
Abstract
High fructose corn syrup (HFCS) is an alternative of liquid sweetener to sucrose that is isomerized by commercial glucose isomerase (GI). One-step production of 55 % HFCS by thermostable GI has been drawn more and more attentions. In this study, a new hyperthermophilic GI from Thermoanaerobacter ethanolicus CCSD1 (TEGI) was identified by genome mining, and then a 1317 bp fragment encoding the TEGI was synthesized and expressed in Escherichia coli BL21(DE3). To improve the activity of TEGI, two amino acid residues, Trp139 and Val186, around the active site and substrate-binding pocket based on the structural analysis and molecular docking were selected for site-directed mutagenesis. The specific activity of mutant TEGI-W139F/V186T was 2.3-fold and the value of k cat/K m was 1.86-fold as compared to the wild type TEGI, respectively. Thermostability of mutant TEGI-W139F/V186T at 90 °C for 24 h showed 1.21-fold extension than that of wild type TEGI. During the isomerization of glucose to fructose, the yield of fructose could maintain above 55.4 % by mutant TEGI-W139F/V186T as compared to 53.8 % by wild type TEGI at 90 °C. This study paved foundation for the production of 55 % HFCS using the thermostable TEGI.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
High fructose corn syrup (HFCS), a liquid mixture of glucose and fructose, has become a major sweetener and food additive used extensively in a wide variety of processed foods and beverages ranging from candy, breads, tins, jams to drinks due to its functional and technological advantages over sucrose [6, 27]. 42 % of HFCS has been widely produced by glucose isomerase (GI). Currently, 55 % of HFCS with better function of sweetness is more popular as compared to 42 % of HFCS [33]. It has been theorized and confirmed that elevated temperature (>85 °C) is required for isomerization by GI to achieve the 55 % of HFCS due to the isomerization reaction equilibrium, which would avoid additional downstream concentration steps [40]. By far, the production of 55 % of HFCS by one step is still not realized because of lack of thermostable GI with high activity [7, 14]. Thus, it is necessary and urgent to screen a novel GI with high activity and stability at elevated temperature.
GIs (EC 5.3.1.5) catalyze the isomerization of d-glucose or d-xylose to d-fructose or d-xylulose, which have been classified into two classes (class I and class II) based on the peptide length [44]. GIs of class II have more than 30–40 amino acids compared with GIs of class I at the N-terminus of the protein [7]. Each monomer contains two domains, one is an N-terminal major domain folded as a (α/β)8-barrel which contains the catalytic pocket and metal binding sites and the other is a C-terminal minor domain folded as a large loop away from the larger domain that attaches the adjacent subunit to form a tightly bound dimer [20, 22]. Recently, some of GIs from class I have been applied into industry to produce 42 % of HFCS at 65 °C, which mainly originated from the Streptomyces murinus (SMGI), Bacillus coagulans (BCGI), Actinoplanes mussousriensis (AMGI) and Streptomyes rubiginosus (SRGI) [14, 23, 46]. However, these reported GIs showed poor activities and thermostabilities at high temperature over 80 °C. For example, SMGI performs well (750 kg fructose/kg enzyme) to produce 42 % of HFCS at 55–65 °C, but its productivity reduce to 273 kg fructose/kg enzyme and half-life was only 100 min at 80 °C, which could not meet the requirements for the 55 % of HFCS production [21]. Some hyperthermophilic GIs from Thermotoga neapolitana (TNGI) and Thermotoga maritime (TMGI) belonging to class II have also been cloned and characterized [5, 8]. The optimum temperatures for TNGI and TMGI reached 95 and 100 °C, respectively. The lifetime productivity of TNGI at 90 °C is 1169 kg fructose/kg enzyme, which is 5.6 times higher than that of SMGI [5]. In recent years, over-production of recombinant hyperthermophilic GIs has been achieved in Escherichia coli strains [1, 2, 4]. Akdağ and Çalık reported the highest production of GI reached 4364.1 U/L using the exponential feeding strategy [1]. To meet the requirement of the industry, it is imminent to exploit novel GIs of class II with better activities and stabilities at higher temperature to produce 55 % of HFCS by one-step conversion [18].
In this study, to produce 55 % of HFCS, genome mining method was used to identify the novel thermophilic GI from class II, and GI from T. ethanolicus CCSD1 (TEGI) was screened from GenBank. After codons optimization, the synthesized TEGI gene was cloned and successfully expressed in E. coli BL21(DE3). To improve the thermostability and catalytic efficiency, W139 and V186 of TEGI were selected for further mutation by site-directed mutagenesis method based on the structure modeling and molecular docking, and the mutant TEGI (TEGI-W139F/V186T) with higher activity and thermostability was obtained. Furthermore, the characteristics of this mutant were investigated in detail. The specific activity of mutant TEGI to d-glucose was significantly increased to 92.1 U/mg. Thermostability of mutant TEGI was more stable than wild type TEGI and showed 68 % residual activity after 24 h incubation at 90 °C. For pH stability, residual activity of mutant TEGI was 82.4 % after 24 h incubation at pH 6.5, higher than that of wild type TEGI. Using this mutant TEGI-W139F/V186T as the biocatalyst, the isomerization reaction could reach equilibrium within 1.5 h and fructose yield of 55.4 % was obtained. The obtained mutant TEGI-W139F/V186T has great potential in the 55 % of HFCS production.
Materials and methods
Chemicals, strains, plasmids and growth conditions
d-Glucose, d-fructose and other chemicals were of analytical grade purity and commercially available. The pET-28b(+) (Novagen, Darmstadt, Germany) and E. coli BL21(DE3) (Invitrogen, Karlsruhe, Germany) were used for cloning and expression of GIs. Recombinant E. coli was incubated at 37 °C in Luria–Bertani (LB) medium (1 % tryptone, 0.5 % yeast extract and 0.5 % NaCl) containing 50 µg/mL kanamycin (Kan).
Database mining and sequence alignment
The GI genes were obtained from the GenBank by the program of Blast X and Blast P (http://blast.ncbi.nlm.nih.gov/Blast.cgi) using amino acid sequence of TMGI as a template. The structure-based multiple sequence alignment of amino acid sequences was carried out by program ClustalX and the sequence alignment was generated using the program ESPRIPT [34, 41].
DNA manipulation, plasmid construction and sequencing
The GI gene from T. ethanolicus CCSD1 (TEGI) was codon optimized by an online codon adaptation tool JCAT (http://www.jcat.de/Start.jsp) and chemically synthesized according to its recorded sequence in NCBI (http://www.ncbi.nlm.nih.gov/), and amplified by polymerase chain reaction (PCR) using primers containing NcoI and XhoI restriction sites (Table S1 in the Supporting Information) [9]. The PCR reaction system (50 µL) consisted of 50 ng synthesized DNA, 50 µM dNTP, 0.5 µM of forward and reverse primers, 5 µL 10× pfu DNA buffer and 2 U pfu DNA polymerase. Amplification was carried out in a thermal cycler (Bio-Rad, CA, USA) under the following conditions: 5 min at 95 °C, 30 cycles of 50 s at 94 °C, 30 s at 58 °C, 1.5 min at 72 °C and 10 min at 72 °C. The PCR product was digested with NcoI and XhoI and ligated into pET-28b(+) to construct the recombinant plasmid, pET28b-TEGI, and then the pET28b-TEGI plasmid was transformed into E. coli BL21(DE3) competent cells [11]. 6× His-tag had been designed to be possessed at the C-terminal of protein for purification by affinity chromatography. Plasmid DNA was isolated using the AxyPrep Plasmid Miniprep Kit (Axygen Biotech Ltd., Hangzhou, China) according to the instructions of the manufacturer. Extraction of DNA from agarose gel was carried out with the AxyPrep DNA Gel Extraction Kit (Axygen Biotech Ltd.). The positive clones were selected and sequenced using ABI 3730XL DNA Analyzer (Sangon Biotech Co. Ltd., Shanghai, China).
Homology modeling and molecular docking
The three-dimensional (3D) structure of TEGI was constructed using Build Homology Models (MODELER) in Discovery Studio 3.0 using the crystal structure of a high homologous protein TTGI (PDB accession code: 1A0C) as template with the homology of 87 % identity to the TEGI. The catalytic tunnel size was analyzed using HotSpot Wizard (http://loschmidt.chemi.muni.cz/hotspotwizard/) to identify the residues essential for protein function, enzyme activity and substrate specificity. Autodock 4.2 was then used to study the interaction between substrate and TEGI, and to predict the spatial structure and catalytic mechanism of TEGI. His101 and Asp104 in TEGI were selected as flexible residues and all the C–OH were set rotatable. The conformation with the lowest binding energy was selected as final conformation. The visualization and structure analysis were performed with PyMOL program version 0.99 (http://www.pymol.org) [12].
Site-directed mutagenesis
The recombinant plasmid pET28b-TEGI was used as template for the site-directed mutagenesis. The synthetic mutagenesis primers were shown in Table S1 in the Supporting Information. The condition of PCR was 98 °C for 3 min, followed by 30 cycles of 98 °C for 10 s, 58 °C for 15 s and 72 °C for 7 min. The final extension at 72 °C lasted for 10 min. The restriction enzyme DpnI was added to the PCR products directly to remove the methylated template for 3 h at 37 °C. Then the products were purified and transformed into E. coli BL21(DE3) for expression. The mutant plasmid from the previous round of mutation was used as template for next round.
Expression and purification of recombinant enzymes
Recombinant E. coli transformants were cultured at 37 °C in 50 mL LB media containing 50 µg/mL kanamycin until its optical density at 600 nm (OD600) reached 0.6, and then 0.1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) was added to induce the expression of the target protein at 28 °C for 12 h. 1 g of the harvested cells was resuspended in 10 mL phosphates buffer (pH 6.5) and lysed by sonication with a Vibra-Cell VC 505 ultrasonic processor (Sonics and Materials Inc., CT, USA), and the obtained soluble supernatant by centrifugation was used as the crude enzyme for further investigation.
The supernatant containing recombinant enzymes were directly applied to the nickel-NTA superflow column (1 × 10 cm2) which had been previously equilibrated with binding buffer. Then, the column was subjected to washing with five folds column volume of the same buffer and was eluted using elution buffer (50 mM Tris–HCl, 500 mM NaCl, 500 mM imidazole, pH 7.5) with a linear gradient of 20–500 mM imidazole. The eluted fusion proteins were collected, which were used for enzyme assay and sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) analysis [29]. Total protein concentration was determined by the bicinchoninic acid (BCA) protein assay kit (Nanjing Keygen Biotechnology Co, Nanjing, China) based on the method reported by Smith et al. [36].
Enzyme assays
d-Glucose was used as substrate and the compositions of reaction mixture were as follows: 10 % d-glucose (w/v), 10 mM Mg2+, 1 mM Co2+, 50 mM phosphates buffer (pH 6.5) and an appropriate amount of the enzyme in a total volume of 10 mL. The isomerization reaction was performed at 90 °C for 10 min and followed by incubating at 0 °C for 10 min to inactivate the enzyme. d-fructose formed in the reaction mixture was measured using a SpectraMax M5 spectrophotometer (Molecular Devices LLC, CA, USA) by the cysteine–carbazole–sulfuric acid method [19]. One unit of the enzyme activity was defined as the amount of enzyme that produced 1 μmol of d-fructose per min under the assay conditions described above.
Determination of kinetic parameters
Kinetic parameters of wild type TEGI and its mutants were calculated from the initial rate of the purified enzyme using d-glucose as substrate at the concentrations ranging from 50 to 400 mM. The double-reciprocal plots of the initial reaction rate against the substrate concentrations were performed [42].
Characterization of purified recombinant enzymes
The optimum temperatures of wild type TEGI and mutant TEGI-W139F/V186T were determined by incubating the enzyme in assay mixtures at temperatures ranging from 60 to 95 °C and determining the residual enzyme activities in 50 mM phosphates buffer (pH 6.5) with 1 mM Co2+, 10 mM Mg2+, 10 % d-glucose (w/v) and 2.4 mg purified enzymes, respectively. For thermostability analysis, samples of wild type and mutant were incubated for 24 h at 90 °C. The residual activity was determined at 90 °C under the standard assay conditions. The non-heated enzyme was taken as the control (100 % of activity).
The optimum pH was determined by assaying the enzyme reaction in 50 mM potassium phosphate buffer (pH 5.0–7.0), 50 mM Tris–HCl buffer (pH 7.0–8.5) and 50 mM glycine sodium hydroxide buffer (pH 8.5–10.0) with 1 mM Co2+, 10 mM Mg2+, 10 % d-glucose (w/v) and 2.4 mg purified enzymes at 90 °C, respectively. To determine the pH stability, the purified enzymes were preincubated in phosphates buffer (pH 6.5) with the concentration of 50 mM at 0 °C for 24 h. The residual activities were determined under the standard assay conditions. The non-preincubated enzyme was taken as the control (100 % activity).
The purified enzymes were incubated overnight at 4 °C in 50 mM phosphates buffer containing 10 mM EDTA, and then dialyzed against 50 mM phosphates buffer containing 2 mM EDTA and finally dialyzed against 50 mM phosphates buffer. The effects of divalent metal ions on enzyme activities were studied by incubating the enzyme in reaction mixtures with 50 mM phosphates buffer (pH 6.5), 10 % d-glucose (w/v) and 2.4 mg dialyzed purified enzymes containing Mg2+, Co2+, Mn2+, Zn2+, Ni2+, Cu2+, Ca2+, Ba2+ and Fe2+ with a final concentration of 10 mM. The residual activity was measured under the standard assay conditions and expressed as a percentage of the activity observed in 1 mM Co2+ and 10 mM Mg2+.
Biocatalysis of d-glucose to d-fructose using mutant TEGI-W139F/V186T
After cultivation, E. coli cells harboring mutant enzyme were harvested by centrifugation at 12,000×g for 10 min at 4 °C. 10 mL of conversion system contained 2 mL crude enzyme (5.69 mg protein/mL), 1 mM Co2+, 10 mM Mg2+, 10 % glucose (w/v) and 50 mM phosphates buffer (pH 6.5). The reaction was carried out on a water bath shaker at 90 °C for 5 h. Samples were taken periodically for HPLC analysis. The bioconversion catalyzed by wild type TEGI was taken as the control.
HPLC analysis
Detection of concentrations of d-glucose and d-fructose was performed with a Waters HPLC system (Waters, Milford, USA) fitted with a Waters 2414 refractive index detector (Waters) using a Hi-Plex Ca column (300 × 7.7 mm) (Agilent Technologies, Waldbronn, Germany) with a guard column. Samples and standards were eluted at 60 °C with ultrapure water (0.5 mL/min). For calculation of the reaction products, d-glucose and d-fructose standards were included in the run. The yield represents the ratio between the formed d-fructose and the initial d-glucose.
Statistical analysis
If not specifically noted, all experiments in this study were performed in triplicate. Analysis of variance (ANOVA) was carried out using the SAS program version 8.1 (SAS Institute Inc., Cary, NC). Least significant differences (LSD) were computed at p < 0.05.
Sequence submission
The genes encoding the wild type TEGI and mutant TEGI-W139F/V186T were respectively deposited in the GenBank database under the accession numbers of KP844881 and KP844882.
Results
Screening thermophilic GIs from GenBank database
To improve the economic feasibility and competitiveness for industrial production of 55 % HFCS, the GI with high activity and thermostability toward glucose is desirable. By comparing the enzymatic characteristics of GIs from Class I and Class II that have been previously reported, which was found that the thermostability of class I of GIs was usually lower than that of class II although some GIs have better productivities [18]. It is reported that the activity of immobilized SMGI from class I was satisfied at moderate temperature (150 U/g, 45 % conversion at 65 °C), but its thermostability still could not meet the industrial requirement [21]. By contrast, the optima temperature of GIs from class II had an advantage over GIs from class I because of their additional 30–40 amino acids at the N-terminus, which makes them more flexible under high temperature. TNGI and TMGI, the representatives of class II of GIs showed their optima temperature at 95 and 100 °C, and half-lives at 80 °C were 1.2 and 11.6 h, respectively [5]. Therefore, in this work, the genome mining approach was used to screen thermophilic GIs from GenBank using TMGI as template [35]. The conserved motif FSVAFWHTF of TMGI was used as a probe, and five highly homologous GI sequences were obtained (Fig. S1 in the Supporting Information). Via deletion of the highly homologous hits that are from the same kind of bacteria with >90 % homology and the published sequences, GIs from Thermotoga petrophila RKU-1 (TPGI, YP_001244714.1) and Thermoanaerobacter ethanolicus CCSD1 (TEGI, WP_003868244.1) were selected. In the amino acid sequence of TEGI, His and Pro exist at a higher frequency than those in TPGI, which provide more rigidity to the polypeptide backbone and enhanced thermal stability of the enzyme. Based on the analysis above, the TEGI was selected for further research.
Cloning and expression of recombinant TEGIs
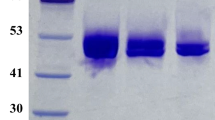
To obtain the overexpression of TEGI, the codons of TEGI gene were optimized and replaced by those with higher frequencies according to the codon usage bias of E. coli, which caused the CAI (Codon Adaptation Index) increased from 0.17 to 1.00, and GC content increased from 36.1 to 48.9 % in TEGI gene sequence. Subsequently, the optimized TEGI gene was synthesized and subcloned to the T-vector. To construct expression vector, the gene encoding TEGI was successfully amplified by PCR and cloned into pET28b(+) between the restriction sites of NcoI and XhoI. 6× His-tag was designed to combine with C-terminal of the target protein, which makes the expressed recombinant TEGI could be purified by Ni2+ affinity chromatography in one step. The recombinant plasmid was identified and confirmed by the restriction endonuclease analysis and DNA sequencing. The positive clone was used for cultivation and induction, the recombinant TEGI was successfully expressed in E. coli after 12 h induction. The SDS-PAGE analysis of intracellular soluble protein indicated that the molecular weight (MW) of TEGI approximated 50 kDa (Fig. 1a). According to the densitometric scanning result, TEGI had a good soluble expression. The activity assay showed that specific activity of TEGI reached 39.9 U/mg and 53.8 % of conversion could be obtained using glucose as substrate.

a SDS-PAGE analysis of expression products and purified enzymes from TEGI. Lane M standard proteins marker of different molecular weights. Lane 1 E. coli BL21(DE3), lane 2 E. coli BL21(DE3)/pET28b(+), lane 3 TEGI without induced by IPTG, 4 TEGI induced by IPTG, lane 5 precipitation of cell extract, lane 6 supernatant of the cell extract, lane 7 TEGI purified by the metal affinity chromatography. b SDS-Page analysis of purified enzymes from wild type TEGI and mutant TEGI-W139F/V186T. Lane 1 wild type TEGI purified by the metal affinity chromatography. Lane 2 mutant TEGI-W139F/V186T purified by the metal affinity chromatography
Site-directed mutagenesis of TEGI based on molecular simulation
To improve the activity and thermostability of the recombinant TEGI, the molecular modification was carried out in this study. The 3D structure of TEGI was constructed with a GI crystal structure from Thermoanaerobacterium thermosulfurigenes (TTGI, PDB: 1A0C) as template by SWISS-MODEL homology modeling and the sequence homology is 87 % between TTGI and TEGI. The quality of the model obtained was assessed by procheck. 93.1 % residues were located in the most favorable regions, 6.7 % in additional allowed regions and 0.3 % in generously allowed regions which indicate that the model is credible. The structure of TEGI contains a TIM barrel consisting of eight α-helices and eight parallel β-strands which is accord with the published structures (Fig. 2a). To investigate the conserved residues around the catalytic pocket, four GI structures (PDB: 1A0C, 4HHL, 4J4K and 4LNC) were used to do the multiple structure alignment by ESPRIPT and the active pocket was further analyzed by Autodock 4.2. Through the multiple structure alignment, Trp and Val were identified as conserved residues at the bottom of the active pocket of TEGI structures (Fig. 3). And it was reported that Trp replaced by another small residue containing a phenyl ring which is located in a β-sheet of TIM barrel could reduce the K m value previously via enlarging the active pocket [26]. Moreover, there is a hydrophobic core in the TIM barrel which would also contribute to the stability of enzyme (Fig. 2b). So Trp139 was replaced by Phe which is a hydrophobic amino acid with the aim to enlarge the active pocket and strengthen the intramolecular hydrophobic interaction to improve the activity and thermal stability (Fig. 2c, d). Val contains a small hydrophobic side chain which is not beneficial for the binding of the hydrophilic d-glucose. So Thr or Ser was selected to replace the Val186 containing a hydrophilic side chain but similar steric hindrance. After first round of site-directed mutagenesis, three mutants TEGI-W139F, TEGI-V186T and TEGI-V186S were obtained. Among them, TEGI-W139F and TEGI-V186T showed improvement in activities compared to the wild type TEGI (Fig. 4). To further improve the activity of TEGI, the second round of site-directed mutagenesis to overlap the two positive mutations Trp139Phe and Val186Thr was carried out, and then the double point mutant TEGI-W139F/V186T was obtained. The activities and thermostabilities of these mutants obtained in these two rounds of site-directed mutagenesis were determined and the double point mutant TEGI-W139F/V186T showed the highest activity of 92.1 U/mg (Fig. 4) and thermostability of 68 % residual activity after 24 h at 90 °C (Table 1). In addition, the K m value was reduced from 421.0 mM (TEGI) to 245.2 mM (TEGI-W139F/V186T) and the catalytic efficiency (k cat/K m) of TEGI-W139F/V186T was improved 1.86-fold as compared to wild type TEGI (Table 2). In consideration of the preferable characteristics, the mutant TEGI-W139F/V186T was selected for further investigation.

The overall structure of TEGI constructed by homology modeling. a The TIM barrel, the β strand were colored by purple, b the hydrophobic core composed by the hydrophobic residue on β strand of TIM barrel, c the position of Trp139 on the β strand, d the position of Phe139 on the β strand and the interaction with the neighbor hydrophobic residues

The analysis of active pocket of glucose isomerase in Protein Data Bank. a 1A0C, b 4HHL, c 4J4K, d 4LNC

Specific activities of the purified TEGI and its mutants under the standard reaction condition (90 °C, pH 6.5)
Purification and biochemical characterization of wild type TEGI and mutant TEGI-W139F/V186T
After completion of the cultivation and induction according to the procedure mentioned in the “Materials and methods”, the E. coli cell biomass reached 1.95 g (DCW)/L and activity reached 1718 U/L for wild type TEGI, while the E. coli cell biomass reached 2.44 g (DCW)/L and activity reached 4535 U/L for mutant TEGI-W139F/V186T. Both wild type TEGI and mutant TEGI-W139F/V186T were purified by Ni2+ affinity chromatography according to the procedures described in “Materials and methods”. Table S2 in the Supporting Information summarized the results of the purification efficiency for wild type TEGI and mutant TEGI-W139F/V186T. The elutes containing wild type TEGI and mutant TEGI-W139F/V186T were analyzed using SDS-PAGE (Fig. 1b).
The effects of temperature on the activities and stabilities of wild type TEGI and mutant TEGI-W139F/V186T were investigated. As shown in Fig. 5a, the wild type TEGI and mutant TEGI-W139F/V186T showed the maximum activities both at 90 °C. The activity of wild type TEGI increased slowly from 60 to 90 °C; however, the activity of mutant TEGI-W139F/V186T increased gradually from 60 to 80 °C but increased sharply from 80 to 90 °C. The activity of both wild type TEGI and mutant TEGI-W139F/V186T began to decrease when the temperature increased above 90 °C. Moreover, the specific activity of mutant TEGI-W139F/V186T (92.1 U/mg) was improved 231 % compared to that of wild type TEGI (39.9 U/mg). The study of thermostability showed that wild type TEGI maintained approximately 56 % of its activity after incubation at 90 °C for 24 h, while the residual activity of mutant TEGI-W139F/V186T could retain more than 68 % at the same conditions, which is 1.21-fold increase compared with wild type TEGI (Fig. 5b). The results confirmed that mutant TEGI-W139F/V186T was more thermostable than the wild type TEGI and other reported GIs (Table 1).

Effects of temperature and pH on the activities and stabilities of wild type and mutant TEGI-W139F/V186T. a Effect of temperature on activities. The reaction was performed at 60–95 °C as the indicated temperatures. b Effect of temperature on stabilities. Wild type TEGI and mutant TEGI-W139F/V186T were incubated for 24 h at 90 °C. The residual activity was measured under standard assay condition. The original activity without incubation was taken as 100 %. c Effect of pH on activities. Wild type TEGI and mutant TEGI-W139F/V186T activities were measured at the indicated pH values in 50 mM buffer: phosphate buffer (pH 5.0–7.0), Tris–HCl (pH 7.0–8.5), Gly-NaOH (pH 8.5–10.0). d Effect of pH on enzyme stability. The purified enzymes were preincubated in phosphate buffer of pH 6.5 with the concentration of 50 mM at 0 °C for 24 h. The residual activity was measured under standard assay condition. The original activity at individual pH was taken as 100 %. All the reactions under various conditions were started by the addition of the enzyme solution that was preincubated with 10 mM Mg2+, 1 mM Co2+ at 90 °C and pH 6.5 for 3 min
To investigate the effect of pH on GIs activity, the reaction was performed in buffers monitored at different pH from 5.0 to 10.0. As shown in Fig. 5c, both wild type TEGI and mutant TEGI-W139F/V186T showed almost the same trends of activities under different pH conditions. The maximum specific activity of the evolved and wild type TEGI was observed at the slightly acidic pH 6.5. Compared to wild type TEGI, the activity of mutant TEGI-W139F/V186T has increased significantly ranging from pH 5.0 to 10.0. Moreover, the specific activity of mutant TEGI-W139F/V186T was about 2.3-fold higher as compared to the wild type TEGI under the optimal temperature and pH. The investigation of pH stability showed that mutant TEGI-W139F/V186T was more stable than wild type TEGI, and 82.4 % of its original activity was maintained when exposed at pH 6.5 for 24 h. However, the residual activity of wild type TEGI was 80.9 % at the same conditions (Fig. 5d). The results of optimal pH and pH stability showed that there is no apparent difference for TEGI before and after genetically engineered.
The effects of various divalent metal ions on the activities of wild type TEGI and mutant TEGI-W139F/V186T are shown in Table 3 in the Supporting Information. Both Co2+ and Mg2+ had stimulating effects on the activities of wild type TEGI and mutant TEGI-W139F/V186T, indicating that the GI activity was Co2+ and Mg2+ dependent. Mn2+ (10 mM) also had slight activation on enzyme activity. These results were identical to those previously reported [15, 28]. The metal ions Ba2+, Ni2+ and Fe2+ with concentration of 10 mM had slight inhibitions on the activities of wild type TEGI and mutant TEGI-W139F/V186T. The stronger inhibitory effects were observed in the presence of Cu2+, Zn2+ and Ca2+, but the tolerance ability of mutant TEGI-W139F/V186T to Ca2+ had been slightly improved compared with wild type TEGI.
The kinetic parameters of the wild type TEGI and mutant TEGI-W139F/V186T with glucose concentrations from 50 to 400 mM were determined according to the Michaelis–Menten model and Lineweaver–Burk plots (Fig. S2 in the Supporting Information). As depicted in Table 2, Michaelis constants (K m) of the wild type TEGI and mutant TEGI-W139F/V186T were 421 and 245 mM. The analysis of the K m values indicated that the mutant TEGI-W139F/V186T showed a higher affinity toward d-glucose than wild type TEGI and lead to a 1.86-fold improvement in catalytic efficiency (k cat/K m). The maximum reaction rates (V max) of wild type TEGI and mutant TEGI-W139F/V186T were 27 and 29.2 mM/(min mg) at 90 °C, and the turnover numbers (k cat) were 22.6 and 24.5 s−1, respectively.
Biocatalysis of d-glucose to d-fructose using mutant TEGI-W139F/V186T
To verify the potential application of mutant TEGI-W139F/V186T in enzymatic production of 55 % HFCS, the isomerization was performed by mutant TEGI-W139F/V186T, and the wild type TEGI was used as the control. The reaction mixture with a final volume of 10 mL in a 50-mL flask proceeded for 5 h at 90 °C with shaking (150 rpm). The time courses of the bioconversion of d-glucose to produce HFCS using the wild type TEGI and mutant TEGI-W139F/V186T are shown in Fig. 6. The molar yield of d-fructose reached 45.9 % within 30 min and increased sharply to reach 54.8 % after 1.5 h using mutant TEGI-W139F/V186T as catalyst, and then the isomerization tended to be steady and finally reached maximum 55.4 % when the reaction extended to 5 h. In contrast, d-fructose was produced with a molar yield of 34.9 % within 30 min and increased rapidly within 3 h to reach 52.9 %, and the isomerization gradually maintained equilibrium after 3 h transformation by wild type TEGI and continuously retained greater than 53 % upon prolonged incubation. Mutant TEGI-W139F/V186T with short equilibrium time attributed to the higher affinity and catalytic efficiency toward d-glucose, and an enhancement of thermostability which was preferable to the commercial GIs reported in the literatures [21, 30]. These results suggested that the mutant TEGI-W139F/V186T had a higher catalytic efficiency which was a potential candidate in the one-step production of 55 % HFCS.

Progress curves of the isomerization of glucose in 10 ml reaction mixture with 50 mM phosphate buffer (pH 6.5), Co2+ (1 mM) and Mg2+ (10 mM) using wild type TEGI and mutant TEGI-W139F/V186T as biocatalyst, respectively
Discussion
Glucose isomerase (GI) is one of the most important industrial enzymes, which is widely used in the production of HFCS, a sweetener for beverages and foodstuffs. The commercial available GI belonging to the class I has been used for manufacturing the 42 % HFCS in a mild temperature, but it is very difficult to make the conversion reach 55 % in one step by currently available GIs [7]. So far, 55 % of HFCS has to be produced by additional downstream processing steps [14]. However, it is confirmed that some GIs of class II from thermophilic bacterium had good thermostability. Therefore, according to the thermodynamic equilibrium of isomerization, the availability of thermostable and thermoactive GIs of class II for 55 % of HFCS production raises the possibility that higher temperature could be used to improve the potential yield of fructose [40]. Currently, screening hyperthermophilic GIs has been a hot issue in production of 55 % HFCS.
A genome mining approach, substitute for the microorganisms screening from soil, following gene cloning was adopted in this study to search for robust GI capable of producing HFCS with high thermostability and catalytic efficiency. Screening GIs containing two advantages of class I and class II can be achieved through this method, which is difficult and time-consuming in conventional way [39]. This promising method for quick discovery of novel biocatalysts had been emerged and applied in recent years which made a great progress in nitrile biocatalysis [17]. Kaplan et al. [24] using genome method found the nitrilase from fungal and made it express successfully in E. coli. As a point of reference, in this study, the gene encoding T. ethanolicus CCSD1 GI with a good activity and thermostability was obtained by the strategy of genome mining method and was successfully expressed in E. coli.
The wild type TEGI has showed great potentiality as biocatalyst candidate for its high conversion (53.8 %) under the high temperature (90 °C). However, to fulfill the requirements of the industrial production of 55 % HFCS, the catalytic activity of the recombinant GI needs to be further improved. The site-directed mutagenesis technique was used to improve the thermostability and activity of TEGI toward d-glucose [45]. The key to the success of site-directed mutagenesis is the analysis of the structure and selection of the mutation site [18]. The model structure of TEGI showed a typical (α/β)8 barrel fold, an additional about 31 amino acids N-terminus whose specific function was not clear and an active center located at the C-terminus of the β-barrel [10]. The active center was two metal binding sites, M1 (the structural metal site) bounds Glu232, Glu268, Asp296, Asp339, and two water molecules, and M2 (the catalytic metal site) bounds Glu268 (shared with M1), His271, Asp307, Asp309, and the catalytic water [25]. Studies on the 3D structure and catalytic mechanism of GI revealed that the catalytic residues including His101, Lys341, Asp339, Asp309 and Lys234 are highly conserved [16, 25]. In particular, His101 was involved in the sugar ring opening after proton acceptance from the O1 atom of the sugar hydroxyl and transfer it to O5 of the ring [3]. Our strategy to improve catalytic efficiency of TEGI was to expand the substrate-binding pocket by introducing small, hydrophobic amino acids or to increase its binding affinity toward the hydrophilic d-glucose by introducing hydrophilic amino acids into TEGI at the positions around the active site [38]. In addition, enhancing the rigidity of protein or stabilizing its structure by introduce hydrogen bond could improve the thermostability of enzyme [18].
As discussed above, Trp139 and Val186 located in the proximity of the catalytic site His101, which were highly conserved, were selected as mutation points (Fig. 7). Trp139 was substituted by a smaller residue Phe (Fig. 7a, b) resulting in an extension of the binding pocket, which decreased the steric hindrance for binding of the substrate with the enzyme. The O5 ring oxygen was close to the position 139, it could be speculated that mutant Phe is more spacious making the ring opening easier, so as to increase the catalytic efficiency [31, 38]. And also, the replacement of Phe increased the intramolecular hydrophobic interaction which improved the thermostability (Table 1). The effect of replacing a hydrophilic Thr in site of Val186 stabilized the transition state by producing hydrogen bond with C6-OH group of d-glucose instead of hydrophilic interaction according to the result of Val186 replaced by Ser and then improved the catalytic efficiency for d-glucose [37]. These two mutant sites were overlapped to generate a double mutant TEGI-W139F/V186T which showed an increased affinity of the enzyme for its substrate d-glucose, as a result of decrease in K m value than single point mutant. And a shorter distance of 3.0 Å between O5 ring oxygen and imidazole nitrogen was observed by molecular docking (Fig. 7). The spacious volume of the substrate-binding pocket could decrease a steric hindrance when glucose ring opening and isomerizing, so the specific activity and k cat/K m were dramatically improved compared to wild type TEGI.

Comparison of the conformation of the wild type TEGI (a) and mutant TEGI-W139F/V186T (b). Amino acid backbones were represented by ribbon cartoon model. The active sites, His 101 and Asp 104, Trp139 and Val186 (for wild type TEGI, a), and Phe139 and Thr186 (for mutant TEGI-W139F/V186T, b) were represented by sticks models. In mutant TEGI-W139F/V186T, a new hydrogen bond (green dashed line) between Thr186 and C6-OH of d-glucose was formed to stabilize the transitional conformation. Replacement of Trp139 with a smaller residue Phe enlarged the binding pocket. The distance between O5 ring oxygen and imidazole nitrogen was shorter (color figure online)
Furthermore, the mutant TEGI-W139F/V186T biocatalyst with the best thermostability displayed a higher conversion that made the reaction reach equilibrium within 1.5 h at 90 °C, the yield of fructose reached 55.4 %, which was superior to the previously reported GIs at similar reaction temperatures. Meanwhile, the higher catalytic efficiency by this mutant will not only economically shorten equilibrium time but also reduce the by-product. All these make mutant TEGI-W139F/V186T a promising biocatalyst utilized in 55 % of HFCS production [7, 18]. Obviously, to produce 55 % HFCS at an industrial scale, a proper immobilization technique may be a powerful tool to improve enzyme properties, and the catalytic conditions need to be optimized, which is undergoing in our lab.
In conclusion, in this study a new GI gene from T. ethanolicus CCSD1 was screened by genome mining and heterologously overexpressed in E. coli. To improve the activity and conversion of d-glucose, TEGI had been engineered by site-directed mutagenesis, and a mutant, TEGI-W139F/V186T with high catalytic activity of 92.1 U/mg, was successfully obtained. The value of k cat/K m and thermostability of this mutant at 90 °C for 24 h was 1.86-fold and 1.21-fold than that of wild type TEGI. Structure analysis showed that the mutant TEGI-W139F/V186T had a more spacious substrate-binding pocket than the wild type TEGI and a more steady conformation between substrate and active site, which improved the catalytic activity. In addition, mutant TEGI-W139F/V186T was applied in biotransformation of glucose with conversion reaching 55.4 % and shortened the reaction equilibrium time within 1.5 h. This study indicated that mutant TEGI-W139F/V186T will be an attractive and competitive candidate for one-step production of 55 % HFCS.
References
Akdağ B, Çalık P (2015) Recombinant protein production by sucrose-utilizing Escherichia coli W: untreated beet molasses-based feeding strategy development. J Chem Technol Biotechnol 90(6):1070–1076
Angardi V, Çalık P (2013) Beet molasses based exponential feeding strategy for thermostable glucose isomerase production by recombinant Escherichia coli BL21 (DE3). J Chem Technol Biotechnol 88(5):845–852
Asboth B, Naray Szabo G (2000) Mechanism of action of D-xylose isomerase. Curr Protein Pept Sci 1(3):237–254
Ata Ö, Boy E, Güneş H, Çalık P (2015) Codon optimization of xylA gene for recombinant glucose isomerase production in Pichia pastoris and fed-batch feeding strategies to fine-tune bioreactor performance. Bioprocess Biosyst Eng 38(5):889–903
Bandlish RK, Hess JM, Epting KL, Vieille C, Kelly RM (2002) Glucose-to-fructose conversion at high temperatures with xylose (glucose) isomerases from Streptomyces murinus and two hyperthermophilic Thermotoga species. Biotechnol Bioeng 80(2):185–194
Barclay T, Markovic MG, Cooper P, Petrovsky N (2012) The chemistry and sources of fructose and their effect on its utility and health implications. J Excip Food Chem 3(2):67–82
Bhosale SH, Rao MB, Deshpande VV (1996) Molecular and industrial aspects of glucose isomerase. Microbiol Rev 60(2):280–300
Brown SH, Sjøholm C, Kelly RM (1993) Purification and characterization of a highly thermostable glucose isomerase produced by the extremely thermophilic Eubacterium, Thermotoga maritima. Biotechnol Bioeng 41(9):878–886
Caruthers MH (1985) Gene synthesis machines: DNA chemistry and its uses. Science 230(4723):281–285
Chang C, Park BC, Lee DS, Suh SW (1999) Crystal structures of thermostable xylose isomerases from Thermus caldophilus and Thermus thermophilus: possible structural determinants of thermostability. J Mol Biol 288(4):623–634
Chung C, Niemela SL, Miller RH (1989) One-step preparation of competent Escherichia coli: transformation and storage of bacterial cells in the same solution. Proc Natl Acad Sci 86(7):2172–2175
DeLano WL (2002) The PyMOL user’s manual. DeLano Scientific, San Carlos 452
Deng H, Chen S, Wu D, Chen J, Wu J (2014) Heterologous expression and biochemical characterization of glucose isomerase from Thermobifida fusca. Bioprocess Biosyst Eng 37(6):1211–1219
DiCosimo R, McAuliffe J, Poulose AJ, Bohlmann G (2013) Industrial use of immobilized enzymes. Chem Soc Rev 42(15):6437–6474
Epting KL, Vieille C, Zeikus JG, Kelly RM (2005) Influence of divalent cations on the structural thermostability and thermal inactivation kinetics of class II xylose isomerases. FEBS J 272(6):1454–1464
Fuxreiter M, Böcskei Z, Szeibert A, Szabó E, Dallmann G, Náray-Szabó G, Asbóth B (1997) Role of electrostatics at the catalytic metal binding site in xylose isomerase action: Ca2+-inhibition and metal competence in the double mutant D254E/D256E. Proteins: Struct Funct Bioinf 28(2):183–193
Gong JS, Lu ZM, Li H, Zhou ZM, Shi JS, Xu ZH (2013) Metagenomic technology and genome mining: emerging areas for exploring novel nitrilases. Appl Microbiol Biotechnol 97(15):6603–6611
Hartley BS, Hanlon N, Jackson RJ, Rangarajan M (2000) Glucose isomerase: insights into protein engineering for increased thermostability. Biochimica et Biophysica Acta (BBA)-Protein Struct Mol Enzymol 1543(2):294–335
Hlima HB, Bejar S, Riguet J, Haser R, Aghajari N (2012) Engineered glucose isomerase from Streptomyces sp. SK is resistant to Ca2+ inhibition and Co2+ independent. J Ind Microbiol Biotechnol 39(4):537–546
Hlima HB, Bejar S, Riguet J, Haser R, Aghajari N (2013) Identification of critical residues for the activity and thermostability of Streptomyces sp. SK glucose isomerase. Appl Microbiol Biotechnol 97(22):9715–9726
Jørgensen O, Karlsen L, Nielsen N, Pedersen S, Rugh S (1988) A new immobilized glucose isomerase with high productivity produced by a strain of Streptomyces murinus. Starch-Stärke 40(8):307–313
Jenkins J, Janin J, Rey F, Chiadmi M, Van Tilbeurgh H, Lasters I, De Maeyer M, Ven Belle D, Wodak SJ (1992) Protein engineering of xylose (glucose) isomerase from Actinoplanes missouriensis. 1. Crystallography and site-directed mutagenesis of metal binding sites. Biochemistry 31(24):5449–5458
Jensen VJ, Rugh S (1987) Industrial-scale production and application of immobilized glucose isomerase. Methods Enzymol 136:356–370
Kaplan O, Bezouška K, Malandra A, Veselá AB, Petříčková A, Felsberg J, Rinágelová A, Křen V, Martínková L (2011) Genome mining for the discovery of new nitrilases in filamentous fungi. Biotechnol Lett 33(2):309–312
Kovalevsky AY, Hanson L, Fisher SZ, Mustyakimov M, Mason SA, Trevor Forsyth V, Blakeley MP, Keen DA, Wagner T, Carrell H (2010) Metal ion roles and the movement of hydrogen during reaction catalyzed by D-xylose isomerase: a joint X-ray and neutron diffraction study. Structure 18(6):688–699
Li XJ, Zheng RC, Ma HY, Huang JF, Zheng YG (2014) Key residues responsible for enhancement of catalytic efficiency of Thermomyces lanuginosus lipase Lip revealed by complementary protein engineering strategy. J Biotechnol 188:29–35
Lima DM, Fernandes P, Nascimento DS, Ribeiro R, De Assis SA (2011) Fructose syrup: a biotechnology asset. Food Technol Biotechnol 49(4):424–434
Liu SY, Wiegel J, Gherardini FC (1996) Purification and cloning of a thermostable xylose (glucose) isomerase with an acidic pH optimum from Thermoanaerobacterium strain JW/SL-YS 489. J Bacteriol 178(20):5938–5945
Liu ZQ, Zhang XH, Xue YP, Xu M, Zheng YG (2014) Improvement of Alcaligenes faecalis nitrilase by gene site saturation mutagenesis and its application in stereospecific biosynthesis of (R)-(−)-mandelic acid. J Agric Food Chem 62(20):4685–4694
Luiten R, Quax W, Schuurhuizen P, Mrabet N (1990) Novel glucose isomerase enzymes and their use. Patent EP0351029 (A1)
Meng M, Lee C, Bagdasarian M, Zeikus JG (1991) Switching substrate preference of thermophilic xylose isomerase from D-xylose to d-glucose by redesigning the substrate binding pocket. Proc Natl Acad Sci 88(9):4015–4019
Mu W, Wang X, Xue Q, Jiang B, Zhang T, Miao M (2012) Characterization of a thermostable glucose isomerase with an acidic pH optimum from Acidothermus cellulolyticus. Food Res Int 47(2):364–367
Parker K, Salas M, Nwosu VC (2010) High fructose corn syrup: production, uses and public health concerns. Biotechnol Mol Biol Rev 5(5):71–78
Robert X, Gouet P (2014) Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res 42(W1):W320–W324
Seffernick JL, Samanta SK, Louie TM, Wackett LP, Subramanian M (2009) Investigative mining of sequence data for novel enzymes: a case study with nitrilases. J Biotechnol 143(1):17–26
Smith P, Krohn RI, Hermanson G, Mallia A, Gartner F, Provenzano M, Fujimoto E, Goeke N, Olson B, Klenk D (1985) Measurement of protein using bicinchoninic acid. Anal Biochem 150(1):76–85
Sriprapundh D, Vieille C, Zeikus JG (2003) Directed evolution of Thermotoga neapolitana xylose isomerase: high activity on glucose at low temperature and low pH. Protein Eng 16(9):683–690
Sriprapundh D, Vieille C, Zeikus JG (2000) Molecular determinants of xylose isomerase thermal stability and activity: analysis of thermozymes by site-directed mutagenesis. Protein Eng 13(4):259–265
Sukumar M, Jeyaseelan A, Sivasankaran T, Mohanraj P, Mani P, Sudhakar G, Arumugam V, Bakthavachalu S, Ganeshan A, Susee M (2013) Production and partial characterization of extracellular glucose isomerase using Thermophilic Bacillus sp. isolated from agricultural land. Biocatal Agric Biotechnol 2(1):45–49
Tewari YB, Goldberg RN (1985) Thermodynamics of the conversion of aqueous glucose to fructose. Appl Biochem Biotechnol 11(1):17–24
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25(24):4876–4882
Tseng S, Hsu JP (1990) A comparison of the parameter estimating procedures for the Michaelis–Menten model. J Theor Biol 145(4):457–464
Wang J, Jin C, Fu R, Shen D (2006) Glucose isomerase mutants, DNA thereof and use thereof. US Patent 7704719 B2
Xu H, Shen D, Wu XQ, Liu ZW, Yang QH (2014) Characterization of a mutant glucose isomerase from Thermoanaerobacterium saccharolyticum. J Ind Microbiol Biotechnol 41(10):1581–1589
Zheng H, Liu Y, Sun M, Han Y, Wang J, Sun J, Lu F (2014) Improvement of alkali stability and thermostability of Paenibacillus campinasensis Family-11 xylanase by directed evolution and site-directed mutagenesis. J Ind Microbiol Biotechnol 41(1):153–162
Zittan L, Poulsen P, Hemmingsen SH (1975) Sweetzyme—a new immobilized glucose isomerase. Starch-Stärke 27(7):236–241
Acknowledgments
This project was financially supported by the National Natural Science Foundation of China (No. 31401527) and the Key Scientific and Technology Programs of Zhejiang Province (No. 2013C02027).
Conflict of interest
The authors declare that they have no financial or non-financial competing interests in the publication of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Liu, ZQ., Zheng, W., Huang, JF. et al. Improvement and characterization of a hyperthermophilic glucose isomerase from Thermoanaerobacter ethanolicus and its application in production of high fructose corn syrup. J Ind Microbiol Biotechnol 42, 1091–1103 (2015). https://doi.org/10.1007/s10295-015-1639-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-015-1639-0