
Abstract
The genes encoding the mevalonate-based farnesyl pyrophosphate (FPP) biosynthetic pathway were encoded in two operons and expressed in Escherichia coli to increase the production of sesquiterpenes. Inefficient translation of several pathway genes created bottlenecks and led to the accumulation of several pathway intermediates, namely, mevalonate and FPP, and suboptimal production of the sesquiterpene product, amorphadiene. Because of the difficulty in choosing ribosome binding sites (RBSs) to optimize translation efficiency, a combinatorial approach was used to choose the most appropriate RBSs for the genes of the lower half of the mevalonate pathway (mevalonate to amorphadiene). RBSs of various strengths, selected based on their theoretical strengths, were cloned 5′ of the genes encoding mevalonate kinase, phosphomevalonate kinase, mevalonate diphosphate decarboxylase, and amorphadiene synthase. Operons containing one copy of each gene and all combinations of RBSs were constructed and tested for their impact on growth, amorphadiene production, enzyme level, and accumulation of select pathway intermediates. Pathways with one or more inefficiently translated enzymes led to the accumulation of pathway intermediates, slow growth, and low product titers. Choosing the most appropriate RBS combination and carbon source, we were able to reduce the accumulation of toxic metabolic intermediates, improve growth, and improve the production of amorphadiene approximately fivefold. This work demonstrates that balancing flux through a heterologous pathway and maintaining steady growth are key determinants in optimizing isoprenoid production in microbial hosts.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Advances in gene discovery and metabolic engineering have enabled the production of complex chemicals and pharmaceuticals using heterologous pathways in genetically engineered microbes (Fortman et al. 2008; Khosla and Keasling 2003; Nakamura and Whited 2003; Peralta-Yahya and Keasling 2010; Steen et al. 2010; Stephanopoulos 2007). Due to the difficulty in predicting the expression of heterologous genes, imbalances in gene expression and enzyme activity often result, which create bottlenecks in the heterologous pathway, reduce product formation, and lead to the accumulation of intermediates that are either toxic to the cell or can inhibit pathway enzymes (Barbirato et al. 1996; Harcum and Bentley 1999; Kizer et al. 2008; Zhu et al. 2002).
One such pathway, the mevalonate-based isoprenoid biosynthetic pathway, was constructed in Escherichia coli to bypass the native deoxyxylose-5-phosphate-based pathway (and any regulation) and to increase the production of isoprenoids (Fortman et al. 2008; Harker and Bramley 1999; Kajiwara et al. 1997; Kim and Keasling 2001; Matthews and Wurtzel 2000). To facilitate the identification of the bottlenecks, the genes encoding the enzymes of the mevalonate pathway were expressed in two operons, each on its own plasmid, based on their position relative to mevalonate: pMevT harbors the genes for the mevalonate pathway from acetyl-CoA to mevalonate and pMBIS harbors the genes for the mevalonate pathway from mevalonate to farnesyl pyrophosphate (FPP) (Fig. 1a). Although this heterologous pathway increased product yields, imbalances in the expression of the genes encoding the pathway enzymes resulted in the accumulation of pathway intermediates, slow growth, and low product titers. A detailed study of the heterologous mevalonate pathway, through substrate feeding experiments, suggested that in vivo accumulation of three mevalonate pathway intermediates (dimethylallyl pyrophosphate [DMAPP], isopentenyl pyrophosphate [IPP], and FPP) inhibits normal cell growth (Martin et al. 2003). Furthermore, through gene titration experiments, we found that mevalonate kinase (MK), which converts mevalonate to mevalonate phosphate, limited flux through the lower part of the mevalonate pathway and, hence, amorphadiene production and that increasing its expression increased amorphadiene production (Anthony et al. 2009).

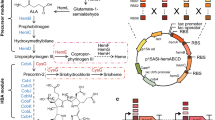
Lower mevalonate pathway constructs used in this study. a Mevalonate pathway consisting of eight genes was initially divided into two operons: upper mevalonate pathway (pMevT) and lower mevalonate pathway (pMBIS). b Using PCR, the five genes of the lower mevalonate pathway were amplified. By designing PCR primers that contain the selected RBS sequences, three RBSs with three different strengths were placed 5′ of the first three genes of the MBIS operon. Only one RBS of medium activity was used with the last two genes, idi and ispA. The nine constructed operons containing ADS and MBIS operon cloned into the pTrc99a backbone. These constructs were transformed into DH1 strain of E. coli along with the pMevT operon and were tested for growth and amorphadiene production. c The 10 constructs containing ADS with RBSs of different activity levels and two of the MBIS operons (MBIS1 and MBIS2). The two MBIS operons selected were the ones with all medium activity and high activity RBSs 5′ of the first three genes of MBIS operon. These 10 operons (pTrc-RBS.ADS-MBIS1 and pTrc-RBS.ADS-MBIS2) were transformed into E. coli DH1 along with pMevT and were tested for growth and amorphadiene production
There are several published methods to alter enzyme levels and, therefore, pathway flux: choice of plasmid origin of replication (Carrier et al. 1998; Jones et al. 2000; Jones and Keasling 1998; Kittleson et al. 2011), promoters of different strengths (Alper et al. 2005; Anthony et al. 2009; Davis et al. 2011; Kelly et al. 2009; Pitera et al. 2007), codon optimization of genes that are poorly expressed (Redding-Johanson et al. 2011), utilization of synthetic scaffolds to co-localize pathway proteins (Dueber et al. 2009), and use of enzymes with different properties (Ma et al. 2011), to name a few. Because of the lack of accurate models to predict protein production from DNA sequence, the evidence that some genes in the pathway were poorly expressed, and a general lack of enzyme kinetic information, we chose to use a combinatorial approach to investigate the effect of ribosome binding site (RBS) strength on gene expression, amorphadiene production, and cell growth. For this purpose, RBSs of various strengths were cloned 5′ of the genes of the lower mevalonate pathway and the amorphadiene synthase (ADS) gene. These RBSs were selected based on their theoretical strengths. The RBS sequences were selected either through literature search or predictions of the RBS calculator (Salis et al. 2009). A combination of these RBS–gene parts was assembled into different constructs, with the same promoter on all constructs. When growth and product titers of E. coli strains expressing these constructs were monitored, significant differences among the various constructs were observed. Further analysis of mevalonate pathway enzymes revealed a correlation between protein levels and RBS strength. Measurement of pathway intermediate concentrations provided additional information on how the changes in protein levels translate to different product titers and growth rates. Through this study, we now have a better understanding of how different parts of the mevalonate pathway work together.
The results presented here demonstrate that balancing flux through a heterologous pathway and maintaining steady growth are key determinants in optimizing isoprenoid production in microbial hosts. The method used and information gained from this study should be valuable for engineering microbial production of other molecules of therapeutic and industrial interest.
Materials and methods
All chemical standards were obtained from Sigma-Aldrich, Carlsbad, CA, USA. All chemicals used were of analytical and reagent grade, and all solvents used were of high-performance liquid chromatography (HPLC) grade.
Strains and plasmids used in this study are listed in Table 1. E. coli DH10B was used for cloning and construction of MBIS constructs. E. coli DH1 (Invitrogen, Carlsbad, CA, USA) was used for expression and production studies. Media components and chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA) and Fisher Scientific (Pittsburgh, PA, USA). All experiments were performed in Terrific Broth (TB) medium with appropriate antibiotics for plasmid selection and glycerol as the carbon source. All strains were grown at 30 °C.
For targeted proteomics experiments, mass spectrometry grade trypsin (Trypsin Gold) was obtained from Promega (Madison, WI, USA) and prepared according to the manufacturer’s instructions.
Operon and plasmid construction
The genes encoding MK (mevK), phosphomevalonate kinase (PMK; pmk), mevalonate pyrophosphate decarboxylase (PMD; pmd), isopentenyl pyrophosphate isomerase (idi), FPP synthase (ispA), and ADS (ads) were polymerase chain reaction (PCR)-amplified with RBSs of various strengths, assembled into operons, and cloned into plasmids, all with the same promoters (Table 1; Fig. 1). The MBIS operon (mevK, pmk, pmd, idi, and ispA) was previously cloned into pBBR1MCS, a low-copy plasmid with an isopropyl β-d-1-thiogalactopyranoside (IPTG)-inducible lac promoter, Plac, creating pMBIS. To increase the expression of every gene in this operon, a higher copy number plasmid with a stronger promoter was used. All of the constructed MBIS operons were cloned into a pTrc99a derivative containing the pBR322 origin, lacI Q, and Ptrc, creating plasmid pTrc-M.ADS-MBIS1-9. RBSs of various strengths were placed 5′ of each of the first three genes of the lower half of the mevalonate-based isoprenoid biosynthetic pathway to construct the new MBIS operons. The RBSs used with the three genes mentioned previously were chosen based on their theoretical strengths (Hui et al. 1984) so that each of these genes had a theoretically strong, medium, or weak RBS in the various constructs. The RBSs that were placed 5′ of ads were also selected based on their predicted theoretical strengths using an RBS calculator (Salis et al. 2009) so that each of the RBSs selected is theoretically stronger than the medium-strength RBS that was originally placed 5′ of ADS (https://salis.psu.edu/software/forward). V1.0 of the RBS calculator was used when these studies were conducted in 2009. The sequence provided for designing these RBSs were the 50 bp prior to the start codon of ADS and the 50 bp following the start codon of ADS (including the start codon, ATG). The BioBrick cloning strategy was used to assemble the various components of the pathway (Shetty et al. 2008). The BioBrick strategy uses restriction sites such that, when two DNA parts are ligated together, the ligation event destroys the site and leaves the same sites at the ends of the assembled parts so that more parts can be added in the same manner. This allows one to use the same enzymes for each assembly, and the procedure can be repeated to put different parts together.
The first three genes of the lower mevalonate pathway (mevK, pmk, and pmd) were amplified by PCR using pMBIS as the template and gene-specific primers that contained the three selected RBSs and restriction sites EcoRI, BglII, BamHI, and XhoI. The last two genes, idi and ispA, were also amplified by PCR. Only one RBS, the medium-strength RBS, was used with these two genes (Fig. 1) because these genes are native to E. coli and, as such, should be easily expressed. Each individual part was cloned into a BioBrick cloning vector, pBca9145 (ColE1 origin of replication, AmpR, and no promoter), to facilitate the construction of MBIS operons. We combined the MBIS operon and ads with its original, medium-strength (M) RBS into one operon using pTrc99a as the backbone. ADS was digested with EcoRI/BamHI restriction enzymes, while the pBca9145-MBIS plasmids were digested with EcoRI/BglII restriction enzymes. The two digested fragments were ligated together to form the pBca9145-M.ADS-MBIS plasmids. Retaining all of the BioBrick restriction sites, the pBca9145-M.ADS-MBIS plasmids were then digested with EcoRI and BamHI to be cloned into EcoRI/BamHI sites of pTrc99a expression vector to form the pTrc-M.ADS-MBIS constructs. Out of 27 possible RBS–gene combinations to form the pTrc-M.ADS-MBIS constructs, nine constructs were chosen to be studied (Fig. 1b). All of the resulting constructs were sequenced to ensure accuracy. RBS sequences are provided in Online Resource 1.
Growth conditions
E. coli DH1 was transformed with the appropriate plasmids and plated onto Luria Broth Agar plates containing appropriate antibiotics. Overnight cultures were started from fresh transformations into 5 mL of Luria Broth (Difco) with antibiotics. Overnight cultures were diluted to an optical density at a wavelength of 600 nm (OD600) of 0.05 in 40 mL of TB (Difco) with 1 or 3 % glycerol and overlaid with 20 % (v/v) dodecane to trap the product, amorphadiene. Experiments were initiated when cultures reached an OD600 of 0.2–0.3 (~2–3 h after subculture) by the addition of 1 mM IPTG. Cultures were incubated for 48–72 h, and at each time point, the OD600 was read and samples from the dodecane phase were taken to determine amorphadiene production. Previous studies have shown that over 97 % of the amorphadiene produced partitions into the dodecane overlay of cultures (Newman et al. 2006).
Gas chromatography–mass selective analysis of amorphadiene
Samples (100 μL) were removed from the dodecane layer of each culture and centrifuged at 13,300×g for 3 min at room temperature. After centrifugation, 10 μL of the organic phase was added to 990 μL of ethyl acetate. To measure amorphadiene levels, 1 μL of the sample was injected into an Agilent 6890 series gas chromatograph (GC) equipped with an Agilent 5973 mass selective (MS) detector and a CycloSil-B (chiral) capillary column (30 m × 250 μm × 0.25 μm thickness; Agilent). The GC oven temperature program was as follows: 100 °C for 45 s, a ramp of 35 °C/min to 200 °C, a ramp of 100 °C/min to 250 °C, and a hold at 250 °C for 1 min. The MS was operated in selected ion monitoring (SIM) mode scanning for the molecular ions at 189 and 204 m/z. Purified amorphadiene was used to generate standard curves for obtaining production titers.
Targeted proteomic analysis
Whole cell lysate samples were prepared for targeted proteomic analysis as follows. The cells were pelleted via centrifugation at 8,000×g (4 °C) for 5 min, the supernatant was decanted, and cells were flash frozen in liquid nitrogen and stored at −80 °C. Protein extraction was done by using chloroform/methanol precipitation (Wessel and Flügge 1984) and sonication in 6 M urea buffer with 500 mM triethylammonium bicarbonate. For the urea sample preparation protocol, cells were resuspended in 600 μL of fresh urea buffer and transferred to a new tube. Samples were sonicated while on ice for 5 s, followed by a 10-s rest period for a total sonication time of 3 min. After sonication, the sample was centrifuged at maximum speed (28,000×g) for 30 min at 4 °C to pellet undissolved cell debris. The supernatant was transferred to a new tube and protein quantitation was done by using the Bradford assay. For chloroform/methanol sample preparation, cells were allowed to thaw on ice and vortexed to homogenize the solution. One hundred microliters of the concentrated cell pellet was placed into a fresh 1.7-mL tube, to which 400 μL of methanol was added and vortexed. Subsequently, 100 μL of chloroform was added and mixed, followed by the addition of 400 μL of liquid chromatography and mass spectrometry (LC-MS) grade water. Samples were vortexed and spun in a benchtop centrifuge at 20,000×g for 1 min. The upper layer was discarded carefully, and 300 μL of methanol was added and mixed. Samples were centrifuged at 20,000×g for 2 min, allowing the protein to collect at the bottom of the tube. The supernatant was removed and protein pellets were allowed to dry in a vacuum concentrator for 30–60 min. Five hundred microliters of 50 mM ammonium bicarbonate in 10 % acetonitrile was added, and samples were placed in a sonic bath intermittently with continuous vortexing to resuspend the pellet. Protein concentration was determined with the Bio-Rad DC Protein Reagent (Bio-Rad, Hercules, CA, USA), according to the manufacturer’s instructions. Approximately 100 μg of protein was taken from each sample and diluted to a final concentration of 1 μg/μL in 100 mM ammonium bicarbonate. To reduce the disulfide bonds in the samples, 5 mM tris(2-carboxyethyl)phosphine was added to the solution and allowed to sit at room temperature for 30 min. The proteins were alkylated with iodoacetic acid (200 mM in 100 mM NaOH) in the dark for 30 min. Following alkylation, trypsin (1 μg/μL) was added to the sample to a final concentration of 1:50 (trypsin/sample) and incubated at 37 °C overnight. Prior to analysis, samples produced via the urea protocol were desalted by using C18 preparative spin columns. The peptides prepared by chloroform/methanol precipitation were used without further purification.
Peptide samples were analyzed on a LC-MS/MS system by using targeted proteomics (Zhu et al. 2002). Briefly, a 4000Q-Trap mass spectrometer (Applied Biosystems, Foster City, CA, USA) was coupled to an Eksigent TEMPO NanoLC-2D via an Applied Biosystems Nanosource II operating in positive ion mode. 4000Q-Trap was operated in selected reaction monitoring (SRM) mode by which the concentrations of targeted proteins were determined relative to bovine serum albumin peptides that were added as an internal standard. Samples were loaded onto a PepMap100 μ-Guard column (Dionex/LC Packings) and separated on PepMap100 C18 analytical column (75 μm i.d., 150 mm length, 100 A, and 3 μm). The column was equilibrated for 2 min with 95 % A (2 % acetonitrile and 0.1 % formic acid) and 5 % B (98 % acetonitrile and 0.1 % formic acid). The column was then ramped up to 70 % B in 15 min during which the peptides were separated. A ramp up to 80 % B in 3 min washed the remaining peptides off the column where it was held for 10 min, after which it was quickly ramped down to 5 % B where it was held for 13 min to allow the column to re-equilibrate. The raw data were processed by using Multiquant (Applied Biosystems, Foster City, CA, USA).
Metabolite extraction
All solvents used for metabolite analyses were of HPLC grade (Honywell Burdick & Jackson, Morristown, NJ, USA). Whole cell lysate samples were prepared for targeted metabolite analysis as follows. The cells (from 10 mL E. coli culture for the first three time points, and 5 mL E. coli culture for the remaining time points) were pelleted via centrifugation at 8,000×g (4 °C) for 5 min. After the supernatant was decanted, metabolite extraction was performed by adding 1 mL of ice-cold methanol to the pellet, followed by mixing. The resulting methanol lysate was then centrifuged at 8,000×g for 5 min, and the methanol supernatant was transferred to a separate tube. This step was repeated once more but with ice-cold water in place of methanol. The final 50:50 methanol/water solution was dried by lyophilization (Labconco Co., Kansas City, MO, USA), reconstituted in 0.5 mL 50:50 methanol/water, and filtered via an Amicon Ultra 5K molecular weight cutoff centrifuge filter (MilliPore, Millipore Inc., Billerica, MA, USA).
Analysis of FPP by liquid chromatography and mass spectrometry
FPP was made up to 200 μM, as the stock solution, in 50:50 methanol/water. The separation of the FPP was conducted on a ZIC-HILIC column (150 mm length, 2.1 mm internal diameter, and 3.5 μm particle size; from Merck SeQuant, and distributed via The Nest Group, Inc., Southborough, MA, USA) using an Agilent Technologies 1200 Series HPLC System (Agilent Technologies, Santa Clara, CA, USA). Injection volumes for chemical standards and metabolites were 2 and 10 μL, respectively. The temperature of the sample tray was maintained at 4 °C using an Agilent FC/ALS Thermostat. The column compartment was set to 32 °C. FPP was eluted isocratically with a mobile phase composition of 36 % of 50 mM ammonium acetate in water and 64 % of acetonitrile. A flow rate of 0.15 mL/min was used throughout.
The HPLC system was coupled to an Agilent Technologies 6210 time-of-flight mass spectrometer (LC-TOF MS) by a 1/6 post-column split. Contact between both instrument setups was established using a LAN card in order to trigger the MS into operation upon the initiation of a run cycle from the MassHunter workstation (Agilent Technologies, Santa Clara, CA, USA). Electrospray ionization (ESI) was conducted in the negative ion mode and a capillary voltage of −3,500 V was utilized. MS experiments were carried out in full scan mode, at 0.85 spectra/s and a cycle time of 1.176 s, for the detection of [M–H]− ions. The instrument was tuned for a range of 50–1,700 m/z. Prior to LC-TOF MS analysis, TOF MS was calibrated via an ESI-L low-concentration tuning mix (Agilent Technologies, Santa Clara, CA, USA). Data acquisition and processing were performed by the MassHunter software package. FPP from E. coli extracts were quantified via seven-point calibration curves ranging from 625 nM to 50 μM. The R 2 coefficients for the calibration curves were ≥0.99.
Analysis of mevalonate by liquid chromatography and mass spectrometry
The chemical standard of mevalonate was prepared by treating mevalonolactone with 2 M potassium hydroxide (Martin et al. 2003). Mevalonate was then dissolved in a solution of 50 % methanol and 50 % water. The separation of mevalonate was conducted on a Carbomix H-NP 10:8 % column (250 mm length, 4.6 mm internal diameter, and 5 μm particle size; Sepax, San Francisco, CA, USA) using an Agilent Technologies 1100 Series HPLC System. A sample injection volume of 1 μL was used throughout. The temperature of the sample tray was maintained at 4 °C by an Agilent FC/ALS Thermostat. The column compartment was set to 50 °C. Mevalonate was eluted isocratically with a mobile phase composition 0.1 % formic acid in water. A flow rate of 0.2 mL/min was used throughout.
The HPLC system was coupled to an Agilent LC-MSD SL mass spectrometer (LC-MS) by a 1/6 post-column split. The LC-MS system was controlled by the Chemstation (Agilent Technologies, Santa Clara, CA, USA) software package. Contact between both instrument setups was established by a LAN card in order to trigger the MS into operation upon the initiation of a run cycle from Chemstation. ESI-MS was conducted in the negative ion mode, and a capillary voltage of −3,500 V was utilized. MS experiments were carried out in the SIM mode, with a dwell time of 995 ms, for the detection of [M–H]− ions. The instrument was tuned for a range of 50–2,000 m/z. The LC-MSD SL was calibrated externally by the Agilent ES tuning mix (Agilent Technologies, Santa Clara, CA, USA). Data acquisition and processing were performed by MassHunter. Mevalonate from E. coli extracts was quantified via seven-point calibration curves ranging from 312.5 nM to 100 μM. The R 2 coefficients for the calibration curves were ≥0.99.
Results
Initial reactions of the lower mevalonate pathway limit carbon flux to amorphadiene
In previous work, we found that a significant amount of mevalonate accumulated in the culture medium of cells expressing the mevalonate pathway, suggesting a limitation of the lower mevalonate pathway (mevalonate to FPP). Therefore, previous work to improve isoprenoid production in E. coli focused on the limiting steps in the conversion of mevalonate to amorphadiene (Anthony et al. 2009). The overexpression of mevK was shown to increase amorphadiene production by twofold to threefold (Anthony et al. 2009). Since the first three genes of the lower mevalonate pathway were from S. cerevisiae, whereas the last two were E. coli’s native genes, we decided to focus our efforts on optimizing the expression of the heterologous genes encoding the enzymes MK, PMK, and PMD. To this end, we constructed plasmids with RBSs of high (H), medium (M), and low (L) strengths (Hui et al. 1984) cloned 5′ of mevK, pmk, and pmd (Fig. 1b). It should be emphasized that the activity level of each RBS is theoretical, and the observed strength of an RBS is context dependent. Nine MBIS operons were constructed using different RBS combinations; idi and ispA, each with a medium-strength RBS, were cloned 3′ of pmd; and ads, with a medium-strength RBS, was cloned 5′ of mevK. All genes were placed under transcriptional control of a single promoter, Ptrc, on a high-copy plasmid. The resultant ADS-MBIS operons were cloned into a pTrc99a backbone, creating plasmid pTrc-M.ADS-MBIS1-9 (Table 1; Fig. 1). E. coli DH1 was transformed with pMevT, which encodes the enzymes (acetoacetyl-CoA synthase, HMG-CoA synthase, and HMG-CoA reductase) that convert acetyl-CoA into mevalonate and the various pTrc-M.ADS-MBIS constructs. Cultures were induced with IPTG, and samples were taken at 24 and 48 h to monitor growth and amorphadiene production. By comparing growth between the different strains, it was possible to check for any growth defect that could have resulted from the accumulation of toxic intermediates. Also, as protein levels changed with different RBSs, we would expect the amorphadiene titer to vary due to differences in carbon flux to amorphadiene.
The strains expressing these nine MBIS synthetic operons showed significant differences in growth and amorphadiene production. Strains harboring MBIS2, MBIS3, MBIS4, and MBIS7 grew poorly up to 24 h and produced low titers of amorphadiene (Fig. 2); the others, which had higher cell density at 24 h, produced higher titers of amorphadiene. It should be noted that, while MBIS4 grew poorly, it had an initially high specific productivity (24 h time point; Fig. 2c). All strains had similar cell densities at 48 h. The strain that produced the most amorphadiene harbored MBIS5, which has a strong RBS 5′ of pmk. Given that our previous gene supplementation studies had shown that poor pmk expression was limiting pathway flux (Anthony et al. 2009), even a slight improvement in expression level could potentially relieve a bottleneck in the conversion of phosphomevalonate to IPP, resulting in higher amorphadiene titers.

Cell growth (a), amorphadiene production (b), and specific amorphadiene production (c) of E. coli DH1 harboring pMevT and pTrc-M.ADS-MBIS1-9 24 h (blue) and 48 h (red) after induction, grown on 1 % glycerol. The first position is for mevK, the second position for pmk, and the third position for pmd. Some of the strains showed growth inhibition at 24 h that was relieved at 48 h. The strains that grew poorly at 24 h produced amorphadiene poorly
Use of a stronger RBS 5′ of ADS in order to improve production
It was previously demonstrated that ADS could be a limiting step in amorphadiene production (Anthony et al. 2009), and in order to achieve optimal production, the ADS level should be high. Additionally, we have shown that, if the sesquiterpene synthase does not utilize the FPP produced by the heterologous pathway at the same rate that it is produced, it will accumulate inside the cell to a level that is toxic (Martin et al. 2003; Withers et al. 2007). However, attempts to engineer ADS for improved amorphadiene production proved to be difficult. Therefore, theoretically stronger RBSs were chosen in order to increase enzyme synthesis. Four synthetic RBS sequences with different activity levels, all theoretically higher than the original RBS cloned 5′ of ADS, were chosen. These RBS-ADS constructs were then cloned into vectors harboring the MBIS1 and MBIS2 operons, creating plasmids pTrc-RBS.ADS-MBIS1 and pTrc-RBS.ADS-MBIS2, respectively (Fig. 1c). The MBIS2 construct was chosen over MBIS5 because it possessed high-strength RBSs for each of the first three genes of the lower mevalonate pathway as opposed to just having a high-strength RBS for PMK, as in the case of MBIS5. Further, MBIS2 displayed severe growth inhibition presumably due to significant FPP accumulation (which was later confirmed, see succeeding paragraphs) and, therefore, had the greatest potential to produce the highest titer of amorphadiene among the nine MBIS constructs. MBIS1 was chosen as the control for comparison as it possessed medium-strength RBSs for each of the first three genes of the lower mevalonate pathway and showed no inhibition of growth. Any increase in ADS should, in theory, reduce the level of FPP accumulation in E. coli, increase amorphadiene production, and relieve growth inhibition. E. coli DH1 was transformed with pMevT and one of the 10 pTrc-RBS.ADS-MBIS plasmids. Cultures were induced with IPTG, and samples were taken at 24 and 48 h to monitor growth and amorphadiene production.
The growth differences among the strains expressing ADS with different strength RBSs were more significant than the growth difference between the strains harboring the two versions of the MBIS operons (MBIS1 or MBIS2; Fig. 3). Strains expressing ADS with RBSs H.2 and H.4 grew to lower densities at 24 and 48 h than strains harboring the other RBS-ADS constructs. These RBSs should be stronger than the original (M) RBS 5′ of ads and, hence, result in elevated ADS that consumes the FPP supply of the cell. Since FPP is required for growth, too much consumption of this intermediate by ADS could also have impacted the cell negatively.

Cell growth (a) and amorphadiene production (b) of E. coli DH1 harboring pMevT and various versions of pTrc-RBS.ADS-MBIS 24 h (blue) and 48 h (red) after induction, grown using 1 % glycerol. RBSs of various strengths (H.1, H.2, H.3, and H.4) were cloned 5′ of ads using two version of the MBIS operon, MBIS1 and MBIS2. Strains with different strengths of RBS 5′ of ads exhibited growth differences. Strains with lower OD produced more amorphadiene compared to the strain with original (M) RBS (0.8 g/L; Fig. 2b)
Amorphadiene production was similar between strains expressing MBIS1 and MBIS2 operons (Fig. 3b). The RBS H.1 appears to be weaker than the original (M) RBS and resulted in decreased amorphadiene levels (520 mg/L) compared to the original (M) RBS (860 mg/L). While RBSs H.2 and H.4 affected growth and resulted in a lower OD, there was moderate improvement in amorphadiene production compared to the original (M) RBS (Figs. 2 and 3). There was no improvement in amorphadiene production in strains expressing the MBIS2 operon compared to strains expressing the MBIS1 operon, even when stronger RBSs were used with ADS.
As there was no residual glycerol for several of the constructs (data not shown), one limitation to amorphadiene production could be the carbon source. It would be impossible to differentiate between the two operons (MBIS1 and MBIS2) if there is not enough carbon available to support higher amorphadiene production than what was already achieved by strains harboring the MBIS1 operon. To determine if a higher titer could be obtained with an increased amount of carbon source, we tested six constructs with a higher glycerol concentration (3 %) in the medium. Cultures were induced with IPTG, and samples were taken at 24 and 48 h to monitor growth and amorphadiene production. The strains tested exhibited the same growth pattern in both 1 and 3 % glycerol (Figs. 3 and 4). E. coli DH1 harboring pTrc-M.ADS-MBIS2 showed growth inhibition at 24 h that was relieved at 48 h. E. coli DH1 harboring either pTrc-H.2.ADS-MBIS1 or pTrc-H.2.ADS-MBIS2 exhibited a slower growth rate and lower OD compared to the control strain (pTrc-M.ADS-MBIS1).

Growth (a), amorphadiene production (b), and specific amorphadiene production (c) of E. coli DH1 harboring pMevT and pTrc-RBS.ADS-MBIS at 24 h (blue) and 48 h (red) after induction, grown using 3 % glycerol. Strains with different strengths of the RBS 5′ of ADS exhibited growth differences. While all the strains studied show moderate improvement in amorphadiene production when glycerol concentration was increased, the strain expressing pTrc-M.ADS-MBIS1 produces a significantly higher amorphadiene concentration than any other strain
Analysis of the amorphadiene concentration from the tested constructs, however, yielded interesting results. At the higher concentration of glycerol (3 %), all strains harboring the MBIS1 operon produced more amorphadiene (e.g., had higher amorphadiene titers) than strains expressing the MBIS2 operon (Fig. 4b). Although amorphadiene production for all strains benefited from the added glycerol, the improvement observed in the case of E. coli harboring constructs with the medium-strength RBS 5′ of ads was significantly greater than for any of the other constructs (approximately threefold higher specific production); such improvement was not observed when a stronger RBS (H.2) was placed 5′ of ads for either MBIS1 or MBIS2. By increasing the carbon source from 1 to 3 %, strains with elevated enzyme levels were expected to synthesize more of the precursor metabolite FPP, which could, in turn, be used to produce more amorphadiene. However, our attempts to increase enzyme levels resulted in reduced growth. This may be attributed to different activities of the enzymes in the heterologous pathway, resulting in the accumulation of pathway intermediates and by-products that inhibited pathway enzymes or were toxic to the cells (and/or growth) or overconsumption of intermediate metabolites (e.g., FPP) that are critical for cell growth. In addition, the amorphadiene titers remained relatively unchanged between 24 and 48 h for strains expressing the MBIS2 operon, while the amorphadiene titers increased from 24 to 48 h for all strains expressing the MBIS1 operon. This observation was more pronounced when specific production is considered (Fig. 4c). This was clearly observed when comparing the two strains harboring pTrc-M.ADS-MBIS1 and pTrc-M.ADS-MBIS2. While the strain harboring pTrc-M.ADS-MBIS2 showed higher specific production at 24 h than the strain harboring pTrc-M.ADS-MBIS1, there was no additional amorphadiene produced between the 24- and 48-h time points for this strain. Based on these results, amorphadiene production appears to have stopped after the 24-h time point for strains expressing the MBIS2 operon, but not for strains expressing the MBIS1 operon. This is most likely caused by the fact that the cells have stopped production of FPP.
Proteomics analysis of gene expression
A proteomics approach was used to quantify the levels of all enzymes in the pathway (Redding-Johanson et al. 2011). We reasoned that determining protein profiles for every enzyme in the mevalonate pathway would help us confirm the hypothesis that a decrease in enzyme levels after 24 h in strains expressing MBIS2 could impede amorphadiene production after this time point. E. coli DH1 harboring pMevT and pTrc-M.ADS-MBIS1 or pMevT and pTrc-M.ADS-MBIS2 (Table 2) were incubated in TB with 3 % glycerol and induced with 0.5 mM IPTG at an OD of 0.25–0.35. Two samples were taken at 24 and 48 h and were prepared for proteomics measurements.
The RBSs used to drive the expression of idi and ispA are the same in the MBIS1 and MBIS2 operons. We expected that, when all of the expression conditions were kept constant for these two genes, both constructs would yield similar protein levels. However, Idi and IspA from MBIS2 were produced at lower levels than those from MBIS1 at both 24- and 48-h time points (Fig. 5a). Another important observation was the increased MK level (by tenfold) in the strain harboring pTrc-M.ADS-MBIS2 despite the generally lower expression of the other genes harbored on this plasmid compared to pTrc-M.ADS-MBIS1. This confirms the preliminary assumption that the predicted high-strength RBS selected for MK in MBIS2 is in fact stronger than that in MBIS1 and results in a significant increase in protein production. From the proteomics data, it is difficult to draw any conclusions with regard to the RBS strength for PMK and PMD. SRM transitions for PMK were validated with His-tag purified PMK protein, indicating that the low observed amount of PMK in the sample was not due to limitations of the mass spectrometry method. Such low PMK levels make any comparison and prediction difficult. The generally lower expression of the pTrc-M.ADS-MBIS2 construct makes it difficult to draw any conclusion on the PMD RBS strength as well.

Relative levels of all enzymes in the mevalonate pathway at 24 h (light bars) and 48 h (dark bars) after induction. a Blue bars represent E. coli DH1 harboring pMevT and pTrc-M.ADS-MBIS1 and green bars represent E. coli DH1 harboring pMevT and pTrc-M.ADS-MBIS2. b Blue bars represent E. coli DH1 harboring pMevT and pTrc-H.2.ADS-MBIS1 and green bars represent E. coli DH1 harboring pMevT and pTrc-H.2.ADS-MBIS2
The proteomics analysis data for strains harboring pTrc-H.2.ADS-MBIS1 and pTrc-H.2.ADS-MBIS2 showed similar protein profiles (Fig. 5b). One objective of studying these two strains was to compare the RBS strength of the original, medium-strength (M) RBS and the higher-strength synthetic RBS activity H.2 (which resulted in higher amorphadiene production compared to medium-strength RBS in 1 % glycerol) 5′ of ads. By comparing the ADS levels between strains harboring pTrc-M.ADS-MBIS1 and pTrc-H.2.ADS-MBIS1, we found that the RBS, H.2, with theoretically higher activity yielded twice as much protein as the original, medium-strength (M) RBS.
For pTrc-H.2.ADS-MBIS1 and pTrc-H.2.ADS-MBIS2, the levels of proteins produced from every gene in the pathway, with the exception of ads and idi, were lower when compared to constructs harboring the original, medium-strength (M) RBS 5′ of ads (Fig. 5). The stronger RBS (H.2) 5′ of ads results in higher ADS levels, but the MBIS1 operon is unable to produce enough FPP to support the demand for both ADS and cellular growth.
Metabolite analysis
There was insufficient kinetics information available for most of the enzymes in our engineered mevalonate pathway to predict the optimal protein level for each of the enzymes in the pathway. We expected to see improved amorphadiene titers by choosing stronger RBSs that yielded higher protein levels. However, it appears that maintaining a balanced pathway, where none of the intermediates accumulate or deplete, is the key determinant in optimizing isoprenoid biosynthesis.
By determining the concentrations of intermediate metabolites, the bottlenecks in the pathway could be identified and it can be verified if accumulation of any of the intermediates was associated with toxicity and growth inhibition. These results could be employed to further optimize the isoprenoid biosynthetic pathway and provide us with a better understanding of how different parts of this pathway work together.
As mentioned previously, the accumulation of FPP is toxic and causes growth inhibition. To determine the FPP concentration that is tolerable by E. coli without causing any growth inhibition, we transformed E. coli DH1 with pMevT and pTrc-MBIS2 (3 % glycerol as the carbon source). The ADS gene was not expressed to allow for accumulation of FPP. Multiple samples were then taken at different time points during the course of the experiment to determine how FPP accumulation affects growth and to measure the intermediate metabolite concentrations along the growth curve.
E. coli harboring pMevT and pTrc-MBIS2 grew normally until induction. After induction, the cells stopped growing and no further increase in OD was observed. It took approximately 10 h for cells to recover and for cell growth to be restored (Fig. 6a). Using LC-TOF MS analysis, we were able to measure only FPP (Fig. 6b) and mevalonate (Fig. 6c) concentrations. Given that the samples were pre-concentrated and considering the sensitivity of this approach, we can claim that not a significant amount of any of the other pathway intermediates (the standards for which we were able to measure) accumulated at any time point during this experiment.

Growth and intracellular metabolite levels of E. coli DH1 harboring various mevalonate pathway constructs, grown on 3 % glycerol. Symbols represent E. coli harboring pMevT and pTrc-M.ADS-MBIS1 (red squares), E. coli harboring pMevT and pTrc-M.ADS-MBIS2 (blue diamonds), or E. coli harboring pMevT and pTrc-MBIS2 (gray circles). a Cell density. b FPP concentration normalized to cell density. c Mevalonate concentration normalized to cell density. E. coli harboring pMevT and pTrc-M.ADS-MBIS1 was induced at 2.3 h (OD600 = 0.30) by the addition of 1 mM IPTG. E. coli harboring pMevT and pTrc-M.ADS-MBIS2 was induced at 2.3 h (OD600 = 0.45) by the addition of 1 mM IPTG. E. coli harboring pMevT and pTrc-M.MBIS1 was induced at 4.5 h (OD600 = 0.32) by the addition of 1 mM IPTG
By comparing the FPP concentration profile (Fig. 6b) and growth curve (Fig. 6a) of E. coli harboring pMevT and pTrc-MBIS2, FPP production appears to start just after induction, and the concentration reaches its maximum when the steady increase in the OD ceases. FPP concentration then begins to decrease and, by the time the cells have resumed their growth, it has diminished to basal levels. After induction, during the time when FPP accumulation occurs in the cell, there was no accumulation of mevalonate. However, as the FPP concentration diminished to the basal level, mevalonate began to accumulate. At the final time point, FPP was not detected, whereas mevalonate had accumulated to very high levels (Fig. 6). These results suggest that E. coli can tolerate approximately 1 μM FPP inside the cell before cell growth is affected and further production of FPP is prevented. This strain showed growth inhibition after induction that was later relieved (Fig. 6a). However, by comparing the FPP concentration profile (Fig. 6b) and growth curve (Fig. 6a) of E. coli harboring pMevT and pTrc-M.ADS-MBIS2, it appears that FPP levels as low as 400 nM significantly hinder cell growth and, by the time the FPP concentration reaches 700 nM, further production of this intermediate appears to stop.
We also examined growth and intracellular FPP and mevalonate concentrations for cells expressing ADS (pMevT with pTrc-M.ADS-MBIS1 or pTrc-M.ADS-MBIS2). E. coli DH1 was transformed with either pMevT and pTrc-M.ADS-MBIS1 or pMevT and pTrc-M.ADS-MBIS2, and 3 % glycerol was provided as the carbon source. Multiple samples were taken at different time points from each culture in order to monitor growth and to analyze intermediate metabolite concentrations. E. coli DH1 harboring pMevT and pTrc-M.ADS-MBIS1 exhibited normal growth and did not show any signs of growth inhibition (Fig. 6a). This strain accumulated FPP to a maximum concentration of 200 nM (Fig. 6b). After 10 h, the FPP concentration decreased; the cells then maintained a steady FPP concentration. At the earlier time points, E. coli DH1 harboring pMevT and pTrc-M.ADS-MBIS2 grew similarly to the strain harboring pMevT and pTrc-M.ADS-MBIS1. However, at approximately 10 h after induction, the growth rate of the MBIS2 strain slowed considerably. It accumulated FPP to a higher concentration (700 nM) during the period when the growth rate was reduced. After the FPP concentration declined and growth resumed, the cells harboring MBIS2 reached a final density comparable to that exhibited by cells harboring MBIS1. At the final time point, only a negligible amount of FPP was detected. At 40 h after inoculation, both strains harboring pTrc-M.ADS-MBIS1 and pTrc-M.ADS-MBIS2 grew at similar rates (Fig. 6a). These strains began accumulating mevalonate in the late exponential phase (Fig. 6c). In the strain harboring pMevT and pTrc-M.ADS-MBIS2, no mevalonate accumulated until the FPP concentration decreased at around 25 h. When the activity through the lower mevalonate pathway decreased so that FPP was not being produced any longer, mevalonate accumulated to high levels. In contrast, the strain harboring pMevT and pTrc-M.ADS-MBIS1 continued to use mevalonate to make FPP and amorphadiene, and therefore, mevalonate accumulation was significantly lower in this strain.
Discussion
Through the optimization of the heterologous mevalonate pathway in E. coli, we have found that overexpression of the first two genes of the lower mevalonate pathway leads to higher amorphadiene titers (Anthony et al. 2009). Furthermore, a considerable amount of mevalonate was secreted by the cells into the culture medium (data not shown), which suggests that the upper mevalonate pathway is not limiting. These observations lead us to conclude that, despite the high titers of amorphadiene produced by our engineered strains, the optimal balance had not been obtained between the upper and lower mevalonate pathway and there was a bottleneck in the lower part of the pathway limiting product formation.
To address this flux imbalance and further optimize the lower mevalonate pathway, we took the approach of controlling the expression level of each enzyme in the lower mevalonate pathway through the use of RBSs of different strengths. We found that changing RBSs in an operon can have a dramatic impact on the levels of proteins and intermediates in the metabolic pathway and the level of the final product. Strains that accumulated high levels of FPP, a toxic intermediate at high cellular concentrations, did not grow well and produced low levels of amorphadiene. FPP accumulation could be a result of either higher flux through the lower mevalonate pathway or low production of ADS, the enzyme that transforms FPP to amorphadiene, or both. Furthermore, we found that a particular RBS can be strong when used with one gene but weak when used with another gene, making prediction of protein production difficult.
From the proteomics data, there is sufficient evidence to suggest that the high-strength RBS 5′ of mevK is stronger than the medium-strength RBS and yields higher protein levels by threefold to tenfold (Fig. 5). As mentioned earlier, in nearly all cases, strains expressing the MBIS2 operon consistently exhibited lower protein levels than strains expressing the MBIS1 operon. This was the case even for genes that had the exact same RBS between the two operons (e.g., idi and ispA). Taking into account the general downregulation of the MBIS2 operon, the actual strength of the high-strength RBS 5′ of mevK could possibly be even higher than what is observed from the proteomics analysis. However, growth inhibition and lower amorphadiene production were observed with increased MK levels in this case. Growth inhibition could be caused by a number of different factors including accumulation of toxic pathway intermediates and overconsumption of cellular resources (amino acids, nucleotides, or RNA polymerases/ribosomes). The strain expressing the construct with high-strength RBS 5′ of the first three genes of the MBIS operon (pTrc-M.ADS-MBIS2) exhibited severe growth inhibition at 24 h and lower amorphadiene levels compared to the control strain harboring pTrc-M.ADS-MBIS1. We hypothesize that the increased enzyme concentration directed more carbon flux through the mevalonate pathway and lead to increased FPP concentration, which in turn inhibited cell growth. It was, therefore, expected that an increased ADS level would lead to the restoration of growth and increased amorphadiene production.
Four RBSs of different strengths were selected by taking into account the sequence of the ADS gene. At least two of these RBSs (H.2 and H.4) resulted in higher amorphadiene titers when compared to the control strain, E. coli DH1 harboring pMevT and pTrc-M.ADS-MBIS1, cultured in TB + 1 % glycerol (Fig. 3b). Furthermore, strains harboring pTrc-H.2.ADS-MBIS1 and pTrc-H.2.ADS-MBIS2 both grew to lower optical densities than the control strain. The only difference between pTrc-M.ADS-MBIS1 and pTrc-H.2.ADS-MBIS1 constructs is the RBS 5′ of ads. The assumption that the synthetic RBS, H.2, is stronger than the original, medium-strength (M) RBS was confirmed by proteomics analysis of these two strains, where a twofold increase in ADS level was observed. The higher ADS level can deplete the cell’s FPP supply and lead to growth inhibition, explaining the lower optical densities when ADS with higher activity RBS was used.
We showed that providing additional carbon by increasing the initial glycerol concentration in the growth medium did not restore growth. Therefore, it can be concluded that limitations in growth and production were not a result of carbon source limitation caused by overconsumption of cellular resources. When the glycerol concentration was increased to 3 %, none of the strains with theoretically strong RBSs and high protein levels yielded the highest amorphadiene titers. In fact, the strain with all medium-strength RBSs (pTrc-M.ADS-MBIS1; Fig. 4) exhibited a significant improvement in amorphadiene production compared to the previously engineered operons we had tested. Also, by increasing the carbon substrate, the amorphadiene production increases by approximately fivefold compared to the same strain when 1 % glycerol was used. This demonstrates the importance of the balanced expression of all genes of the heterologous mevalonate pathway.
From the metabolite profiling studies, it appears that E. coli can accumulate a certain level of FPP before its growth was inhibited, after which cells appear to regulate or shut down growth and, thus, FPP production (Fig. 6a, b). By determining the intermediate metabolite concentration profile and comparing the FPP concentration between the two strains expressing pTrc-M.ADS-MBIS1 and pTrc-M.ADS-MBIS2, the growth profile for these two strains can be explained. When observing the metabolite levels, E. coli harboring pTrc-MBIS2 accumulated up to 1 μM of FPP, at which point the growth rate slowed and a sharp decrease in FPP concentration was observed. From these experiments, a concentration of 1 μM appears to be the maximum intracellular concentration of FPP that can be tolerated by the cell. Growth was restored when the FPP concentration dropped below 200 nM. The strain harboring pTrc-M.ADS-MBIS1 accumulated FPP to a maximum concentration of approximately 200 nM, and the concentration of FPP was maintained below 200 nM during the entire time course of the experiment (Fig. 6a). This concentration was tolerated well by the cell (Fig. 6b). Consequently, the strain grew at a normal rate and produced an amorphadiene titer of 3.6 g/L. E. coli harboring pTrc-M.ADS-MBIS2 accumulated FPP to a peak concentration of approximately 700 nM. This concentration was higher than the FPP threshold concentration of 200 nM and was not tolerated well by the cell (Fig. 6b). This strain grew normally in the early exponential phase, during which the FPP concentration was well below 200 nM. As the FPP concentration increased, the growth rate significantly slowed. Eventually, growth was restored when the FPP level diminished to below 200 nM.
In the case of the strain harboring pTrc-M.ADS-MBIS2, ADS used only a portion of the available FPP, allowing the cells to maintain a lower intracellular concentration of this metabolite compared to the strain harboring pTrc-MBIS2. This allowed the strain to grow slightly faster and reach a higher cell density compared to the strain harboring pTrc-MBIS2.
Results presented in this study suggest that choosing the appropriate RBSs for the enzymes in a metabolic pathway can substantially impact growth and product formation: we have shown that the MK level in the strain harboring the MBIS2 construct is significantly increased despite the generally lower production of all of the enzymes encoded by this operon and the strain harboring this operon accumulated more FPP. We have also successfully increased the ADS level. We hoped that, by providing more ADS, we could take advantage of this increased flux and convert the excess FPP to more amorphadiene. However, while our attempts to increase both the flux through mevalonate pathway and ADS level have yielded the desired outcome, the combined effect was not predictable.
We focused on changing the RBS strength of only the first three genes of the lower mevalonate pathway and ads. However, the optimal concentration of each of the enzymes of the lower mevalonate pathway is not known. As higher throughput cloning and screening methods become widely available, it should be possible to test many more combinations of RBS strength for all the genes in the pathway to achieve optimal protein and metabolite balance.
We have shown here that RBS strength can dramatically alter the production of enzymes in a heterologous metabolic pathway and, thus, the flux through that pathway. Because it is difficult to predict the optimal strength of RBSs for genes of interest, combinatorial assembly of the pathway genes with many RBS variants is necessary to find the best combination that will maximize pathway flux. We anticipate that these methods will become common for pathway optimization as combinatorial methods for gene assembly become more high throughput and widely available. Additionally, the data from these resulting constructs could be used to improve models of gene expression.
References
Alper H, Fischer C, Nevoigt E, Stephanopoulos G (2005) Tuning genetic control through promoter engineering. Proc Natl Acad Sci U S A 102:12678–12683. doi:10.1073/pnas.0504604102
Anthony JR, Anthony LC, Nowroozi F, Kwon G, Newman JD, Keasling JD (2009) Optimization of the mevalonate-based isoprenoid biosynthetic pathway in Escherichia coli for production of the anti-malarial drug precursor amorpha-4,11-diene. Metab Eng 11:13–19. doi:10.1016/j.ymben.2008.07.007
Barbirato F, Grivet JP, Soucaille P, Bories A (1996) 3-Hydroxypropionaldehyde, an inhibitory metabolite of glycerol fermentation to 1,3-propanediol by enterobacterial species. Appl Environ Microbiol 62:1448–1451
Carrier T, Jones KL, Keasling JD (1998) mRNA stability and plasmid copy number effects on gene expression from an inducible promoter system. Biotechnol Bioeng 59:666–672
Davis JH, Rubin AJ, Sauer RT (2011) Design, construction and characterization of a set of insulated bacterial promoters. Nucleic Acids Res 39:1131–1141. doi:10.1093/nar/gkq810
Dueber JE, Wu GC, Malmirchegini GR, Moon TS, Petzold CJ, Ullal AV, Prather KLJ, Keasling JD (2009) Synthetic protein scaffolds provide modular control over metabolic flux. Nat Biotechnol 27:753–759. doi:10.1038/nbt.1557
Fortman JL, Chhabra S, Mukhopadhyay A, Chou H, Lee TS, Steen E, Keasling JD (2008) Biofuel alternatives to ethanol: pumping the microbial well. Trends Biotechnol 26:375–381. doi:10.1016/j.tibtech.2008.03.008
Hanahan D (1983) Studies on transformation of Escherichia coli with plasmids. J Mol Biol 166:557–580
Harcum SW, Bentley WE (1999) Heat-shock and stringent responses have overlapping protease activity in Escherichia coli. Implications for heterologous protein yield. Appl Biochem Biotechnol 80:23–37
Harker M, Bramley PM (1999) Expression of prokaryotic 1-deoxy-d-xylulose-5-phosphatases in Escherichia coli increases carotenoid and ubiquinone biosynthesis. FEBS Lett 448:115–119
Hui A, Hayflick J, Dinkelspiel K, de Boer HA (1984) Mutagenesis of the three bases preceding the start codon of the beta-galactosidase mRNA and its effect on translation in Escherichia coli. EMBO J 3:623–629
Jones KL, Keasling JD (1998) Construction and characterization of F plasmid-based expression vectors. Biotechnol Bioeng 59:659–665
Jones KL, Kim SW, Keasling JD (2000) Low-copy plasmids can perform as well as or better than high-copy plasmids for metabolic engineering of bacteria. Metab Eng 2:328–338. doi:10.1006/mben.2000.0161
Kajiwara S, Fraser PD, Kondo K, Misawa N (1997) Expression of an exogenous isopentenyl diphosphate isomerase gene enhances isoprenoid biosynthesis in Escherichia coli. Biochem J 324(Pt 2):421–426
Kelly JR, Rubin AJ, Davis JH, Joseph H, Ajo-Franklin CM, Cumbers J, Czar MJ, de Mora K, Glieberman AL, Monie DD, Endy D (2009) Measuring the activity of BioBrick promoters using an in vivo reference standard. J Biol Eng 3:4. doi:10.1186/1754-1611-3-4
Khosla C, Keasling JD (2003) Metabolic engineering for drug discovery and development. Nat Rev Drug Discov 2:1019–1025. doi:10.1038/nrd1256
Kim SW, Keasling JD (2001) Metabolic engineering of the nonmevalonate isopentenyl diphosphate synthesis pathway in Escherichia coli enhances lycopene production. Biotechnol Bioeng 72:408–415
Kittleson JT, Cheung S, Anderson JC (2011) Rapid optimization of gene dosage in E. coli using DIAL strains. J Biol Eng 5:10
Kizer L, Pitera DJ, Pfleger BF, Keasling JD (2008) Application of functional genomics to pathway optimization for increased isoprenoid production. Appl Environ Microbiol 74:3229–3241. doi:10.1128/AEM.02750-07
Ma SM, Garcia DE, Redding-Johanson AM, Friedland GD, Chan R, Batth TS, Haliburton JR, Chivian D, Keasling JD, Petzold CJ, Lee TS, Chhabra SR (2011) Optimization of a heterologous mevalonate pathway through the use of variant HMG-CoA reductases. Metab Eng 13:588–597. doi:10.1016/j.ymben.2011.07.001
Martin VJJ, Pitera DJ, Withers ST, Newman JD, Keasling JD (2003) Engineering a mevalonate pathway in Escherichia coli for production of terpenoids. Nat Biotechnol 21:796–802. doi:10.1038/nbt833
Matthews PD, Wurtzel ET (2000) Metabolic engineering of carotenoid accumulation in Escherichia coli by modulation of the isoprenoid precursor pool with expression of deoxyxylulose phosphate synthase. Appl Microbiol Biotechnol 53:396–400
Nakamura CE, Whited GM (2003) Metabolic engineering for the microbial production of 1,3-propanediol. Curr Opin Biotechnol 14:454–459
Newman JD, Marshall J, Chang M, Nowroozi F, Paradise E, Pitera D, Newman KL, Keasling JD (2006) High-level production of amorpha-4,11-diene in a two-phase partitioning bioreactor of metabolically engineered Escherichia coli. Biotechnol Bioeng 95:684–691. doi:10.1002/bit.21017
Peralta-Yahya PP, Keasling JD (2010) Advanced biofuel production in microbes. Biotechnol J 5:147–162. doi:10.1002/biot.200900220
Pitera DJ, Paddon CJ, Newman JD, Keasling JD (2007) Balancing a heterologous mevalonate pathway for improved isoprenoid production in Escherichia coli. Metab Eng 9:193–207. doi:10.1016/j.ymben.2006.11.002
Redding-Johanson AM, Batth TS, Chan R, Krupa R, Szmidt HL, Adams PD, Keasling JD, Lee TS, Mukhopadhyay A, Petzold CJ (2011) Targeted proteomics for metabolic pathway optimization: application to terpene production. Metab Eng 13:194–203. doi:10.1016/j.ymben.2010.12.005
Salis HM, Mirsky EA, Voigt CA (2009) Automated design of synthetic ribosome binding sites to control protein expression. Nat Biotechnol 27:946–950. doi:10.1038/nbt.1568
Shetty RP, Endy D, Knight TF (2008) Engineering BioBrick vectors from BioBrick parts. J Biol Eng 2:5. doi:10.1186/1754-1611-2-5
Steen EJ, Kang Y, Bokinsky G, Hu Z, Schrimer A, McClure A, Del Cardayre SB, Keasling JD (2010) Microbial production of fatty-acid-derived fuels and chemicals from plant biomass. Nature 463:559–562. doi:10.1038/nature08721
Stephanopoulos G (2007) Challenges in engineering microbes for biofuels production. Science 315:801–804. doi:10.1126/science.1139612
Wessel D, Flügge UI (1984) A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal Biochem 138:141–143
Withers ST, Gottlieb SS, Lieu B, Newman JD, Keasling JD (2007) Identification of isopentenol biosynthetic genes from Bacillus subtilis by a screening method based on isoprenoid precursor toxicity. Appl Environ Microbiol 73:6277–6283. doi:10.1128/AEM.00861-07
Zhu MM, Lawman PD, Cameron DC (2002) Improving 1,3-propanediol production from glycerol in a metabolically engineered Escherichia coli by reducing accumulation of sn-glycerol-3-phosphate. Biotechnol Prog 18:694–699. doi:10.1021/bp020281
Acknowledgments
This work was supported by the Joint BioEnergy Institute (http://www.jbei.org) through a contract between Lawrence Berkeley National Laboratory and the US Department of Energy, Office of Science, Office of Biological and Environmental Research (DE-AC02-05CH11231) and the Synthetic Biology Engineering Research Center (http://www.synberc.org) through a grant from the National Science Foundation (BES-0439124). We thank Chris Anderson (Department of Bioengineering, University of California, Berkeley, CA, USA) for the gift of pBca9145.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 53.3 kb)
Rights and permissions
About this article
Cite this article
Nowroozi, F.F., Baidoo, E.E.K., Ermakov, S. et al. Metabolic pathway optimization using ribosome binding site variants and combinatorial gene assembly. Appl Microbiol Biotechnol 98, 1567–1581 (2014). https://doi.org/10.1007/s00253-013-5361-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-013-5361-4