
Abstract
Corynebacterium crenatum SYPA5-5, an l-arginine high-producer obtained through multiple mutation-screening steps, had been deregulated by the repression of ArgR that inhibits l-arginine biosynthesis at genetic level. Further study indicated that feedback inhibition of SYPA5-5 N-acetylglutamate kinase (CcNAGK) by l-arginine, as another rate-limiting step, could be deregulated by introducing point mutations. Here, we introduced two of the positive mutations (H268N or R209A) of CcNAGK into the chromosome of SYPA5-5, however, resulting in accumulation of large amounts of the intermediates (l-citrulline and l-ornithine) and decreased production of l-arginine. Genetic and enzymatic levels analysis involved in l-arginine biosynthetic pathway of recombinants SYPA5-5-NAGKH268N (H-7) and SYPA5-5-NAGKR209A (R-8) showed that the transcription levels of argGH decreased accompanied with the reduction of argininosuccinate synthase and argininosuccinase activities, respectively, which led to the metabolic obstacle from l-citrulline to l-arginine. Co-expression of argGH with exogenous plasmid in H-7 and R-8 removed this bottleneck and increased l-arginine productivity remarkably. Compared with SYPA5-5, fermentation period of H-7/pDXW-10-argGH (H-7-GH) reduced to 16 h; meanwhile, the l-arginine productivity improved about 63.6 %. Fed-batch fermentation of H-7-GH in 10 L bioreactor produced 389.9 mM l-arginine with the productivity of 5.42 mM h−1. These results indicated that controlling the transcription of argGH was a key factor for regulating the metabolic flux toward l-arginine biosynthesis after deregulating the repression of ArgR and feedback inhibition of CcNAGK, and therefore functioned as another regulatory mode for l-arginine production. Thus, deregulating all these three regulatory modes was a powerful strategy to construct l-arginine high-producing C. crenatum.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
l-arginine, a conditionally essential amino acid which acts as a precursor of nitric oxide (NO), is widely used as an essential ingredient in food flavoring and pharmaceutical industry [2, 17]. Bacterial fermentation from natural carbon sources is the major approach for l-arginine production at the industrial scale [12, 26]. With the development of “omics” technology and swift appearance of various accessible genetic tools, considerable attention has recently been given to construct more efficient production strains, as well as to elucidate the unknown regulatory mechanisms involved the l-arginine metabolism processes [17].
Similar to most of the amino acids, the mutant strains of Corynebacterium sp. was employed in industrialized production of l-arginine [26]. Corynebacterium crenatum, a facultative anaerobic gram-positive organism, is extensively used as a cell factory for amino acids and organic acids production [4, 32]. C. crenatum SYPA5-5 was isolated from several mutation-screening steps against l-arginine analogs (e.g., d-arginine, homoarginine and arginine hydroxamate) in our previous study, which could accumulate 30 g L−1 l-arginine after cultivated for 96 h in optimized medium using glucose as main carbon source [32].
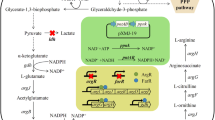
As well as Corynebacterium glutamicum, C. creanatum possesses the so-called economical cyclic l-arginine biosynthetic pathway [5, 37]. Figure 1a shows the de novo biosynthetic pathway which initiates by acetylation of glutamate and proceeds via three enzymatic steps which form further acetylated intermediates (N-acetyl-ornithine). The ornithine acetyltransferase (OAT) could use acetyl group of N-acetyl-ornithine to acetylate glutamate with l-ornithine released. Three more reactions (OTC, ASS, ASL) are followed to finish the assembly of the guanidine group on l-arginine from carbamoylphosphate and the amino group of l-aspartate. Feedback inhibition on N-acetylglutamate kinase (NAGK) by l-arginine appears to be a rate-limiting step in this cyclic pathway [39]. Meanwhile, the genes encoding these enzymes are clustered in chromosome including two large transcripts corresponding to argCJBDFR and argGH, and their transcription levels are regulated by ArgR repressor encoding by argR gene (Fig. 1b) [14, 21, 38].

The pathway and regulatory modes involved in l-arginine biosynthesis in C. crenatum SYPA5-5. a The de novo l-arginine biosynthetic pathway. Outline of the pathway, gene name, enzyme name, EC number and the co-factor needed in biosynthesis were displayed for each step. The dash arrow represents the CcNAGK that was feedback inhibited by terminal product. b The arg cluster encoding the enzymes for l-arginine biosynthesis including two larger transcripts of argCJBDFR and argGH. The transcription of arg cluster was repressed by ArgR repressor, which was deregulated by introducing nonsense mutation (C109T) in argR through an iterative procedure of random mutation and selection in previous study
Previously, chemical mutagenesis and selection with l-arginine analogs led to isolation of l-arginine producer in which NAGK activity was released from the feedback inhibition [39]. In the post-genomic era, “genome-based strain reconstruction” was an efficient method to identify mutations benefit to amino acids production and breeding efficient producers, which was also successfully used for construction of l-arginine producer [19]. The mutations in argB (A26V, M31V) of classical l-arginine producers was identified by sequence analysis to deregulate feedback inhibition of C. glutamicum NAGK, and introducing these mutations into C. glutamicum ATCC13032 could make it accumulate large amount of l-arginine and l-citrulline when combined with deletion of argR gene [13]. Recently, a new in vivo ultrahigh-throughput screening approaches by using metabolite sensors could also yielded feedback-resistant NAGK variants [23]. In our previous studies, sequence analysis of arg cluster of SYPA5-5 showed that the nonsense mutation in argR (C109T) made arg cluster release from ArgR repression through random mutagenesis [34]. Further study by introducing the point mutation (E19R, H26E, R209A, H268N or G287D), respectively, in CcNAGK increased 50 % inhibitory l-arginine concentration (I0.5) significantly in vitro [33], which deregulated the feedback inhibition of CcNAGK by l-arginine in SYPA5-5 during the fermentation [35]. However, moderate expression of a feedback-resistant enzyme, rather than its over-expression by multicopy plasmid, has been proven more useful for increasing amino acids production [7]. Therefore, two of the positive mutations (R209A or H268N) of CcNAGK were introduced into the chromosome of SYPA5-5, respectively, in this study. Unexpectedly, the recombinants accumulated large amount of l-citrulline and l-ornithine as intermediate metabolites of l-arginine biosynthetic pathway.
Similarly, Park et al. proposed effective and systematic metabolic engineering strategies to improve l-arginine production by C. glutamicum ATCC 21831, involving random mutagenesis, removal of regulatory repressor (ArgR and FarR), optimization of NADPH level, disruption of l-glutamate exporter, reverting back of mutated argF (G166C) to its original gene sequence and over-expression of carAB [20]. However, during construction of the l-arginine high-producer, l-citrulline, a simultaneously secreted by-product of l-arginine, was accumulated. In view of revealing the metabolic block between l-ornithine, l-citrulline and l-arginine and its synthetic mechanism, this work analyzed the genetic and enzymatic levels involved in l-arginine biosynthesis. The results showed that transcription levels of argG and argH genes and the corresponding specific enzyme activities of ASS and ASL in recombinants were all decreased compared with SYPA5-5. Then, the metabolic obstacle of accumulating l-citrulline and l-ornithine, caused by the down-regulation of argGH transcription levels after deregulation of the feedback inhibition of NAGK in ArgR deficient C. crenatum, was removed by co-expression of argGH, and l-arginine productivity increased greatly.
Materials and methods
Bacterial strains, plasmids, oligonucleotides
All bacterial strains, plasmids and oligonucleotides used in this study and their relevant characteristics and sources are listed in Table 1 and Table S1 in the supplementary material. C. crenatum SYPA5-5 [32], deposited in CGMCC with the No. 0890, was chosen as the parental strain.
Culture conditions
Escherichia coli was cultured aerobically on Luria–Bertani (LB) [24] at 37 °C as 10 mL cultures in a 50-mL shake flask on a rotary shaker at 220 rpm. SYPA5-5 and its recombinant derivatives were cultivated aerobically at 30 °C on a reciprocating shaker at 120 rpm.
Before l-arginine fermentation, cells were pre-activated in plate with broth medium (containing 20 g L−1 glucose) at biochemical incubator for 24 h, and then transformed into 30 mL seed medium (30 g L−1 glucose, 10 g L−1 yeast extract, 20 g L−1 (NH4)2SO4, 1 g L−1 KH2PO4, 0.5 g L−1 MgSO4·7H2O, 1.5 g L−1 urea) in a 250-mL shake flask cultured for about 15 h (OD562nm = 15–18). For batch fermentation in shake flask, 1 mL seed culture was inoculated into 20 mL fermentation medium (150 g L−1 glucose, 10 g L−1 yeast extract, 50 g L−1 (NH4)2SO4, 1.5 g L−1 KH2PO4, 0.5 g L−1 MgSO4·7H2O, 0.02 g L−1 FeSO4·7H2O, 0.02 g L−1 MnSO4·H2O, 8 × 10−5g L−1 biotin, 5 × 10−4 g L−1 l-histidine, 30 g L−1 CaCO3) in a 250-mL shake flask. For fed-batch fermentation of recombinant strains in 10-L bioreactor, 300 mL seed culture was transformed to 6 L optimized fermentation medium [100 g L−1 glucose, 14 g L−1 yeast extract, 20 g L−1 (NH4)2SO4, 1.5 g L−1 KH2PO4, 0.5 g L−1 MgSO4·7H2O, 0.02 g L−1 FeSO4·7H2O, 0.02 g L−1 MnSO4·H2O, 8 × 10−5g L−1 biotin, 5 × 10−4g L−1 l-histidine, pH 7.0]. The agitation speed was kept at 600 rpm, pH was maintained at 7.0 by flowing aqueous ammonia and the glucose was fed when residual concentration below 10 g L−1. The plasmid-carrying strains were cultured with kanamycin (25 μg mL−1) or ampicillin (100 μg mL−1) depending on the characteristics of plasmids.
Site-directed mutagenesis
Site-directed mutagenesis was carried out essentially via overlap extension PCR [1]. The partial fragments on each side of the mutant site were separately amplified with two pairs of primers (argJBDF-RM209/268, FM209/268-argJBDR) using the genome of SYPA5-5 as template. There was some overlap between primers FM209/268 and RM209/268, respectively. The 5ʹ-end fragment was amplified by forward primer argJBDF as well as reverse primer RM209/268, and the 3ʹ-end fragment by forward primer FM209/268 as well as reverse primer argJBDR (Table 1). The two PCR fragments were purified and used as the templates of another round of PCR with argJBDF and argJBDR as the primers to obtain the fragments with corresponding point mutations for homologous recombination.
Construction of plasmids and strains
SYPA5-5 with chromosomal mutations were performed by the suicide vector pK18mobsacB which could fulfill markerless replacement of genes through homologous recombination [22]. The fragments with point mutations of H268N or R209A were verified by sequence and ligated with pK18mobsacB to obtain the recombinant plasmids pK18mobsacB-argB H268N and pK18mobsacB-argB R209A. After transforming them into SYPA5-5, respectively, by electroporation [27], the desired mutants were firstly selected on sorbitol plates containing kanamycin (25 μg mL−1) and finally selected in LB plate with 10 % (w/v) sucrose. Then, the recombinants were screened and confirmed by PCR and DNA sequencing.
The argGH fragment was amplified via PCR using argGH-F and argGH-R as primers (Table 1) and the genome of SYPA5-5 as template. The fragment was purified, ligated with pMD-19-T simple vector and transformed in E. coli JM109. The strain with recombinant plasmid was verified by sequencing. Correct fragment was digested and inserted into pDXW-10 [31] to obtain the recombinant plasmid of pDXW-10-argGH. After transforming it into SYPA5-5, H-7 and R-8, respectively, by electroporation, positive clones were separately picked out in LB plate containing kanamycin (25 μg mL−1).
Determination of enzyme activities
Corynebacterium crenatum from exponential phase was harvested by centrifugation (10 min, 8000×g, 4 °C), washed and suspended with 50 mM Tris–HCl pH 7.5, and then broken at 4 °C by ultrasonication with the ultrasonic cell disruptor (Scientz Biotechnology, JY92-II, Ningbo, China) for 15 min at 300 W. After centrifugation (15 min, 20000×g, 4 °C), the supernatant was used for enzyme assay. Protein concentration was determined by the Coomassie brilliant blue G-250 dye-binding method of Bradford with bovine serum albumin as the standard [3].
NAGK activity was measured with the hydroxylamine-containing colorimetric assay of Haas and Leisinger, which detected the formation of acetylglutamyl hydroxamate (extinction coefficient 456 M−1 cm−1) at 540 nm [11]. The final assay system (total volume 1 mL) contained 200 mM Tris–HCl (pH 8.0), 40 mM N-acetylglutamate, 40 mM MgCl2, 40 mM ATP, 400 mM NH4OH·HCl. The mixture was incubated at 37 °C for 30 min, terminated by addition of 2 mL stopping solution [4 % (w/v) FeCl3 and 5 % (w/v) trichloroacetic acid in 1 M HCl], and followed by detecting at 540 nm after centrifugation. One unit of the enzyme activity was defined as the amount of enzyme generating one micro mole acetylglutamyl hydroxamate per minute under the condition described above.
Measurement of argininosuccinate synthase (ASS) activity was carried out in two steps [15]. Phosphate was released from ATP hydrolysis catalyzed by ASS in first step with a final system (total 0.9 mL) containing 20 mM Tris–HCl (pH 7.8), 2 mM ATP, 2 mM citrulline, 2 mM aspartate, 6 mM MgCl2, 20 mM KCl, 0.2 U pyrophosphatase. The mixture was incubated at 37 °C for 15 min, and followed by the second step with addition of 6 % H2SO4, 7.5 mM ammonium molybdate, 30 mM ascorbic acid. After incubating at 30 °C for 30 min, the supernatant was analyzed for absorption at 820 nm. Aliquots of solution containing 1–10 μg of Pi were used to make the standard curve. One unit of the enzyme activity was defined as the amount of enzyme generating 1 µmol of Pi per minute under the condition described above.
Measurement of argininosuccinase (ASL) activity was depending on l-arginine synthesis. The final system contained 50 mM Tris–HCl (pH 7.5), 2 mM argininosuccinate, and moderate enzyme. The mixture was incubated at 37 °C for 20 min and product of l-arginine was analyzed by HPLC. One unit of the enzyme activity was defined as the amount of enzyme generating 1 µmol of l-arginine per minute under the condition described above.
Quantitative real-time PCR (qRT-PCR)
SYPA5-5 and its recombinant derivatives were harvested by centrifugation (10 min, 8000×g, 4 °C) in the exponential growth phase during fermentation and prepared total RNA by RNA extract kit (Thermo Scientific, USA). The first strand cDNA was synthesized by PrimeScript®RT reagent Kit (Takarra, Japan), and qRT-PCR was performed using SYBR®Premix Ex TaqTM II (TliRNaseH Plus) on a Bio-Rad CFX96 Manager PCR system (Bio-Rad, USA) using the cycling parameters: 95 °C for 5 min, followed by 40 cycles of denaturation at 95 °C for 10 s, annealing at 60 °C for 20 s and extension in 72 °C for 20 s. The primers of qRT-PCR are listed in Supplementary Table S1. The method of 2−△△Ct was applied to analyze the data and normalized to the transcription level of 16S rRNA [16].
Analysis of fermentation parameters
Cell growth was monitored by measuring the optical density at 562 nm (OD562nm). Cell dry weight (CDW) was calculated as a ratio of 0.375 g L−1 per OD562nn [32].
For quantification of substrate consumption and product formation, about 0.5 mL of samples were taken from the fermentation culture every time and centrifuged (12000×g, 5 min). The resulting supernatants were used for determination of glucose, (NH4)2SO4 and amino acids concentration. Glucose was measured by the method of DNS according to Miller [18]. Phenol-hypochlorite method was used to determine the concentration of (NH4)2SO4 [29]. The concentration of amino acids were detected by HPLC (DIONEX, UltiMate 3000, America) equipped with a venusil AA column (Agela technologies, China). Before analysis of amino acids, the samples were derived by phenyl-isothiocyanate and detected at 254 nm (UV) [8]. All assays were performed by triplicate cultures.
Detection of intracellular l-arginine concentration
Cells were harvested every 24 h during fermentation, mixed with pre-cooling quenching agent (glycerol: 13.5 g L−1 NaCl = 3:2) immediately [28], centrifugated at 4 °C (8000×g, 10 min), washed twice with 0.9 % NaCl, and then subjected to sonication. The supernatant was harvested by centrifugation (8000×g, 10 min, 4 °C) and mixed with isometric 10 % trichloroacetic acid to subside the intracellular soluble protein. Then amino acids concentration of the supernatant was analyzed by HPLC as described above. The intracellular l-arginine concentration was calculated considering a cytoplasmic volume of 1.7 µL mg−1 dry cells [25].
Results
Construction of feedback inhibition deregulated H-7 and R-8
The favorable mutations (H268N or R209A) in CcNAGK, which significantly decreased the sensitivity to l-arginine and kept the specific activity [33], was integrated in SYPA5-5, respectively, through substituted argB gene by its mutant in SYPA5-5. Recombinants of SYPA5-5-CcNAGKH268N (designated as H-7) and SYPA5-5-CcNAGKR209A (designated as R-8) were screened on sucrose agar plate after twice homologous recombination.
To examine the expression levels of CcNAGKH268N and CcNAGKR209A in the recombinants, CcNAGK activities with different concentrations of l-arginine were determined (Fig. 2). As expected, the CcNAGK mutants showed constant specific activities compared to the wild type and remained about 98.2 and 94.3 % activities, respectively, in presence of 15 mM l-arginine (the maximum intracellular l-arginine concentration was 14.5 mM over the fermentation course, Supplementary Fig S1). These results indicated that feedback inhibition of CcNAGK in H-7 and R-8 was successfully deregulated.

Residual activities of CcNAGK in SYPA5-5 (filled squares), H-7 (filled circles) and R-8 (filled triangles) were analyzed by adding different concentrations of l-arginine. The specific activities were 0.62 ± 0.021, 0.79 ± 0.024, and 0.77 ± 0.046 U mg−1, respectively, without adding l-arginine. (All assays were performed by triplicate cultures, standard deviations of the biological replicates were represented by error bars.)
Deregulation of CcNAGK feedback inhibition increases l-ornithine and l-citrulline accumulation
The recombinants H-7 and R-8 were investigated for shake flask fermentation using SYPA5-5 as control. The fermentation profiles over the experimental course were monitored and shown in Fig. 3. Unexpectedly, l-arginine yields of recombinants were lower than SYPA5-5 (Fig. 3a), accompanied by accumulation of large amounts of l-citrulline and l-ornithine as the intermediates of l-arginine biosynthetic pathway (Fig. 3c, d). Meanwhile, glucose and (NH4)2SO4 consumption rates of recombinants were decreased (Fig. 3e, f), which resulted in lower biomass (Fig. 3b) and l-arginine yields (Fig. 3a). After culturing for 88 h, H-7 and R-8 produced 139.4 and 129.9 mM l-arginine, 41.7 and 47.7 mM l-citrulline, 16.8 and 15.0 mM l-ornithine, respectively. Compared with SYPA5-5, H-7 and R-8 showed the reduction of l-arginine production about 16.2 and 21.9 %, the increase of l-citrulline concentration about 3.9-fold and 4.6-fold and the increase of l-ornithine concentration about 1.5-fold and 1.2-fold, respectively. Furthermore, compared with SYPA5-5, cumulative reduction of the major by-product of l-lysine was 67.8 and 75.2 % in H-7 and R-8, respectively (Supplementary Fig. S2). These results suggested that deregulation of feedback inhibition of CcNAGK in SYPA5-5 significantly affected the metabolic flux redistribution involved in l-arginine biosynthetic pathway.

l-arginine (a), biomass (b), l-citrulline (c), l-ornithine (d), glucose consumption (e), and (NH4)2SO4 consumption (f) of SYPA5-5 (filled squares), H-7 (filled circles) and R-8 (filled triangles), respectively, during a representative shake flask batch fermentation. (All assays were performed by triplicate cultures, standard deviations of the biological replicates were represented by error bars.)
Transcription analysis of genes involved l-arginine biosynthetic pathway of H-7 and R-8
To reveal the generative mechanism of l-citrulline and l-ornithine accumulation and l-arginine production reduction, the transcription levels of arg gene cluster were determined in two recombinants, using SYPA5-5 as control. The results showed that the transcription levels of argCJBDF transcript did not show obvious differences (Fig. 4). However, transcription levels of argG and argH decreased more than 80 and 65 % (Fig. 4; Table 2), respectively. In accordance with that, the specific activities of ASS and ASL, encoded by argG and argH, decreased more than 50 % (Table 2). However, transcription levels of central metabolism genes, such as TCA cycle (gltA, icd, kgd, acn, fumC and gdh), anaplerotic pathway (pyc), glycolytic (pgi, pfk, fda, gap, pgk, pyk, aceE), glucose uptake system (ptsG) and pentose phosphate pathway (zwf and gnd), were all up-regulated (Supplementary Table S2).These results indicated that the reduction of transcription levels of argGH transcript, which affected the enzyme expression responsible for the downstream metabolism of l-ornithine and l-citrulline, was the reason for accumulation of the intermediates.

Transcription analysis of the genes involved in l-arginine biosynthetic pathway of recombinants H-7 (white bars) and R-8 (dark gray bars) using SYPA5-5 (black bars) as control. (All assays were performed by triplicate cultures, standard deviations of the biological replicates were represented by error bars.)
Co-expression of argGH transcript in H-7 and R-8 improved l-arginine production
The fermentation profiles and relative genes transcription analysis of recombinants H-7 and R-8 indicated that a bottleneck of the carbon flux from l-citrulline to l-arginine in biosynthetic pathway was a crucial node needed to break through. To increase the expression of argGH, we constructed the recombinant plasmid pDXW-10-argGH and transformed it into H-7, R-8 and SYPA5-5, resulting in H-7/pDXW-10-argGH (H-7-GH), R-8/pDXW-10-argGH (R-8-GH), and SYPA5-5/pDXW-10-argGH (5-GH), respectively. As expected, the transcription levels of argGH and the specific activities of ASS and ASL were increased remarkably in all recombinants (Table 2).
As shown in Fig. 5, after culturing for 64 h, fermentation was terminated when the nitrogen source (NH4)2SO4 was completely consumed by H-7-GH and R-8-GH (Supplementary Table S3), and the yields of the major amino acids were detected. Compared with SYPA5-5, 5-GH showed about 9.5 % increase of l-arginine production, 78.6 % reduction in l-citrulline formation, and a lower l-ornithine formation. Compared with H-7 and R-8, H-7-GH and R-8-GH showed about 1.4–1.6 folds increase of l-arginine production, about 77–80 % reduction in l-citrulline formation and about 76–80 % reduction in l-ornithine formation, respectively. As expected, co-expression of argGH significantly decreased the accumulation of l-citrulline and l-ornithine simultaneously, which disrupted the metabolism bottleneck of H-7 and R-8 and increased their l-arginine yields dramatically. Significantly, in H-7-GH and R-8-GH, the increased Yp/x (amino acids yield on biomass) of l-arginine (about 7.3–7.5 mM g −1(DCW) , l-arginine yields on biomass of H-7-GH or R-8-GH minus that of H-7 or R-8, respectively) were dramatically higher than the decrease of total Yp/x of l-ornithine and l-citrulline (about 2.9–3.1 mM g −1(DCW) , total of l-citrulline and l-ornithine yields on biomass of H-7 or R-8 minus that of H-7-GH or R-8-GH, respectively), while Yp/x of 5-GH only increased 1.32 mM g −1(DCW) (about 0.68 mM g −1(DCW) from l-ornithine and l-citrulline) compared to SYPA5-5 (Table 3). These results revealed that co-expression of argGH in H-7 and R-8 significantly strengthened l-arginine biosynthetic pathway, which showed the positive effect of feedback-resistant CcNAGK on l-arginine production.

The concentration of l-arginine (black bars), l-citrulline (dark gray bars), l-ornithine (white bars), l-lysine (grid shed bars) and l-isoleucine (sparse shed bars) produced by recombinants and their parental strains after culturing for 64 h in shake flask fermentation. The values were averages of at least three independent measurements and the standard deviations were within ±10 %
Batch fermentation of H-7-GH
In above results, recombinants H-7-GH and R-8-GH both improved their l-arginine production; however, H-7-GH showed the preferable ability to produce this target amino acid. Thus, the fermentation characteristics of H-7-GH were studied in the original fermentation medium by this work and shown in Fig. 6 and Table 4. The biomass of H-7-GH was 13.6 g L−1, decreased about 26.4 % compared to SYPA5-5 after culture for 64 h (Fig. 6b; Table 4). Due to its lower growth rate, the glucose consumption rate of H-7-GH was also slower (Fig. 6c). However, (NH4)2SO4 consumption rate of H-7-GH was higher than SYPA5-5 (Fig. 6d), which resulted in higher l-arginine synthetic rate (Fig. 6a) and decreased fermentation duration (Table 4). After culturing for 64 h, only 45 % of the added glucose was consumed by H-7-GH; however, the Yp/s (l-arginine yield on glucose, about 0.69 mmol mmol −1(glucose) ) and l-arginine productivity (3.08 mmol h−1) were greatly improved, and were 97.7 and 54.2, 30.5 and 22.2 % higher than SYPA5-5 and 5-GH, respectively (Table 4).

l-arginine production (a), biomass (b) glucose consumption (c) and (NH4)2SO4 consumption (d) of SYPA5-5 (filled squares), 5-GH (filled circles), H-7-GH in original culture (filled diamonds), H-7-GH in optimized culture (open diamonds), respectively, during shake flask batch fermentation. (All assays were performed by triplicate cultures, standard deviations of the biological replicates were represented by error bars.)
The l-arginine productivity of H-7-GH was obviously limited by the lower growth rate. Then, we optimized the concentration of yeast extract which acts as the most important factor for growth in fermentation medium (Supplementary Fig. S3). When it was raised from 10 to 14 g L−1, the biomass, l-arginine production and l-arginine productivity of H-7-GH were 20.0 g L−1, 247.0 mM and 3.86 mM h−1, increased 47.0, 25.1, 20.2 % compared to culture in original medium (Fig. 6; Table 4). Meanwhile, biomass of H-7-GH in the optimized medium was comparable to SYPA5-5 in the original medium which acted as the optimal condition for its l-arginine production; however, the l-arginine productivity and Yp/s were increased 63.6 and 69.5 %, respectively (Table 4). The Yp/s of H-7-GH in the optimized medium was lower than in the original medium, probably due to higher biomass need large amount of glucose. These results strongly suggested that co-expressing of argGH and deregulating feedback-regulation of CcNAGK could efficiently improve l-arginine productivity in SYPA 5-5. In summary, breaking through the metabolic bottleneck of argGH after deregulating repression of ArgR and feedback inhibition of CcNAGK should be the priority on metabolic engineering of l-arginine-producing strains.
Fed-batch fermentation of H-7-GH in 10-L bioreactor
To study the relevance for an industrial application, we established fed-batch fermentation with H-7-GH in optimized fermentation medium which improve the concentration of yeast extract and decrease of initial concentration of glucose and (NH4)2SO4 to reach higher cell densities. As shown in Fig. 7, fed-batch culture of the H-7-GH resulted in higher biomass of 26.2 g L−1, higher l-arginine production of 389.9 mM, and higher productivity of 5.42 mM h−1 after culturing for 72 h when compared with the shake flask fermentation (Fig. 6; Table 4). However, the Yp/s (0.35 mmol mol −1(glucose) ) was lower than shake flask fermentation, probably because of higher biomass requiring large amount of glucose, which was also found in the fermentation of pyruvate [30]. These results demonstrated that metabolically engineered H-7-GH was capable of efficiently producing l-arginine at appropriate medium and fed-batch fermentation.

Growth (filled circles), glucose consumption (filled squares), and l-arginine production (filled triangles) of H-7-GH during a fed-batch fermentation in a 10-L bioreactor. At least three independent fermentations were performed, showing comparable results
Discussion
A variety of regulatory mechanisms are imposed to control the metabolic flux into l-arginine biosynthesis at the genetic and enzymatic levels [17]. Two main regulatory modes, operating at the genetic level via ArgR repressor and at enzymatic level via allosteric regulation of CcNAGK, have been reported to be performed in Corynebacterium sp. [34, 35]. Breeding of l-arginine producers through random mutation against l-arginine analogs as well as using rational design based on genomics and proteomics databases were expected to disrupt these regulatory modes [13, 39].
Undergoing an iterative procedure of random mutation and selection, nonsense mutation (A109T) in argR was identified in SYPA5-5 which makes the l-arginine biosynthesis operon (arg cluster) release from the repression of ArgR. After deregulating allosteric inhibition of CcNAGK by introducing either one of the point mutation (H268N or R209A) into its chromosome, the two main regulatory modes involved in l-arginine biosynthesis were removed simultaneously, and then led to accumulation of l-citrulline and l-ornithine and decrease of l-arginine production. Interestingly, during reconstructed l-arginine high-producer based on comparative sequence analysis by Ikeda et al., after introducing feedback-resistant CgNAGK into the chromosome of C. glutamicum ∆argR, the resulting strain could accumulate total amount of l-arginine and l-citrulline about 80 mM at the ratio of 63:37 [13]. Meanwhile, when point mutation A26V was introduced in NAGK of C. glutamicum AR4 (∆argR, argB M31V) to further alleviate the feedback inhibition by l-arginine, resulted in slower consumption of carbon sources and decrease of l-arginine production. These phenomena, similar with our results, were not cause enough concern. In present study, the metabolic flux redistribution in l-arginine biosynthesis pathway prompted us to investigate the mechanisms in detail.
The decrease of transcription levels of argGH accompanied by the reduction of ASS and ASL activities resulted in a metabolic obstacle from l-citrulline to l-arginine, and reasonably explained the accumulation of intermediates. In addition, after fermenting for 64 h, the total yield of l-arginine, l-citrulline and l-ornithine on biomass (Yp/x) of recombinants H-7 and R-8 (8.5–9.4 mM g −1(DCW) ) compared to SYPA5-5 (8.1 mM g −1(DCW) ) did not show advantages (Table 3), which strongly suggested that the total metabolic flux towards l-arginine was also controlled after deregulating repression by ArgR and feedback inhibition of CcNAGK. Taking these phenomena together, we presumed that another regulatory mode involved in l-arginine biosynthesis works after breaking these two main regulatory modes by regulation of argGH transcription. The results of further over-expressing argGH through exogenous plasmid pDXW-10 with an unregulated promoter (tac-M) [31] to remove this regulatory mode proved the vital function of regulation of argGH transcription for l-arginine biosynthesis.
Recent studies showed that FarR, one of the transcription regulators belongs to GntR family, involves in ribosomal protein, carbon and energy metabolism, transport system and also participates in regulation of arg cluster, which has a weak binding site upstream of argC and strong binding site upstream of argG [10]. Moreover, deletion of FarR was proved to benefit l-arginine production in C. glutamicum [20]. The transcription of argGH cluster was a common regulatory node and must have been relevant to global regulation depending on some global regulatory factors. Accordingly, the comparative genes expression analysis of central metabolism (Supplementary Table S2) of recombinants H-7 and R-8 indicated that global response was also induced, which explained the changes of their growth rates. However, different type of factors involved in l-arginine biosynthesis might induce the global response through multilevel complex regulatory network in response to the growth and metabolism [9, 17]. Therefore, the regulatory mechanisms involved in control of the transcription of argGH might be various and complicated that remain unclear, and its interaction with global response were unknown yet.
These studies demonstrated that down-regulation of argGH transcription levels was another regulatory mode for controlling l-arginine biosynthesis after deregulating the repression of ArgR and allosteric inhibition of CcNAGK, and disrupting all three regulatory modes could improve l-arginine productivity remarkably. Thus, this work provided a fresh perspective in exploring new regulatory mechanisms involved in l-arginine metabolism process. Further studies can be performed for elucidating repressors that participated in regulation of argGH operon transcription and constructing the higher producer by assembling the other positive strategies, such as over-expression of bifunctional argJ [6], l-arginine transporter lysE [36] and acb cluster or improved the supplement of NADPH and carbamoylphosphate by strengthening the pentose phosphate pathway and overexpressed carAB, respectively [20].
References
An Y, Ji J, Wu W, Lv A, Huang R, Wei Y (2005) A rapid and efficient method for multiple-site mutagenesis with a modified overlap extension PCR. Appl Microbiol Biotechnol 68:774–778
Appleton J (2002) Arginine: clinical potential of a semi-essential amino acid. Altern Med Rev 7:512–522
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Chen X, Jiang S, Zheng Z, Pan L, Luo S (2012) Effects of culture redox potential on succinic acid production by Corynebacterium crenatum under anaerobic conditions. Process Biochem 47:1250–1255
Cunin R, Glansdorff N, Pierard A, Stalon V (1986) Biosynthesis and metabolism of arginine in bacteria. Microbiol Rev 50:314
Dou W, Xu M, Cai D, Zhang X, Rao Z, Xu Z (2011) Improvement of l-arginine production by overexpression of a bifunctional ornithine acetyltransferase in Corynebacterium crenatum. Appl Biochem Biotechnol 165:845–855
Elišáková V, Pátek M, Holátko J, Nešvera J, Leyval D, Goergen J-L, Delaunay S (2005) Feedback-resistant acetohydroxy acid synthase increases valine production in Corynebacterium glutamicum. Appl Environ Microbiol 71:207–213
Fierabracci V, Masiello P, Novelli M, Bergamini E (1991) Application of amino acid analysis by high-performance liquid chromatography with phenyl isothiocyanate derivatization to the rapid determination of free amino acids in biological samples. J Chromatogr B Biomed Appl 570:285–291
Gerosa L, Kochanowski K, Heinemann M, Sauer U (2013) Dissecting specific and global transcriptional regulation of bacterial gene expression. Mol Syst Biol 9:658
Hänßler E, Müller T, Jeßberger N, Völzke A, Plassmeier J, Kalinowski J, Krämer R, Burkovski A (2007) FarR, a putative regulator of amino acid metabolism in Corynebacterium glutamicum. Appl Microbiol Biotechnol 76:625–632
Haas D, Leisinger T (1975) N-Acetylglutamate-5-phosphotransferase of Pseudomonas aeruginosa. Eur J Biochem 52:365–375
Ikeda M (2003) Amino acid production processes. In: Microbial production of l-amino acids. Springer, Hidelberg, 1–35
Ikeda M, Mitsuhashi S, Tanaka K, Hayashi M (2009) Reengineering of a Corynebacterium glutamicum l-arginine and l-citrulline producer. Appl Environ Microbiol 75:1635–1641
Kalinowski J, Bathe B, Bartels D, Bischoff N, Bott M, Burkovski A, Dusch N, Eggeling L, Eikmanns BJ, Gaigalat L (2003) The complete Corynebacterium glutamicum ATCC 13032 genome sequence and its impact on the production of l-aspartate-derived amino acids and vitamins. J Biotechnol 104:5–25
Katewa SD, Katyare SS (2003) A simplified method for inorganic phosphate determination and its application for phosphate analysis in enzyme assays. Anal Biochem 323:180–187
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408
Lu CD (2006) Pathways and regulation of bacterial arginine metabolism and perspectives for obtaining arginine overproducing strains. Appl Microbiol Biotechnol 70:261–272
Miller G (1959) Use of DNS reagent for the measurement of reducing sugar. Anal Chem 31:426–428
Ohnishi J, Mitsuhashi S, Hayashi M, Ando S, Yokoi H, Ochiai K, Ikeda M (2002) A novel methodology employing Corynebacterium glutamicum genome information to generate a new l-lysine-producing mutant. Appl Microbiol Biotechnol 58:217–223
Park SH, Kim HU, Kim TY, Park JS, Kim SS, Lee SY (2014) Metabolic engineering of Corynebacterium glutamicum for l-arginine production. Nat Commun 5:4618
Sakanyan V, Petrosyan P, Lecocq M, Boyen A, Legrain C, Demarez M, Hallet J-N, Glansdorff N (1996) Genes and enzymes of the acetyl cycle of arginine biosynthesis in Corynebacterium glutamicum: enzyme evolution in the early steps of the arginine pathway. Microbiology 142:99–108
Schäfer A, Tauch A, Jäger W, Kalinowski J, Thierbach G, Pühler A (1994) Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene 145:69–73
Schendzielorz G, Dippong M, Grunberger A, Kohlheyer D, Yoshida A, Binder S, Nishiyama C, Nishiyama M, Bott M, Eggeling L (2014) Taking control over control: use of product sensing in single cells to remove flux control at key enzymes in biosynthesis pathways. ACS Synth Biol 3:21–29
Sezonov G, Joseleau-Petit D, D’Ari R (2007) Escherichia coli physiology in Luria–Bertani broth. J Bacteriol 189:8746–8749
Trotschel C, Deutenberg D, Bathe B, Burkovski A, Kramer R (2005) Characterization of methionine export in Corynebacterium glutamicum. J Bacteriol 187:3786–3794
Utagawa T (2004) Production of arginine by fermentation. J Nutr 134:2854S–2857S
Van der Rest M, Lange C, Molenaar D (1999) A heat shock following electroporation induces highly efficient transformation of Corynebacterium glutamicum with xenogeneic plasmid DNA. Appl Microbiol Biotechnol 52:541–545
Villas-Boas SG, Bruheim P (2007) Cold glycerol-saline: the promising quenching solution for accurate intracellular metabolite analysis of microbial cells. Anal Biochem 370:87–97
Weatherburn M (1967) Phenol-hypochlorite reaction for determination of ammonia. Anal Chem 39:971–974
Wieschalka S, Blombach B, Eikmanns BJ (2012) Engineering Corynebacterium glutamicum for the production of pyruvate. Appl Microbiol Biotechnol 94:449–459
Xu D, Tan Y, Huan X, Hu X, Wang X (2010) Construction of a novel shuttle vector for use in Brevibacterium flavum an industrial amino acid producer. J Microbiol Methods 80:86–92
Xu H, Dou W, Xu H, Zhang X, Rao Z, Shi Z, Xu Z (2009) A two-stage oxygen supply strategy for enhanced l-arginine production by Corynebacterium crenatum based on metabolic fluxes analysis. Biochem Eng J 43:41–51
Xu MJ, Rao ZM, Dou WF, Jin J, Xu ZH (2012) Site-directed mutagenesis studies on the l-arginine-binding sites of feedback inhibition in N-acetyl-l-glutamate kinase (NAGK) from Corynebacterium glutamicum. Curr Microbiol 64:164–172
Xu M, Rao Z, Dou W, Xu Z (2013) The role of ARGR repressor regulation on l-arginine production in Corynebacterium crenatum. Appl Biochem Biotechnol 170:587–597
Xu M, Rao Z, Dou W, Yang J, Jin J, Xu Z (2012) Site-directed mutagenesis and feedback-resistant N-acetyl-l-glutamate kinase (NAGK) increase Corynebacterium crenatum l-arginine production. Amino Acids 43:255–266
Xu M, Rao Z, Yang J, Dou W, Xu Z (2013) The effect of a LYSE exporter overexpression on l-arginine production in Corynebacterium crenatum. Curr Microbiol 67:271–278
Xu Y, Labedan B, Glansdorff N (2007) Surprising arginine biosynthesis: a reappraisal of the enzymology and evolution of the pathway in microorganisms. Microbiol Mol Biol Rev 71:36–47
Yim SH, Park MY, Lee HS, Song ES, Lee MS (2008) Analysis of gene expression in arginine biosynthetic gene cluster of Corynebacterium glutamicum. Gene Genom 30:261–270
Yoshida H, Araki K, Nakayama K (1979) Mechanism of l-arginine production by l-arginine-producing mutants of Corynebacterium glutamicum. Agric Biol Chem 43(1):105–111
Acknowledgments
This work was supported by the High Tech Development Program of China (No. 2012AA022102), the National Natural Science Foundation of China (31300028) and the Jiangsu Provincial National Basic Research Program (BK20130137).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no financial or commercial conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
10295_2015_1692_MOESM2_ESM.docx
Supplementary Fig. S1 Analysis of intracellular (filled circles) and extracellular (filled squares) l-arginine concentration during the fermentation course. (All assays were performed by triplicate cultures, standard deviations of the biological replicates were represented by error bars.) (DOCX 194 kb)
10295_2015_1692_MOESM3_ESM.docx
Supplementary Fig. S2 l-lysine concentration of SYPA5-5, H-7 and R-8, respectively after culturing for 88 h in shake flask batch fermentation. (All assays were performed by triplicate cultures, standard deviations of the biological replicates were represented by error bars.) (DOCX 89 kb)
10295_2015_1692_MOESM4_ESM.docx
Supplementary Fig. S3 Biomass (black bars), l-arginine concentration (gray bars) and glucose concentration (white bars) of H-7-GH fermented in different concentration of yeast extract. (DOCX 428 kb) (DOCX 145 kb)
Rights and permissions
About this article

Cite this article
Zhao, Q., Luo, Y., Dou, W. et al. Controlling the transcription levels of argGH redistributed l-arginine metabolic flux in N-acetylglutamate kinase and ArgR-deregulated Corynebacterium crenatum . J Ind Microbiol Biotechnol 43, 55–66 (2016). https://doi.org/10.1007/s10295-015-1692-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-015-1692-8