
Abstract
l-Ornithine is a non-protein amino acid with extensive applications in the food and pharmaceutical industries. In this study, we performed metabolic pathway engineering of an l-arginine hyper-producing strain of Corynebacterium crenatum for L-ornithine production. First, we amplified the L-ornithine biosynthetic pathway flux by blocking the competing branch of the pathway. To enhance L-ornithine synthesis, we performed site-directed mutagenesis of the ornithine-binding sites to solve the problem of l-ornithine feedback inhibition for ornithine acetyltransferase. Alternatively, the genes argA from Escherichia coli and argE from Serratia marcescens, encoding the enzymes N-acetyl glutamate synthase and N-acetyl-l-ornithine deacetylase, respectively, were introduced into Corynebacterium crenatum to mimic the linear pathway of L-ornithine biosynthesis. Fermentation of the resulting strain in a 5-L bioreactor allowed a dramatically increased production of L-ornithine, 40.4 g/L, with an overall productivity of 0.673 g/L/h over 60 h. This demonstrates that an increased level of transacetylation is beneficial for L-ornithine biosynthesis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
L-Ornithine is an important chemical ingredient with applications in the food and pharmaceutical industries as a dietary supplement, as it is known to be efficacious in the treatment of liver diseases and wound healing [10]. It can also be used as a precursor for the synthesis of other amino acids, such as arginine and citrulline, as well as putrescine, an important diamine used as a nylon precursor [29]. L-Ornithine can be produced by fermentation using auxotrophic mutants, such as citrulline- or arginine-requiring mutants of Acinetobacter lwoffii and Corynebacterium sp. obtained by the traditional mutagenesis [1, 6, 17]. However, the yield of L-ornithine is low, and the growth of the auxotrophic mutants is unstable owing to the development of the new auxotrophic mutant.
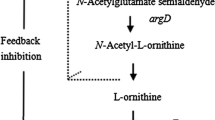
L-Ornithine is an intermediate in the L-arginine biosynthesis pathway; it is produced by the so-called linear pathway in Escherichia coli [19] and by the cyclic pathway in Corynebacterium glutamicum [32]. Recent studies have demonstrated that C. glutamicum might be more advantageous as an L-ornithine overproducer [15]. In C. glutamicum, the L-ornithine biosynthetic pathway is cyclic due to L-ornithine acetyltransferase (OATase, encoded by argJ; EC 2.3.1.35), which catalyzes the conversion of N-acetyl-L-ornithine and L-glutamate to L-ornithine and N-acetyl-L-glutamate (NAG). NAG kinase (NAGK, encoded by argB; EC 2.7.2.8) and then phosphorylates NAG in the second step of the pathway. In addition to OATase and NAGK, argC-encoded N-acetyl-L-glutamate 5-semialdehyde dehydrogenase and argD-encoded N-acetyl-L-ornithine aminotransferase are critical for the conversion of L-glutamate to L-ornithine (as shown in Fig. 1). These four genes are generally involved in one of the L-arginine synthesis clusters, named argCJBD in Corynebacterium strains. Unlike in the cyclic pathway of Corynebacterium spp., there is no OATase in Escherichia coli, and its function is instead performed by N-acetylglutamate synthase (NAGS; encoded by argA; EC 2.3.1.1) and N-acetyl-L-ornithine deacetylase (NAOD; encoded by argE; EC 3.5.1.16). NAGS catalyzes the first committed step of the linear pathway, which is strongly inhibited by arginine but unreactive to ornithine [36]. The enzyme NAOD catalyzes the fifth step of the ornithine biosynthetic pathway, converting N-acetyl-L-ornithine to L-ornithine, an important step for bacterial growth. However, in some bacteria, there are several factors that affect the production of L-ornithine, such as the key enzymes inhibited by L-ornithine and the shortage of precursors and cofactors.

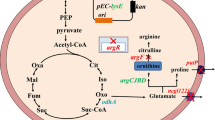
Schematic representation of the L-ornithine biosynthesis pathway of C. crenatum and of the metabolic engineering steps performed in this study. The yellow box represents the targeted modifications of the genes. Red arrows indicate amplification, and the multiple sign indicates gene deletion. Genes and enzymes: argF, encoding ornithine carbamoyltransferase; argA, N-acetyl-glutamate synthase; argE, N-acetyl-L-ornithine deacetylase; argJ, ornithine acetyltransferase; proB, glutamate kinase
Currently, metabolic engineering for amino acid production has focused on carbon flux optimization, including eliminating side pathways, enhancing precursors, and increasing the supply of cofactors and building blocks [18]. In recent years, owing to the increased understanding of L-ornithine biosynthesis and its regulation, many methods of metabolic engineering aiming to enhance L-ornithine production have been carried out [12, 14,15,16, 30, 43]. Hwang et al. reported deletion of argF, argR, proB, and inactivation of the phosphoenolpyruvate carboxykinase activity in a C. glutamicum strain, increasing the L-ornithine yield to 179.14 mg/L [12]. Adaptive evolution and argF/proB/speE triple deletion in C. glutamicum led to a mutant capable of producing L-ornithine at 24.1 g/L [15]. Recently, Kim et al. applied overexpression of argCJBD and optimization of NADPH to produce 51.5 g/L L-ornithine in C. glutamicum YW03 [16]. Subsequently, Zhang et al. reported overexpression of LysE in C. glutamicum S9114, with deletion of argF, ncgl1221, argR, and putP, and attenuation of oxoglutarate dehydrogenase, which resulted in an efficient L-ornithine production titer of 18.4 g/L [43]. In our previous work, we isolated an L-arginine producing strain of C. crenatum (subspecies of C. glutamicum) from a soil sample, and through the breeding of a series of mutants, the mutant strain SYPA 5–5 produced L-arginine at 30.6 g/L [41]. In addition, the repression of the L-arginine biosynthesis operon by the regulator arginine repressor was eliminated by argR lethal mutation [42], and the proB gene involved in byproduct synthesis (L-proline) had been knocked out in our previous study [22]. Moreover, H-9 was used for L-arginine production, with L-arginine feedback inhibition dysregulated by an introduced E19Y point mutation, thus increasing L-arginine production [45, 46]. We initially attempted to enhance L-ornithine accumulation by deleting the side pathway and alleviating the ornithine inhibition of OATase but instead recorded unexpectedly low enzyme activity. Thus, to enhance L-ornithine biosynthesis, in our next attempt, we successfully co-expressed the E. coli argA and S. marcescens argE genes in C. crenatum to mimic a linear transacetylation pathway. To our knowledge, this is the first reported improvement of L-ornithine production in C. crenatum by introducing an artificial linear transacetylation pathway.
Materials and methods
Strains, plasmids, and primers
The argA and argE genes, encoding N-acetylglutamate synthase and N-acetyl-L-ornithine deacetylase, respectively, were identified in E. coli BL21(DE3), Pseudomonas aeruginosa, Serratia marcescens, Pseudomonas putida KT2440, Klebsiella pneumoniae, and Bacillus subtilis 168G, and used as PCR templates. The argJ gene from C. crenatum SYPA5-5, encoding OATase, was also used as a template for PCR. E. coli JM109 and BL21(DE3) were used for gene cloning and expression, respectively. The shuttle vector pDXW10 was used for gene expression in Cc-QF-1, the C. crenatum SYPA5-5 derivate. The suicide vector pK18mobsacB was used for deleting unmarked genes in C. glutamicum [38]. All the strains and plasmids used in this work are listed in Table 1.
Structure simulation
The relatively accurate 3D structure of CcOATase was obtained via the SWISS-MODEL database (http://swissmodel.expasy.org/). Molecular docking was performed using AutoDock 4.2 software (Scripps Institute, California, USA) [26]. The structural changes in the L-ornithine-binding sites were analyzed using PyMOL software [7].
Site-directed mutagenesis of CcOATase and EcNAGS
Site-directed mutagenesis of argJ and argA was carried out by overlapping PCR using the C. crenatum argJ and E. coli BL21 (DE3) argA gene amplicons as templates. To construct recombinant expression vectors containing pDXW10-argAHY, multi-mutated argAHY was generated using overlapping PCR. The successful introduction of desired mutations was confirmed by DNA sequencing, and the desired sequences ligated into pDXW10 were then transformed into E. coli BL21(DE3) for expression. The recombinant plasmids were transformed into C. crenatum using the electroporation method described by Tauch [39].
Expression and purification of proteins
Recombinant E. coli BL21(DE3) cells were cultured in Luria–Bertani (LB) medium at 37 °C until OD = 1.0 (approximately 3 h), induced with 1 mM IPTG, and then incubated for 10 h at 16 °C. The C. crenatum cells were inoculated into Brain Heart Infusion (BHI) medium, cultured at 30 °C for 16 h and 180 rpm, and then harvested by centrifugation. The cells were resuspended in phosphate saline buffer (pH 7.4) and disrupted on ice by sonication to obtain the intracellular protein. Recombinant CcOATase, NAGS, and NAOD proteins were purified using a His-Trap HP affinity column as described by Sambrook [35]. The protein purity was determined by SDS-PAGE (12% acrylamide) [11], and purified protein was used for activity assays.
Enzyme activity assays
OATase activity was measured using a ninhydrin procedure described by Liu [21]. The standard assay contained 100-mM Tris/HCl, pH 7.4, 60 mM L-glutamate, and 8 mM N-acetyl-L-ornithine in a final volume of 0.1 mL. After 2 min of preincubation at 37 °C, the reaction was initiated by adding the enzyme and incubating for 10 min at 37 °C. Afterward, reaction products were measured via the absorbance at 490 nm.
NAGS activity was assayed as described in [9]. The assay contained 100 mM L-glutamate, 0.2 mM acetyl-CoA, 0.2 mM DTNB, 40 mM TEA, and pH 8.0. The reaction was started at 25 °C by adding enzymes and was monitored spectrophotometrically for absorbance at 412 nm.
NAOD was assayed as described in [13]. In this assay, N-acetyl-L-ornithine (NAO) hydrolysis was measured spectrophotometrically at 25 °C by monitoring the peptide bond cleavage producing L-ornithine and acetate. The extent of hydrolysis was calculated by monitoring the decrease in absorbance at 214 nm.
One enzyme unit is defined as the amount of enzyme producing 1 µmol of product per minute. Protein concentration was determined by the Bradford method [4] using bovine serum albumin as a standard. All assays were performed in triplicate.
Analysis of biochemical properties of enzymes
Optimal pH and pH stability [25]: the optimal pH values of N-acetylglutamate synthase and N-acetyl-L-ornithine deacetylase were determined using the following buffers containing the compounds for the assay: citric acid-Na2HPO4, pH 5.0–6.5; sodium phosphate, pH 6.6–7.5; Tris–HCl, pH 8.0–8.5; glycine-NaOH, pH 8.6–9.3; and sodium carbonate, pH 9.4–10.9; and the pH stability was confirmed by incubation at different pH levels at 4 °C.
Optimal temperature and thermal stability [5]: The effects of temperature on N-acetylglutamate synthase and N-acetyl-L-ornithine deacetylase were determined by measuring purified enzyme activity between 20 and 55 °C. To test the enzyme thermal stability, the enzymes were incubated at various temperatures (4, 20, 25, 30, 35, 40, 45, 50, and 55 °C) in the presence of 50-mM PBS buffer (pH 7.4) for 20 h.
Metal ion preference [13]: The purified enzyme solution was first dialyzed against 10-mM PBS buffer (pH 7.4) for 12 h at 4 °C. Enzyme activity was then evaluated as described above in the presence of the divalent cations: (Cu2+, Ca2+, Co2+, Li+, K+, Fe2+, Mg2+, Ba2+, Mn2+, Fe3+, and Zn2+) and EDTA at 1 mM. The assay mixture lacking ions served as the control.
RNA preparation and transcriptional analysis
Total RNA was extracted as described in [22]. The RT-PCR assays were performed as described in our previous report [42]. The primer sequences used in RT-PCR are shown in the supplementary material (Table S1). All assays were performed in triplicate.
L-Arginine/L-ornithine feedback inhibition experiments
To investigate the effect of L-arginine/L-ornithine concentration on EcNAGS/CcOATase activity, we added 0–50 mM of L-arginine/L-ornithine to the enzymatic reaction mixture, and then, the activity of the mutated NAGS/OATase was determined. The feedback inhibition curve was constructed by changing the L-arginine/L-ornithine concentration. IR0.5 is defined as the concentration of L-arginine/L-ornithine that causes 50% inhibition. The activity of the enzyme without L-arginine/L-ornithine was defined as 100%.
Growth medium and conditions for L-ornithine production
Escherichia coli BL21(DE3) was used as the cloning host for construction of the recombinant plasmid and was cultured in LB medium. If required, 25 µg/mL kanamycin (Km) was added for selection. In this study, Cc-QF-0, the SYPA5-5 derivate was used as the original strain for the development of mutants, and the main L-arginine producer was obtained by multiple random mutagenesis. BHI medium was used to propagate C. crenatum. For the L-ornithine fermentation assay, single clones of the mutants were grown on BHI agar plates for 16 h. Subsequently, a colony was inoculated and grown in 10 mL of BHI medium in a test tube at 30 °C for 16 h, and 5 mL of the culture was transferred to 50 mL of the seed medium in a 250-mL normal shaker flask. Each liter of the seed medium contained 25 g glucose, 10 g yeast extract, 5 g beef extract, 15 g (NH4)2SO4, 1 g MgSO4·7H2O, 1 g KH2PO4, 3 g K2HPO4, 200
µg thiamine, 0.5 mM L-arginine, 10 mg MnSO4·H2O, and 10 mg FeSO4·7H2O. After 16 h of cultivation at 30 °C and 180 rpm, the appropriate amount of culture was transferred into 30 mL of fermentation medium in 250-mL baffled shaker flasks and also into a 5-L bioreactor (BIOTECH-5BG, Baoxing Co., China). Each liter of the fermentation medium consisted of 120.0 g glucose, 15.0 g corn steep liquor, 15 g (NH4)2SO4, 1 g KH2PO4, 3 g K2HPO4, 50 µg biotin, 0.5 mM L-arginine, 10 mg MnSO4·H2O, 10 mg FeSO4·7H2O, and 20 g CaCO3. Each liter of the 5-L fermentation medium consisted of 150.0 g glucose, 25.0 g corn steep liquor, 40 g (NH4)2SO4, 1 g KH2PO4, 3 g K2HPO4, 50 µg biotin, 0.5 mM L-arginine, 10 mg MnSO4·H2O, and 10 mg FeSO4·7H2O. The initial pH was adjusted to 7.0. All shaker-flask cultures were performed at 30 °C and 180 rpm, and 500-µL samples were collected every 6 h for further analysis.
Analytical procedures
Cell growth was monitored by measuring the OD600 using a spectrophotometer (UNICOTM-UV2000, Shanghai, China) after dissolving CaCO3 in 0.125 M HCl, and the dry cell weight (DCW) was determined based on a pre-calibrated relationship (1 OD600 = 0.375 g/L DCW). For quantification of substrate consumption and production, 2 mL samples of the culture were harvested and centrifuged (8000×g, 10 min, and 4 °C). Glucose, glutamate, and lactate levels were evaluated using an SBA-40C bioanalyzer (developed by the Biology Institute of the Shandong Academy of Sciences). The amino acid concentrations were analyzed by HPLC (high-pressure liquid chromatography) on an Agilent 1100 LC system (Agilent Technologies, Waldbronn, Germany), following the procedure described in Xu [41]. All assays were performed in triplicate.
Results and discussion
Construction of L-ornithine-producing C. crenatum by deleting proB and argF
Based on the mutated strain, argF was deleted to block the conversion of L-ornithine to L-citrulline and L-arginine. The resulting strain, Cc-QF-1, became an L-arginine and L-proline auxotroph, but the disruption of these two genes did not affect cell growth during shaker-flask cultivation because of the complex nitrogen source present in the seed medium. However, L-ornithine production was found to be best when supplementing with 0.5 mM L-arginine. Under this condition, only the 4.2 g/L titer of L-ornithine obtained from the Cc-QF-1 strain (Table 4) was thought to be due to the stronger feedback inhibition of CcOATase by ornithine in C. crenatum.
Site-directed mutagenesis of ornithine acetyltransferase from C. crenatum
Ornithine acetyltransferase (OATase) encoded by argJ in C. crenatum is an important functional enzyme that catalyzes the transfer of the acetyl group from N-acetylornithine to L-glutamate, resulting in the formation of L-ornithine and N-acetylglutamate [23, 32]. The data indicated that CcOATase suffers from product inhibition by ornithine with an apparent Ki value of 5.2 mM (Table S2). The Cc-QF-2 strain, which overexpressed CcargJ in the Cc-QF-1 strain, showed only a minor increase in the L-ornithine titer to 5.9 g/L compared to Cc-QF-1 (Table 4). This result might explain why feedback inhibition of CcOATase plays a key role in the biosynthesis of L-ornithine in C. crenatum.
The amino acid sequence of CcOATase was submitted to the SWISS-MODEL workspace [2, 3] to obtain a relatively accurate 3D structure model. With the 57.62% similarity of CcOATase and MtbOATase (OATase from Mycobacterium tuberculosis), the crystal structure of MtbOATase (PDB ID: 3IT6), with a resolution of 2.4 Å, was used as a template for modeling the structure of ornithine acetyltransferase complexed with ornithine from C. crenatum (Fig. S1). Homologous alignment and analysis of the structure model indicated that the side chain of the catalytic residues T150 and M176 was potential L-ornithine-binding sites [37]. Thus, site-saturation mutagenesis was applied to these two residues. The specific activities and IR0.5 values were determined for wild-type CcOATase and the mutant enzymes T150X and M176X. The specific activity of the wild-type CcOATase is 112.6 U/mg, and its IR0.5 is 5.2 mM. Unfortunately, the mutant CcOATase enzymes did not exhibit the positive effect that we expected (Table S2). We found that in the mutants with substitutions of the other 19 residues at positions T150 and M176, the specific activities were reduced by at least 50%, especially in T150P, T150F and M176F, M176W, which showed dramatic reductions. The IR0.5 of the mutant T150S was similar to that of CcOATase (5.2 mM), though the IR0.5 values of other mutants with substitutions at the residue T150 (with Gln, Asn, Arg, or Val) increased approximately fivefold, and the IR0.5 of mutants with substitutions at the residue M176 (with Asp, Ser, or Ile) also increased approximately fivefold.
The decreased specific activities of the CcOATase variants were partly due to the lower substrate affinity resulting from the replacement of amino acid residues T150 and M176. The residue T150, located at the entrance of the substrate-binding pocket of CcOATase, is near the substrate-binding sites. It became evident that the site-directed mutants possessed dramatically reduced specific activities. These results suggested that different sizes and shapes of the side chains of residues 150 and 176 had a significant impact on the substrate affinity and catalytic efficiency of CcOATase and indicated that these two residues could be not only the product binding site but also the substrate-binding sites and thus not effective for alleviating L-ornithine inhibition with regard to site-directed mutagenesis.
Screening of N-acetylglutamate synthase (encoded by argA) and N-acetyl-L-ornithine deacetylase (encoded by argE)
Furthermore, to enhance the pathway flux of L-ornithine biosynthesis (Fig. 1) and to determine which enzymes are most effective in increasing the transacetylation activity for L-ornithine biosynthesis in C. crenatum, the N-acetylglutamate synthase, and N-acetyl-L-ornithine deacetylase from different microorganisms were first characterized. N-acetylglutamate synthase, the first enzyme in L-arginine biosynthesis, catalyzes the acetyl-CoA-dependent acetylation of the amino group of glutamic acid [20]. N-acetyl-L-ornithine deacetylase catalyzes the reaction of N-acetyl-L-ornithine to L-ornithine [40]. All enzymes for ornithine synthesis were obtained through BLAST searches of the genomic sequences. The original argA genes were identified from E. coli BL21(DE3), P. aeruginosa, P. putida, and S. marcescens, and the different argE genes were identified from E. coli BL21(DE3), S. marcescens, K. pneumoniae, and B. subtilis. A feedback-resistant artificial linear pathway was introduced into C. crenatum for L-ornithine biosynthesis.
The genes from different sources were sub-cloned into the plasmid pDXW10 and expressed under the tacM promoter in E. coli BL21(DE3). Then, the recombinant N-acetylglutamate synthase and N-acetyl-L-ornithine deacetylase were purified and characterized as summarized in Table 2. It was found that the E. coli BL21(DE3) NAGS recombinant enzyme had a higher activity than the P. aeruginosa, P. putida, and S. marcescens recombinant enzymes. Similarly, the recombinant argE (encoding NAOD) from S. marcescens showed higher activity than the NAOD genes from E. coli BL21(DE3), K. pneumoniae, and B. subtilis. The specific activities of the recombinant EcNAGS and SmNAOD at 25 °C were 50.5 and 580.5 U/mg, respectively. As shown in Table 2, the optimal temperature of the recombinant EcNAGS was 37 °C, while PpNAGS and PaNAGS exhibited a similar optimal temperature (40 °C), the highest recorded value measured to date. The optimal pH of the recombinant EcNAGS was 9.0, slightly different from NAGS from other organisms, which exhibited an optimal pH of 8.0. In addition, the EcNAGS exhibited good stability at pH values ranging from 6.0 to 9.0. However, the stability decreased rapidly when the pH was below 6.0 or above 10.0. The activity of recombinant NAOD in the presence of different metal ions is shown in Table 2. The results indicated that adding Mn2+, Li+, and Mg2+ increased the activity of recombinant NAOD almost 1.5-fold compared to that without the addition of ions.
The recombinant NAGS derived from E. coli BL21(DE3) exhibited the highest specific activity. Nevertheless, it was inhibited by approximately 50% at a concentration of 0.02 mM L-arginine (Fig. 2). In this study, the process of L-ornithine fermentation required fortification with 0.5 mM L-arginine, and we, therefore, performed multiple mutagenesis with pDXW10-argAHY to relieve the feedback inhibition of arginine. According to the previous studies [24, 31], in mutant argA, the His residue in the 15th position was replaced with the amino acid Tyr (H15Y), and the Tyr residue in the 19th position was replaced with Cys (Y19C). The results in Fig. 2 showed that IR0.5 of mutant argAHY increased approximately 200-fold, and the specific activity of the mutant was similar to that of the native EcNAGS (50.5 U/mg).

Influence of arginine concentration on NAGS enzyme activity in the WT and mutant forms of the enzyme. The results are expressed as a percentage of the activity of the same enzyme form in the absence of arginine. The standard assay containing 4 mM AcCoA and 30 mM glutamate was used (see “Materials and methods”). Error bars are based on three biologically independent experiments
Co-expression of exogenous argA and argE in C. crenatum
Here, the argA and argE co-expression system was used to mimic the linear route of the L-ornithine biosynthesis pathway. First, to increase transacetylation in the L-ornithine production pathway, tandem argAHY mutants and argE genes were inserted into the shuttle expression vector pDXW10 using their original RBS. Then, the constructed plasmid was transferred into E. coli BL21(DE3) and Cc-QF-1. Indeed, the overexpression of the two enzymes was not achieved very well (Table 3). Currently, scientists have reported that promoters, ribosome-binding sites (RBSs) and terminators enable gene expression to be adapted in host cells [8, 27, 28]. This approach has already been applied successfully for overexpressing foreign genes in C. glutamicum. It has been reported that continuous genetic modules for the shikimic acid pathway can be constructed with synthetic biology logistics in C. glutamicum [44]. Thus, efforts were made to upregulate the expression level of the argA gene encoding the enzyme NAGS by RBS substitution. The theoretical strength predicted by RBS Calculator [33] (https://www.denovodna.com/software/doLogin) of the natural RBS of the argA gene is 1162 au. Thus, RBSs with strengths of 3000, 4000, 5000, and 6500 au designed by the RBS Calculator were used to replace the natural RBS of the argAHY gene in the expression plasmid, and the start codon GTG of argAHY was simultaneously replaced with ATG. Four reconstructed plasmids were generated and overexpressed in E. coli BL21(DE3). First, as shown in Fig. 3a, the NAGS activities were increased by enhancing the RBS strength. Therefore, this strategy was applied to argE, for which the original RBS (1890 au) was replaced by RBSs with strengths of 3500, 4500, 5000, and 6000 au as designed by the RBS Calculator, and the start codon GTG of argE was simultaneously replaced with ATG. Four reconstructed plasmids were generated and overexpressed in E. coli BL21(DE3). As shown in Fig. 3, this approach effectively controlled the specific NAGS and NOAD activities through the RBS substitution. Among these strains, the highest NAGS and NAOD activities were observed in pDXW10-argA4000 (100.58 U/mg) and pDXW10-argE5000 (798.98 U/mg), respectively.

Effects of RBS replacement on the specific activities of enzymes. a Comparison of NAGS-specific activity in E. coli BL21(DE3) and mutant strains. b Comparison of NAOD-specific activity in E. coli BL21(DE3) and mutant strains. Error bars are based on three biologically independent experiments
The genes EcargAHY and SmargE were inserted in tandem into the pDXW10 plasmid, and then, the recombinant plasmids were expressed in E. coli BL21(DE3). First, as shown in Table 3, both enzyme activities increased in E. coli BL21(DE3). Afterwards, pDXW10-EcargAHY-SmargE recombinant plasmids were transferred into Cc-QF-1 via electroporation. Thus, the mutated Cc-QF-3 and Cc-QF-4 strains were obtained. To check the expression levels of EcNAGS and SmNAOD, SDS-PAGE experiments were performed. Figure 4 shows that the target protein bands exhibited molecular weights of approximately 39 and 52 kDa, respectively, indicating that the heterologous genes had been overexpressed in C. crenatum SYPA5-5, carried by multiple copies of the plasmid pDXW10. In addition, the activities of EcNAGS, SmNOAD, and CcOATase were evaluated by comparison with those of C. crenatum strains. The results indicated that tandem EcargA and SmargE genes increased OATase activity (fourfold higher than that of Cc-QF-1) and acetyl utilization in C. crenatum (shown in Table 3). Subsequently, RT-PCR analysis was performed to observe the relationships between argA, argE, and argJ involved in L-ornithine biosynthesis. The results showed that overexpression of the genes EcargA and SmargE could enhance the CcargJ mRNA transcription level (increased 80%) in C. crenatum compared with Cc-QF-1 (as shown in Fig. 5c). In shaker-flask experiments, the recombinant strain Cc-QF-4 allowed the production of 12.6 g/L of L-ornithine at 72 h after inoculation, which was higher than that obtained with the strains Cc-QF-1 (4 g/L), Cc-QF-2 (5.9 g/L), and Cc-QF-3 (8.2 g/L), as shown in Table 4. Amino acid analysis of the fermentation liquor showed that the concentration of lysine and isoleucine was obviously decreased in L-ornithine flask cultures compared with the other strains’ lysine (Table S3a). The results obtained in this section demonstrate the remarkable effects of EcargA and SmargE in improving CcOATase activity and producing an L-ornithine-engineered strain. Therefore, in this case, optimization of RBS strength and replacement of the start codon for EcargA and SmargE overexpression in C. crenatum are an effective way to enhance the level of acetyl cycle utilization; thus, the production of L-ornithine was substantially increased.

SDS-PAGE analysis of the overexpression of CcargJ, EcargA ,and SmargE in recombinant Cc-QF-1. M, protein marker; Lane 1, Cc-QF-1; Lane 2, Cc-QF-2; Lane 3, Cc-QF-4

Comparison of L-ornithine production and gene transcriptional levels between Cc-QF-1, Cc-QF-2, and Cc-QF-4. a Cell concentration and glucose concentration, b L-ornithine concentration and L-ornithine production rate. Open and filled triangles, Cc-QF-1 recombinant; Open and filled squares, Cc-QF-2 recombinant; open and filled circles, Cc-QF-4 recombinant. c Comparison of argA, argE, and argJ transcriptional levels in the strains Cc-QF-1, Cc-QF-2 and Cc-QF-3. The transcriptional levels of genes were determined by RT-PCR and are presented as relative normalized expression. Error bars are based on three biologically independent experiments
Production of L-ornithine in 5-L bioreactor cultivation
The production performance of the strains Cc-QF-1, Cc-QF-2, and Cc-QF-4 was investigated in a 5-L bioreactor fermentation. The fermentation curves, including the concentrations of cells, glucose, and L-ornithine, and the L-ornithine production rate, are shown in Fig. 5. The growth rates of the three strains were similar throughout the entire fermentation period (shown in Fig. 5a), indicating that overexpression of the CcargJ gene and introduced exogenous genes did not affect cell growth in C. crenatum. It was also observed that compared with the initial strain, the glucose consumption remained fairly consistent at the beginning of fermentation but accelerated markedly after 24 h, as shown in Fig. 5a. The increased consumption of glucose could have resulted from the increased transacetylation in the biosynthetic pathway of L-ornithine and the superior L-ornithine synthesis of the recombinant strain compared to the initial strain. L-ornithine yield and productivity were similar between the strains before 24 h. Then, the recombinant strains showed a slight advantage in L-ornithine production between 24 and 36 h. At the end of fermentation, L-ornithine yield increased markedly, to 40.4 g/L, with an overall productivity of 0.673 g/L/h in the recombinant strain Cc-QF-4, which was approximately 28.2 and 41.8% higher than those of Cc-QF-2 and Cc-QF-1, respectively (Fig. 5b). This result indicates that L-ornithine yield can be markedly increased by introducing an artificial linear transacetylation pathway. We also tracked CcargJ mRNA transcription levels and CcOATase activity during fermentation; compared with the wild-type strain, introduction of EcargA and SmargE increased the intracellular transcription level of CcargJ mRNA (Fig. 5c) and increased the enzyme activity (Table 3). Regarding the acetyl cycle level, the increased L-ornithine flows of carbon metabolism may be the main reason for the difference in L-ornithine production in Cc-QF-4. Amino acid analysis of the fermentation liquor showed that the concentrations of lysine (0.98 g/L) and isoleucine (0.87 g/L) were obviously decreased compared with the wild-type strain (lysine, 2.34 g/L; isoleucine, 1.97 g/L), and proline was undetectable (Table S3b). As expected, lactate acid was not detected in either strain, supporting the idea that the accumulation of acid during shaking culture was due to the restricted oxygen supply. These data suggest that improvement of the acetyl cycle level is effective in increasing the L-ornithine production and yield on glucose.
Conclusion
In summary, we reported a metabolically engineered C. crenatum strain capable of efficiently producing L-ornithine by combining the strategies of deleting competing pathways and introducing an artificial linear transacetylation pathway. The resulting strain produced L-ornithine at 40.4 g/L, which is 41.8% higher than that (23.5 g/L) of the original strain. This study demonstrates the huge potential of C. crenatum to overproduce not only L-ornithine but also L-citrulline and L-arginine from renewable resources such as glucose.
References
Amund OO, Mackinnon G, Higgins IJ (1983) Increased l-ornithine production by an arg mutant of Acinetobacter lwoffi. Eur J Appl Microbiol Biotechnol 17:252–253
Arnold K, Bordoli L, Kopp J, Schwede T (2006) The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22:195–201. https://doi.org/10.1093/bioinformatics/bti770
Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, Kiefer F, Cassarino TG, Bertoni M, Bordoli L, Schwede T (2014) SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res 42:W252–W258. https://doi.org/10.1093/nar/gku340
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Casteele MVD, Demarez M, Legrain C, Glansdorff N, Piérard A (1990) Pathways of arginine biosynthesis in extreme thermophilic archeo- and eubacteria. J Gen Microbiol 136:1177–1183
Choi DK, Ryu WS, Choi CY, Park YH (1996) Production of l-ornithine by arginine auxotrophic mutants of Brevibacterium ketoglutamicum in dual substrate-limited continuous culture. J Ferment Bioeng 81:216–219
Delano WL (2002) The PyMOL Molecular Graphics System. https://pymol.org
Dyakonov T, Muir A, Nasri H, Toops D, Fatmi A (2013) Characterization of 582 natural and synthetic terminators and quantification of their design constraints. Nat Methods 10:659–664
Errey JC, Blanchard JS (2005) Functional characterization of a novel argA from Mycobacterium tuberculosis. J Bacteriol 187:3039–3044. https://doi.org/10.1128/JB.187.9.3039-3044.2005
Han PS, Fishel RS, Efron DT, Williams JZ, Fishel MH, Barbul A (2002) Effect of supplemental ornithine on wound healing. J Surg Res 106:299
Hermann SG (2006) Tricine-SDS-PAGE. Nat Protoc 1:16–22
Hwang JH, Hwang GH, Cho JY (2008) Effect of increased glutamate availability on l-ornithine production in Corynebacterium glutamicum. J Microbiol Biotechnol 18:704
Javidmajd F, Blanchard JS (2000) Mechanistic analysis of the argE-encoded N-acetylornithine deacetylase. Biochemistry 39:1285
Jiang LY, Chen SG, Zhang YY, Liu JZ (2013) Metabolic evolution of Corynebacterium glutamicum for increased production of l-ornithine. BMC Biotechnol 13:47–47
Jiang LY, Zhang YY, Li Z, Liu JZ (2013) Metabolic engineering of Corynebacterium glutamicum for increasing the production of l-ornithine by increasing NADPH availability. J Ind Microbiol Biotechnol 40:1143–1151
Kim SY, Lee J, Lee SY (2015) Metabolic engineering of Corynebacterium glutamicum for the production of l-ornithine. Biotechnol Bioeng 112:416–421. https://doi.org/10.1002/bit.25440
Kinoshita S, Nakayama K, Udaka S (2006) The fermentative production of l-ornithine: preliminary report. J Gen Appl Microbiol 3:276–277
Lee JH, Wendisch VF (2017) Production of amino acids - Genetic and metabolic engineering approaches. Bioresour Technol 245:1575
Lee YJ, Cho JY (2006) Genetic manipulation of a primary metabolic pathway for l-ornithine production in Escherichia coli. Biotechnol Lett 28:1849–1856. https://doi.org/10.1007/s10529-006-9163-y
Leisinger T, Haas D (1975) N-Acetylglutamate synthase of Escherichia coli regulation of synthesis and activity by arginine. J Biol Chem 250:1690–1693
Liu Y, Heeswijck RV, Høj P, Hoogenraad N (1995) Purification and characterization of ornithine acetyltransferase from Saccharomyces cerevisiae. Eur J Biochem 228:291–296
Man Z, Xu M, Rao Z, Guo J, Yang T, Zhang X, Xu Z (2016) Systems pathway engineering of Corynebacterium crenatum for improved l-arginine production. Sci Rep 6:28629. https://doi.org/10.1038/srep28629
Marc F, Weigel P, Legrain C, Almeras Y, Santrot M, Glansdorff N, Sakanyan V (2010) Characterization and kinetic mechanism of mono- and bifunctional ornithine acetyltransferases from thermophilic microorganisms. Febs J 267:5217–5226
Zhao G, Haskins N, Jin Z, N MA, Tuchman M, Shi D (2013) Structure of N-acetyl-l-glutamate synthase/kinase from Maricaulis maris with the allosteric inhibitor l-arginine bound. Biochem Biophys Res Commun 437:585–590. https://doi.org/10.1016/j.bbrc.2013.07.003
Mcgregor WC, Gillner DM, Swierczek SI, Liu D, Holz RC (2013) Identification of a histidine metal ligand in the argE-Encoded N-acetyl-l-ornithine deacetylase from Escherichia coli. Springerplus 2:482
Morris GM, Ruth H, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ (2009) Software news and updates AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 30:2785–2791
Mutalik VK, Guimaraes JC, Cambray G, Lam C, Christoffersen MJ, Mai QA, Tran AB, Paull M, Keasling JD, Arkin AP, Endy D (2013) Precise and reliable gene expression via standard transcription and translation initiation elements. Nat Methods 10:354–360. https://doi.org/10.1038/nmeth.2404
Nowroozi FF, Baidoo EE, Ermakov S, Redding-Johanson AM, Batth TS, Petzold CJ, Keasling JD (2014) Metabolic pathway optimization using ribosome binding site variants and combinatorial gene assembly. Appl Microbiol Biotechnol 98:1567–1581
Qian ZG, Xia XX, Sangyup L (2009) Metabolic engineering of Escherichia coli for the production of putrescine: a four carbon diamine. Biotechnol Bioeng 104:651–662
Qin J, Zhou YJ, Krivoruchko A, Huang M, Liu L, Khoomrung S, Siewers V, Jiang B, Nielsen J (2015) Modular pathway rewiring of Saccharomyces cerevisiae enables high-level production of l-ornithine. Nat Commun 6:8224. https://doi.org/10.1038/ncomms9224
Rajagopal BS, Deponte J, Tuchman M, Malamy MH (1998) Use of inducible feedback-resistant N-acetylglutamate synthetase (argA) genes for enhanced arginine biosynthesis by genetically engineered Escherichia coli K-12 Strains. Appl Environ Microbiol 64:1805
Sakanyan V, Petrosyan P, Lecocq M, Boyen A, Legrain C, Demarez M, Hallet JN, Glansdorff N (1996) Genes and enzymes of the acetyl cycle of arginine biosynthesis in Corynebacterium glutamicum: enzyme evolution in the early steps of the arginine pathway. Microbiology 142(Pt 1):99–108. https://doi.org/10.1099/13500872-142-1-99
Salis HM, Mirsky EA, Voigt CA (2009) Automated design of synthetic ribosome binding sites to control protein expression. Nat Biotechnol 27:946–950. https://doi.org/10.1038/nbt.1568
Salvatore F, Cimino F, d’Ayello-Caracciolo M, Cittadini D (1964) Mechanism of the protection by l-ornithine-l-aspartate mixture and by l-arginine in ammonia intoxication. Arch Biochem Biophys 107:499–503. https://doi.org/10.1016/0003-9861(64)90307-8
Sambrook J, Fritsch EF, Maniatis T (2001) Molecular cloning. A laboratory manual. Anal Biochem 186:182–183
Sancho E (2009) Mechanism of arginine regulation of acetylglutamate synthase, the first enzyme of arginine synthesis. Febs Lett 583:202–206
Sankaranarayanan R, Cherney MM, Garen C, Garen G, Niu C, Yuan M, James MN (2010) The molecular structure of ornithine acetyltransferase from Mycobacterium tuberculosis bound to ornithine, a competitive inhibitor. J Mol Biol 397:979–990. https://doi.org/10.1016/j.jmb.2010.02.018
Schäfer A, Tauch A, Jäger W, Kalinowski J, Thierbach G, Pühler A (1994) Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene 145:69
Tauch A, Kirchner O, Löffler B, Götker S, Pühler A, Kalinowski J (2002) Efficient electrotransformation of Corynebacterium diphtheriae with a mini-replicon derived from the Corynebacterium glutamicum plasmid pGA1. Curr Microbiol 45:362–367
Velasco AM, Leguina JI, Lazcano A (2002) Molecular evolution of the lysine biosynthetic pathways. J Mol Evol 55:445–449. https://doi.org/10.1007/s00239-002-2340-2
Xu H, Dou W, Xu H, Zhang X, Rao Z, Shi Z, Xu Z (2009) A two-stage oxygen supply strategy for enhanced l-arginine production by Corynebacterium crenatum based on metabolic fluxes analysis. Biochem Eng J 43:41–51. https://doi.org/10.1016/j.bej.2008.08.007
Xu M, Rao Z, Dou W, Xu Z (2013) The role of ARGR repressor regulation on l-arginine production in Corynebacterium crenatum. Appl Biochem Biotechnol 170:587–597. https://doi.org/10.1007/s12010-013-0212-4
Zhang B, Yu M, Zhou Y, Li Y, Ye BC (2017) Systematic pathway engineering of Corynebacterium glutamicum S9114 for l-ornithine production. Microb Cell Fact 16:158. https://doi.org/10.1186/s12934-017-0776-8
Zhang B, Zhou N, Liu YM, Liu C, Lou CB, Jiang CY, Liu SJ (2015) Ribosome binding site libraries and pathway modules for shikimic acid synthesis with Corynebacterium glutamicum. Microb Cell Fact 14:71. https://doi.org/10.1186/s12934-015-0254-0
Zhang J, Xu M, Ge X, Zhang X, Yang T, Xu Z, Rao Z (2016) Reengineering of the feedback-inhibition enzyme N-acetyl-l-glutamate kinase to enhance l-arginine production in Corynebacterium crenatum. J Ind Microbiol Biotechnol 44:271–283. https://doi.org/10.1007/s10295-016-1885-9
Zhao Q, Luo Y, Dou W, Zhang X, Zhang X, Zhang W, Xu M, Geng Y, Rao Z, Xu Z (2016) Controlling the transcription levels of argGH redistributed l-arginine metabolic flux in N-acetylglutamate kinase and ArgR-deregulated Corynebacterium crenatum. J Ind Microbiol Biotechnol 43:55–66
Acknowledgements
This work was supported by the National Natural Science Foundation of China (31770058, 31570085), the Jiangsu Province Science Fund for Distinguished Young Scholars (BK20150002), the Research Project of the Chinese Ministry of Education (113033A), the Fundamental Research Funds for the Central Universities (JUSRP51708A), the national first-class discipline program of Light Industry Technology and Engineering (LITE2018-06) and the 111 Project (111-2-06).
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Shu, Q., Xu, M., Li, J. et al. Improved l-ornithine production in Corynebacterium crenatum by introducing an artificial linear transacetylation pathway. J Ind Microbiol Biotechnol 45, 393–404 (2018). https://doi.org/10.1007/s10295-018-2037-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-018-2037-1