of them. To determine whether the presence of the analytes in the cocktail mixes would mask, enhance, or not affect the analysis of the polypeptide, the following two experiments were conducted as follows:
-
(a)
Five previously weighed negative control chicken muscle tissue samples were fortified with each of the cocktail mixes to a final concentration of 100 μg/kg, and
-
(b)
Five previously weighed negative control chicken muscle samples were fortified with the polypeptides at their LOQs and each of the cocktail mixes to a final concentration of 100 μg/kg.
Matrix effects (ME)
To understand the effect that the chicken muscle tissue matrix would have on the mass detector responses under ESI (+ve) conditions in order to determine to what extent they would influence and impact quantitative analysis, the detector responses for each of the analytes in neat solvents were compared to those of matrix-matched (chicken muscle) standards. Matrix-matched samples were prepared by extracting blank (i.e., drug-free) tissue samples following the method described and adding the analytical standards to the extract just prior to re-constitution with solvent for LC-mass spectral analyses. This was accomplished by plotting the calibration curves of detector response versus concentration of polypeptide over the analytical range for chemical standards (CS) in neat solvent (i.e., without matrix) and comparing their slopes with those of the matrix-matched (MM) samples over the same analytical range. This permitted the calculation of the Matrix Effect (% ME) from Eq. 2:
Extraction efficiency (EE) of the method for the seven polypeptide residues
Having established that there was a significant matrix suppression effect on the MS detector signal for each of the seven polypeptides in this study under ESI (+ve) conditions, we calculated the extraction efficiency for each polypeptide over the analytical range from Eq. 3:
A matrix fortified (MF) sample is one in which the analytes of interest are fortified to a blank (drug-free) sample prior to extraction and clean-up following the described method.
The Process Efficiency (% PE) which measures the net effect of extraction efficiency plus matrix effect on the final response of the analyte extracted from the matrix was calculated from Eqs. 3 or 4:
Quantitative analysis
Because it had been demonstrated that there were significant, but varying matrix effects on the detector signals for each polypeptide, matrix fortified calibration standards were determined to be the most appropriate calibration set to use since they would inherently correct for both recoveries and account for the matrix effects. We therefore proceeded to generate the following linear equations obtained over a period of at least 6 days from a minimum of six matrix fortified calibration standards data sets monitoring the triple-charge transition ion for each polypeptide under ESI (+ve) conditions for the quantitative analysis of each of the polypeptides:
with coefficients of determination, R 2 ≥0.995 for all the polypeptides. From these regression Eqs. 5–11, the limits of detection (LOD) and limits of quantification (LOQ) for the method were calculated and experimentally verified.
The limit of confirmation (LOC)
These were experimentally determined for each polypeptide by examination of the confirmation criteria on the analysis of matrix-fortified chicken tissue samples at sequentially diminishing concentrations to that defined concentration at, and below which, the confirmation criterion failed.
Accuracy and precision
Intra-day and inter-day precision (calculated as % RSD) and % accuracy [calculated as the ratio of the difference in concentration of polypeptide fortified to sample (known) from the experimentally measured concentration to that of the known (fortified) concentration multiplied by 100] were measured for each polypeptide. The intra-day precision and accuracy of the method was determined by running three independent replicates at each of three concentrations (i.e., 50.0, 75.0, and 100 ng/g). The experiments were repeated on two to three other consecutive days at each of the same three concentrations to estimate the inter-day precision and accuracy of the method. The results of these experiments are summarized in Table 3.
Stability studies
The method was used to determine the stability of:
-
(a)
the polypeptides under frozen storage conditions (−20 °C) for samples fortified at 75.0, 150, and 225 μg/kg over a 5–12-week period;
-
(b)
the polypeptides to four freeze-thaw cycle experiments over a 4-week storage period; and
-
(c)
the chemical standards and matrix extracts were studied by repeatedly injecting one chemical standard in neat solvent and two chicken muscle tissue extracts over two to three cycles of a typical analysis time (7.8 h).
Results and discussion
Figure 1a shows the chemical structures of the polymixins B1, B2, E1, and E2, bacitracin (Fig. 1b), and enduracidin (Fig. 1c) together with their molecular weights ranging from 1155 to 1203. The polypeptides generate double-charge and triple-charge ions readily under ESI (+ve) conditions (Table 2). Figure 2a displays a LC-mass chromatogram of an extract obtained from a drug-free chicken muscle sample. Figure 2b shows the LC-mass chromatogram for the seven polypeptides fortified to drug-free chicken muscle tissue at the defined concentrations for the method and extracted using the described procedure: polymixin E2 at 50 ng/g, polymixin B2 at 50 ng/g, polymixin E1 at 50 ng/g, polymixin B1 at 50 ng/g, enduracidin A at 50 ng/g, enduracidin B at 35 ng/g, and bacitracin A at 50 ng/g, respectively. The elution profile of the seven polypeptides on this column was polymixin E2, polymixin B2, polymixin E1, polymixin B1, enduracidin A, enduracidin B, and bacitracin A with retention times of 3.39, 3.76, 4.12, 4.54, 7.44, 8.43, and 9.04 min, respectively. It can be seen from the chromatogram that all the seven polypeptides fortified to the sample at the specified concentrations were chromatographically resolved from one another and from other tissue components. It is also significant to note that the chromatographic conditions used in our method permitted a rapid analysis of all seven polypeptides under a 15-min LC run time without the addition of ion-pairing agents as reported in the Kaufmann paper [14]. In that paper which showed a 14-min LC run time, only the chromatographic retention profiles for polymixin B1 and B2 were shown. The sharper peaks obtained for polymixin B2, it was claimed, was because of the addition of TFA to the mobile phase even though that practice resulted in even further suppression of the mass detector signal for polymixin B1. These conditions were not observed in our method which did not resort to the use of ion-pairing reagents. Additionally, the interference peaks that we had reported in an earlier publication to elute before and after bacitracin that made it difficult to sensitively analyze for bacitracin [4] which were also observed and reported in previous publications by others [13, 15] were not seen with our method that used a mobile phase that did not include TFA as ion-pairing agent. The triple-charge ions from the polypeptides were selected for quantitative analysis and the double-charge transition ion products to accurately identify a polypeptide.
Method validation
Once all the characteristic performance parameters for the method had been determined, the method was subjected to a validation study to determine whether the method’s measured parameters could be independently verified and also whether it meets the fit-for-purpose criteria for use in a regulatory control program.
Selectivity
The results for all these analyses were negative indicating that the method is selective and is able to detect truly negative (i.e., polypeptide-free) samples as negative.
Interference experiments
The samples from experiments “a” and “b” were extracted and analyzed following the described method. All the samples in experiment “a” tested negative while all the samples from experiment “b” tested positive. The results demonstrated that the method’s ability to detect truly negative samples will not be affected by the presence of these other potential interfering compounds and that the presence of these compounds in a chicken muscle tissue will not mask, enhance, or suppress the detector responses required for the quantitative analysis of the seven polypeptides included in this study.
Matrix effects
From Eq. 2, a ME >100 indicates signal enhancement while a ME <100 indicates signal suppression. The results of this analysis (data not shown) indicated that chicken muscle tissue significantly suppresses the mass detector signals for all the polypeptides and confirmed our notion and those of others [14] not to use neat standard solutions of these polypeptides for their quantitative LC-MS/MS analysis.
Extraction (EE) and process efficiency (PE) of the method
The % Extraction Efficiencies calculated for polymixin E1 (colistin A), polymixin E2 (colistin B), enduracidin A, enduracidin B, polymixin B1, polymixin B2, and bacitracin A calculated from Eq. 3 were 41, 44, 40, 40, 41, 40, and 52 %, respectively. The Process Efficiency (% PE) which measures the net effect of extraction efficiency plus matrix effect on the final response of the analyte extracted from the matrix was calculated from Eqs. 3 and 4 as 23, 24, 22, 28, 25, 24, and 39 % for polymixin E2, polymixin B2, polymixin E1, polymixin B1, enduracidin A, enduracidin B, and bacitracin, respectively.
LODs, LOQs, and LOCs
LOQs calculated from the linear regression equations developed in Eqs. 5–11 were as follows: 39.0, 50.0, 71.0, 74.0, 66.0, 50.0, and 30.0 μg/kg for polymixin E1, polymixin E2, polymixin B1, polymixin B2, enduracidin A, enduracidin B, and bacitracin A, respectively. The corresponding LODs for the polypeptides are 12, 15, 21, 22, 20, 15, and 9 μg/kg for polymixin E1, polymixin E2, polymixin B1, polymixin B2, enduracidin A, enduracidin B, and bacitracin A, respectively. The method’s limit of confirmation (LOC) is defined as the concentration where the weakest diagnostic ion no longer appears at an acceptable signal-to-noise level or where the false-negative rate becomes excessive. The LOC is estimated to show that the method is sufficiently sensitive for its purpose and to predict transferability of the method to other laboratories. The LOCs for polymixin E2, polymixin E1, polymixin B2, polymixin B1, enduracidin A, enduracidin B, and bacitracin A are 20, 25, 30, 20, 10, 15, and 12 μg/kg, respectively.
Accuracy and precision
The three concentrations selected for evaluation of the accuracy and precision of the method were 50, 75, and 100 μg/kg. The validation process was to compare the selectivity, LOQs, choice of matrix function for quantification, precision, and accuracy parameters measured for the method by the originators of the method during method development and characterization and assess the ability of the method to accurately and consistently detect and quantify the analytes of interest in “unknown samples.” While the incorporation of stable isotope labeled internal standards would have contributed to the accuracy of the method, it was impossible to find stable isotope labeled standards for these polypeptides. Therefore, to determine if, in the absence of stable isotope internal standards, the variability in precision known to be associated with the sample processing of biological samples such as chicken tissue was minimized, we calculated the % CV of the slopes of the calibration curves that were generated from six different lots of drug-free chicken samples over a period of 3 days and found them all to be ≤3–4 %. These results were consistent with the criteria recommended by Matuszewski and enabled us to conclude that our method can be considered to be free from the relative matrix effects [16].
For the validation study, an experienced analyst who was not originally involved in the development and measurement of the characteristic performance parameters for the method was provided a copy of the standard operating procedure for the method and instructed to use the method to analyze a set of samples including negative control samples and negative control samples fortified in triplicate at three selected concentrations for each of the polypeptides. All the polypeptides showed within-day and between-day precision of <14 % calculated as % RSD. The accuracy determined for all the samples analyzed was ≤±20 % without any systematic bias. The results of the independent evaluation of the claims of the analytical method for precision and accuracy showed that there were no statistically significant differences between the parameters determined at characterization by the originators of the method and those determined independently through the validation process (Table 3).
Stability studies
The results of the stability studies (data not shown) showed that the polypeptide-containing chicken muscle tissue samples are stable when stored at −20 °C for 8 weeks. The polypeptides were determined to degrade after one freeze-thaw cycle after a week of storage, and it is therefore not recommended to freeze-thaw polypeptide-containing muscle tissue samples; the sample extracts and standards held in the analytical chamber at 10 °C were found to be stable after 31 injections (7.8 h analysis time representing the maximum time for a full 30-sample load) suggesting that these standards and extracts may be stored in the laboratory at 10 °C and re-injected again within a period of 7.8 h should a repeat injection/analysis be required for one reason or other.
Application of the method
In the absence of incurred residue material, samples were prepared in duplicate by the Laboratory Quality Manager, coded, randomized, and provided “blind” to the validation analyst for extraction and analysis. The results shown in Table 4 indicated that the method can be used to accurately identify, confirm, and quantify polypeptides present in an “unknown” real chicken muscle sample either individually and/or as the sum of the major components of the polypeptide as required by international regulations. The method was accurate (no false-negative or false-positive results) without any systematic bias and met the “fit-for-purpose” criterion for use in a regulatory program to monitor the use of these polypeptides in poultry production. The method has since been demonstrated to be suitable for the analysis of these polypeptides in porcine, bovine, turkey muscle tissues, and milk (unpublished data).
Conclusion
A method with a LOQ of 50.0 μg/kg for polymixin E2, 39.0 μg/kg for polymixin E1, 74.0 μg/kg for polymixin B1, 71.0 μg/kg for polymixin B2, 66.0 μg/kg for enduracidin A, 50.0 μg/kg for enduracidin B, and 30.0 μg/kg for bacitracin was developed for the determination and confirmation of seven polypeptide drug residues in chicken muscle. It was validated and demonstrated to be fit for use in a regulatory control program and determined to be suitable for the determination of polymixin E and bacitracin residues in chicken muscle tissue for which the Codex Alimentarius Commission and several countries including the EU have proposed MRLs of 150 and 500 μg/kg, respectively, for chicken muscle tissue. With a LOC of 20, 25, 30, 20, 10, 15, and 12 μg/kg, the method would also be suitable for regulatory screening of the use of enduracidin in Japan and Korea. Therefore, our method, which does not use ion-pairing reagents in the mobile phase, permits us to use the same instrument to conduct different analysis for which the use of ion-pairing reagents would demand significant washing/cleaning of our LC lines that may contribute to significant instrument downtime prior to switching the instrument for the analysis of other analytes for which the presence of TFA would be deleterious. This multi-residue polypeptide method, the first of its kind to be reported for the analysis of these seven polypeptide residues in chicken muscle tissues, will, hopefully, demonstrate to our trading partners including the European Union that Canada is now more than able to ensure market access of its poultry meat products to the international global market. With a 30-place vacuum manifold, it is possible for an analyst to analyze 30 samples in a day.
References
Council Regulation 2821/98, Commission regulation 2788/98 (1998) Off J Eur Commun Legis L347:32
Commission of the European Communities, Council Directive 70/524/EEC (1994) Off J Eur Commun Legis L270:1
Lee S, Gedir R, Boison JO (2009) A determinative and confirmatory method for residues of the metabolites of carbadox and olaquindox in porcine tissues. Anal Chim Acta 637:128–134
Lee S, Matus J, Gedir R, Boison J (2011) A validated LC-MS method for the determination of bacitracin drug residues in edible pork tissues with confirmation by LC-tandem mass spectrometry. J Liq Chromatogr Relat Technol 34:2699–2722
Boison J, Lee S, Gedir R (2009) Analytical determination of virginiamycin drug residues in edible porcine tissues by LC-MS with confirmation by LC-MS/MS. J AOAC Int 92:329–339
Dickson LC, O’Byrne C, Chan W (2012) A quantitative method for residues of macrolide antibiotics in porcine kidney by liquid chromatography-tandem mass spectrometry. J AOAC Int 95(2):567–575
Ikai Y (1995) Chemical analysis of peptide antibiotics. In: Oka H, Nakazawa H, Harada K, MacNeil JD (eds) Chemical analysis of antibiotics used in agriculture, 1st edn. AOAC International, Arlington, VA, USA, pp 407–437, 22201–3301
Horie M, Hoshino Y, Nose N, Nakazawa H, Fujita M, Takabatake E (1985) Determination of enramycin in chicken and swine muscles by high performance liquid chromatography. Food Hyg Saf Sci Shokuhin Eiseigaku Zasshi Jpn 26(4):337–342
Nagase M, Fukamachi K (1987) Determination of virginiamycin in swine, cattle and chicken muscle by HPLC with fluorescence detection. Bunseki Kagaku 36:297–300
Moats WA, Leskinen L (1988) Determination of virginiamycin residues in swine tissue using high performance liquid chromatography. J Agric Food Chem 36:1297–1300
Saito K, Horie M, Hoshino Y, Nose N, Nakazawa H, Fujita M (1989) Determination of virginiamycin in chicken and swine tissues by high performance liquid chromatography. Eisei Kagaku 35:63–67
Decolin D, Leroy P, Nicolas A, Archimbault P (1997) Hyphenated liquid chromatographic method for the determination of colistin residues in bovine tissues. J Chromatogr Sci 35:557–564
Wan EC-H, Sin DW-M, Wong Y-C (2006) Detection of residual bacitracin A, colistin A and colistin B in milk and animal tissues by liquid chromatography tandem mass spectrometry. Anal Bioanal Chem 385:181–188
Kaufman A, Widmer M (2013) Quantitative analysis of polypeptide antibiotic residues in a variety of food matrices by liquid chromatography coupled to tandem mass spectrometry. Anal Chim Acta 797:81–88
Sin DWM, Ho C, Wong Y-C, Ho S-K, Ip AC-B (2005) Analysis of major components of residual bacitracin and colistin in food samples by liquid chromatography tandem mass spectrometry. Anal Chim Acta 535:23–31
Matuszewski BK (2006) Standard line slopes as a measure of a relative matrix effect in quantitative HPLC-MS bioanalysis. J Chromatogr B 830:293–300
Acknowledgment
The authors acknowledge Jana Kinar of the Canadian Food Inspection Agency (CFIA) Laboratory in Saskatoon for technical assistance in the laboratory.
Conflict of interest
Mention of a company name or brand name does not constitute an endorsement by the Canadian Food Inspection Agency Laboratory in Saskatoon above others of a similar nature or product.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Boison, J.O., Lee, S. & Matus, J. A multi-residue method for the determination of seven polypeptide drug residues in chicken muscle tissues by LC-MS/MS. Anal Bioanal Chem 407, 4065–4078 (2015). https://doi.org/10.1007/s00216-015-8644-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-015-8644-z