
Abstract
A series of site-directed mutant glucose isomerase at tryptophan 139 from Thermoanaerobacterium saccharolyticum strain B6A were purified to gel electrophoretic homogeneity, and the biochemical properties were determined. W139F mutation is the most efficient mutant derivative with a tenfold increase in its catalytic efficiency toward glucose compared with the native GI. With a maximal activity at 80 °C of 59.58 U/mg on glucose, this mutant derivative is the most active type ever reported. The enzyme activity was maximal at 90 °C and like other glucose isomerase, this mutant enzyme required Co2+ or Mg2+ for enzyme activity and thermal stability (stable for 20 h at 80 °C in the absence of substrate). Its optimum pH was around 7.0, and it had 86 % of its maximum activity at pH 6.0 incubated for 12 h at 60 °C. This enzyme was determined as thermostable and weak-acid stable. These findings indicated that the mutant GI W139F from T. saccharolyticum strain B6A is appropriate for use as a potential candidate for high-fructose corn syrup producing enzyme.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
d-Glucose isomerase (GI, EC 5.3.1.5), which is also referred to as xylose isomerase [15], is an intracellular enzyme which can convert d-glucose to d-fructose. This enzyme has been used commercially because of its capacity to produce high-fructose corn syrup (HFCS) [2]. Owing to the industrial significance of the enzyme, GIs from various microorganisms have been studied, and the corresponding genes were cloned and sequenced [1, 5, 11, 17–19]. These enzymes are generally thermostable and require metal ions for their activity and stability [16, 21].
In the industrial process for HFCS production, high temperature is helpful to increase the isomerizaiton reaction rate and allow the glucose–fructose equilibrium shift toward fructose [3]. On the other hand, industrial production of HFCS requires a slightly acidic pH range to prevent sugar browning and the formation of by-products [4]. However, the majority of commercially available GIs lost most of their catalytic activities under acidic conditions and the pH optima for enzyme activity are in the range 7.5–9.0. Therefore, thermostable glucose isomerases with slightly acidic pH optima have better industrial application value. For these reasons, many thermostable and weak-acid stable GIs were isolated and identified from various microorganisms [8, 21], or obtained by molecular modification using modern genetic and protein engineering [9, 23]. Furthermore, the relationship between structure and function of GIs, especially the mechanisms of thermostability and weak-acid stability, was widely studied [6, 7].
Therrnoanaerobacteriurn saccharolyticum strain B6A (formerly Thermoanaerobacter strain B6A) is a thermophilic anaerobic bacterium capable of actively degrading xylan [13]. These thermophilic microorganisms produce intrinsically thermostable GI, which have evolved and adapted to the extreme environment of their natural habitats. The organism’s genomic sequence was sequenced and deposited as GenBank accession number L09699 [18]. The GI produced by T. saccharolyticum strain B6A has been purified and its physicochemical and catalytic properties have been determined [17]. The enzyme activity was maximal at 80 °C and the GI displayed apparent pH optima at pH 7.0–7.5.
The importance of tryptophan at position 139 to thermophilic glucose isomerase from Clostridium thenmosulfurogenes has been investigated [20]. The research indicated that the mutation of tryptophan 139 enhanced the catalytic efficiency of the enzyme toward glucose greatly, which makes the high-efficiency producing HFCS by GI become possible. The result also provides evidence that alteration of wide-type GI to preferring GI for industrial application can be achieved using genetic and protein engineering. In the present work, site-saturation mutagenesis was used to generate a series of site-directed mutant glucose isomerase at tryptophan 139 from T. saccharolyticum strain B6A, to obtain a thermostable and weak-acid stable GI, which was desirable for industrial application.
Materials and methods
Bacterial strains and media
Thermoanaerobacterium saccharolyticum strain B6A (American Type Culture Collection, Manassas, VA, USA) was grown in LB medium with shaking at 180 rpm and at 37 °C. E. coli strain HB101, used as the host cell for protein expression, was grown in LB medium with 50 mg/l ampicillin at 37 °C.
Cloning of glucose isomerases
The gene encoding glucose isomerases was amplified by PCR from genomic DNA of T. saccharolyticum strain B6A, which was purified by Chelex-100 chelating resin (Bio-Rad). The sequence of the oligonucleotide primers designed for gene cloning was based on the putative GI gene sequence from Thermoanaerobacterium saccharolyticum strain B6A-RI (GenBank accession number L09699): forward (5′-AGATTACCTAGGTACATATGAATAAATATTTTGAGAACGT-3′, AvrII and NdeI sites underlined) and reverse (5′-ATTTCCGATGGCGCGCCTTATTCTGCAAACAAATACTGA-3′, AscI site underlined). The gene was amplified by a mixture of Pfu DNA polymerase and Taq DNA polymerase to add an A-tail on the 3′ end of the PCR product for the next ligation with the cloning vector.
The PCR product was purified by standard agarose gel electrophoresis (1 % gel) and ligated into pGEM®-T Easy Vector from Promega (Madison, WI, USA). After transformation of the ligation mix into chemically competent E. coli HB101 cells, plasmids were isolated using a Rapid Plasmid Miniprep System (Marligen Biosciences Inc., USA) and the presence of a correct insertion was confirmed by sequencing. BigDye® Terminator v3.1 Cycle Sequencing Kit was used for pre-treatment prior to automated sequencing by ABI Prism 377.
Site-directed mutagenesis
The point mutations were introduced into the target gene by a series of PCR [14]. The glucose isomerase gene cloned into pGEM®-T Easy vector was used as template for the PCR. Forward and reverse primers were designed to amplify the two crossover sections containing the desired point mutation and are listed in Table 1. These primers were synthesized by Tech Dragon Co. Ltd., Hong Kong. Mutations were verified by DNA sequencing using ABI Prism 377 (Applied Biosystems).
Expression and purification of glucose isomerase
The correct mutant plasmids were transformed into chemically competent E. coli HB101 cells for protein expression, and the cells would grow on the LB plates with ampicillin and the positive colonies were grown in LB medium containing ampicillin at 37 °C. After overnight growth, cells collected by centrifugation were resuspended in 20 mM PBS buffer with 5 mM MgCl2 and 250 μM CoCl2, pH 6.5. The cells were lysed by sonication at 33 % amplitude with 10-s pulses (with a 1-s delay between pulses) on ice, with a Sonicator Cell Disruptor, converter model W200 R, equipped with a 3-mm probe (Heat systems-ultrasonics, Inc., Plainview). The sonicated material was centrifuged at 12,000g for 10 min at 4 °C to obtain a cell-free protein extract.
The protein extract was put into 80 °C water bath for 10 min and then was centrifuged at 12,000g for 10 min. The supernatant was transferred into metal chelating affinity chromatography column charged with 0.5 ml of 0.1 M nickel chloride solution in advance and then equilibrated with 5 ml of washing solution (10 mM pH 6.0 PBS buffer plus 8 M urea and 0.5 M NaCl). The column was then washed with 10 ml washing solution. Afterwards, the bound protein was eluted with elution solution (10 mM pH 7.5 PBS buffer with 8 M urea, 0.1 M NaCl and 50 mM EDTA) and was collected for next step. For activity assay the residual EDTA was removed by dialysis against 20 mM pH 6.5 PBS buffer with 5 mM MgCl2 and 250 μM CoCl2. Finally, the elution was further purified by gel-filtration chromatography using G75 Superdex size exclusion column (HiLoad TM 16/60 Superdex75 prep grade, Pharmacia) pre-equilibrated with 2 CV of gel-filtration elution buffer (20 mM pH 6.5 PBS with 5 mM MgCl2 and 250 μM CoCl2), analyzed by SDS–polyacrylamide gel electrophoresis, and stored at 4 °C. Protein concentrations were determined using a Bio-Rad protein assay kit (Bio-Rad), with bovine serum albumin as the standard.
Enzyme assays
Glucose isomerase activity was routinely assayed using anhydrous d-(+)-glucose (from Sigma-Aldrich Corp) as substrate. The enzyme (0.1–0.2 mg/ml) was incubated in 20 mM pH 6.5 PBS buffer with 5 mM MgCl2, 250 μM CoCl2 and 1 M glucose at 80 °C for 10 min. The reaction was terminated by cooling the sample in ice–water mixture. The amount of produced fructose in samples was determined by cysteine–carbazole–sulphuric acid method [12]. Absorbances were measured at 560 nm using Perkin Elmer MBA2000 spectrophotometer. One unit of isomerase activity is defined as the amount of the enzyme needed to produce 1 μmol product per min under the assay conditions.
Thermostability and heat-induced enzyme precipitation
To determine the effect of temperature on GI activity, the assay mixtures were incubated at the temperatures of interest in MJ Research PTC 200 Thermal Cycler for 10 min. For thermostability determination, the enzyme samples were firstly diluted with corresponding buffer to equal concentration. 200 μl diluted enzyme was added into a 1.5-ml Eppendorf tube and heated in water bath at special temperature for different time. After heat treatment the enzyme was centrifuged at 12,000g for 15 min to remove protein precipitation. The supernatant was used to determine residual protein concentration and activity. The first-order rate constant, k, of irreversible thermoinactivation was obtained by linear regression in semi-log coordinates. Enzyme half-life was calculated from the equation: t 1/2 = ln 2/k.
pH stability assay
To determine the effect of pH on GI activity, the substrates 1 M glucose were prepared with 5 mM MgCl2 and 250 μM CoCl2 in different pH buffers. The NaAc/HAc buffer covered the range of pH 4.0–5.5, the phosphate buffer for the range of pH 6.0–8.0 and the Tris–HCl buffer for pH 8.5–9.0. For pH stability assay, the enzyme was diluted with corresponding different pH buffers to equal protein concentration. The diluted samples were exposed in various pHs for 12 h at 60 and 80 °C for stability detection.
Effect of metal ions on enzyme activity and stability
To evaluate the effects of metal ions on enzyme activity and stability, metal ions were removed from the purified GI by dialysis against 20 mM PBS buffer (pH 6.5) overnight with several changes of buffer. The effects of various metal ions on enzyme activity were determined by adding different concentrations of MgCl2, MnCl2 and CoCl2 to the dialyzed enzyme preparation and assay the GI activity as described above. The effects of metal ions on enzyme thermal stability were also determined by measuring residual enzyme activity under optimum assay conditions after a 24-h preincubation at 80 °C in the presence of various concentrations of metal ions.
Conversion ratio of glucose to fructose
A mimic industrial substrate for production of HFCS was constructed to test conversion rate of isomerization by glucose isomerase. The substrate was 50 % (w/v) industrial glucose in pH 7.5 PBS buffer with 5 mM MgCl2 and 250 μM CoCl2. The liquid enzyme was mixed with mimic substrate in different E/S ratios in 1.5-ml eppendorf tubes and incubated in water bath of different temperatures for different time. The products were cooled down on ice immediately to stop isomerisation reaction and diluted 400-fold with ddH2O. The fructose amount in diluted samples was measured by cysteine–carbazole–sulphuric acid method [12].
Results
Cloning and site-directed mutagenesis of GI
The GI gene was cloned from T. saccharolyticum strain B6A. After electrophoresis, the PCR amplification product of GI gene formed a specific band about 1,300 bp, which agreed well with expected result (Fig. 1a). The site-directed mutations of tryptophan 139 were introduced to the GI gene by PCR due to the importance of this residue to the enzyme catalytic efficiency of GI [20]. Every pairs of DNA fragments containing mutant site were reassembled to full-length genes (Fig. 1b). All the mutant GI genes were sequenced to verify the correct introduction of the point mutations. The recovered native and mutant DNA fragment were inserted into pGEM®-T Easy Vector by T-A ligation and then were transformed into E. coli strain HB101. The plasmids were purified and sequenced to verify that the recombinant plasmids pGEM®-T-(native or mutant GIs) were constructed correctly.

The agarose gel electrophoresis analysis of a PCR product of amplified native GI gene from B6A (lane 1), and b the reassembled full-length mutant GI amplified using the GI primers (lane 1). The length of band in both is about 1.3 kb, and both lane 2 are GeneRuler™ DNA Ladder
Overproduction and purification of GI
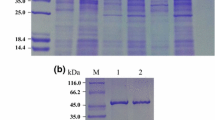
The native and mutant GIs were expressed in E. coli HB101 and were purified as described in “Materials and methods”. The expressed product was identified by SDS-PAGE, and specific protein bands at about 50 kDa were found during different incubation time (Fig. 2), which matched the molecular weight size of the predicted recombinant protein. As shown, the exogenous GI protein was expressed to the maximum amount after incubation for about 16 h.

SDS-PAGE analysis of overproduction of native GI during different incubation time. Lane 1 5 μg BSA as the standard, lane 2 BenchMark® Prestained Protein Ladder; lane 3–10: the overproduction of GI in E. coli HB101 at different incubation time. The band of GI is about 50 kDa (MW = 50,447 Da)
The crude extract was firstly purified by heat treatment at 80 °C for 10 min. The relative total activity after heat treatment showed that GI activity was not affected by the heat treatment (data not shown), and most of host proteins were removed by the heat treatment while the GI protein remained (Fig. 3). The native and mutant GI were finally purified to electrophoretic homogeneity by metal chelating affinity chromatogram and gel filtration. The purified native GI after every step was analyzed by SDS-PAGE (Fig. 3).

SDS-PAGE analysis of purified native T. saccharolyticum GI. Lane 1 BenchMark® Prestained Protein Ladder; lane 2 crude extract of native GI, lane 3 partially purified native GI by heat treatment, lane 4 further purified native GI by metal chelating affinity chromatography, lane 5 further purification by gel filtration, lane 6 5 μg BSA as the standard
The relative enzyme activities of wild and mutant glucose isomerases were assayed at 80 °C with purified enzyme and are listed in Table 2. Total 17 mutants were obtained except for two failure constructions, W139P and W139R. Most of the successful mutants have higher activity than the wide-type more or less. Several mutants, the substitution with cysteine, phenylalanine, asparagine, serine and threonine, were considered to be better than the native GI, and the best mutant W139F reaches 2.4-fold increase in specific activity at 80 °C. This mutant was selected for the further characterization.
Effects of temperature and pH
The temperature dependence of the native GI and mutant W139F was determined in the presence of 5 mM MgCl2 and 250 μM CoCl2 after 10 min incubation at various temperatures. As shown in Fig. 4a, both the native and mutant GIs exhibited optimum activity at 90 °C, and higher temperatures above 90 °C deactivated both enzymes by a drastic decline, although the mutant GI is the comparatively slighter one. The significant difference between both GIs is that the specific activity of W139F reached 2.4-fold of native GI at 80 °C. For the industrial application of GI, it is desirable to react at temperature above 60 °C [4]. Further, the thermal stability of GI and W139F was investigated. As shown in Fig. 4b, the mutant W139F retained 50 % of its initial activity after 20 h of exposure at 80 °C, while the half-life of native GI is 12 h. In addition, the existence of substrate glucose would enhance the thermal stability of both GIs [10]. The half-lives of native GI and W139F were increased from 12 to 18 h and from 20 to 25 h, respectively (data not shown). The results showed that the mutant GI was relatively thermostable and preferred to be the candidate for industrial application.

Effect of temperature on the activity (a) and stability (b) of native T. saccharolyticum GI (filled circle) and mutant W139F (filled triangle). The optimal temperature was determined by assaying the enzyme activity in 20 mM PBS buffer (pH 6.5) in the presence of 5 mM Mg2+ and 250 μM Co2+ at various temperatures. The temperature stability was investigated by exposing the enzyme at 80 °C for different time intervals at pH 6.5 in the presence of 5 mM Mg2+ and 250 μM Co2+. Residual activity was measured under standard conditions (80 °C, pH 6.5). Activity measured with non-incubated enzyme was set to 100 %. Half-lives of native GI and mutant W139F are 12 and 20 h, respectively
The effect of pH on the native GI and W139F was investigated. As shown in Fig. 5a, both glucose isomerases were active at pH from 5.0 to 9.0, and the maximum activity was obtained at pH 7.0. After heat treatment at 60 and 80 °C for 12 h by exposing the enzyme in pH 6.0–8.0, the residual activity of W139F and native GI were assayed (Fig. 5b). The data indicated that the mutant GI was more thermal stable at alkaline pH than acidic environment, and the wide-type GI showed a similar result (Fig. 5b, inset). Almost 100 % of mutant GI activity was remained after heat treatment at 80 °C for 12 h at pH above 7.5. Further, more than 80 % activity of the purified W139F was retained at pH 6.0–8.0 after incubation at 60 °C for 12 h while the activity of native GI was retained for only 2 h. In addition, the data point which is more than 100 % indicated that the control enzyme stored at 4 °C (pH 6.5) is not stable. This phenomenon may be another evidence of cold denaturation of thermophilic protein [22]. At last, these results showed that the mutant GI was active and stable under weakly acidic conditions, although the pH optima did not change compared with the native GI.

Effect of pH on the activity (a) and thermal stability (b) of native GI (filled circle) and mutant W139F (filled triangle). The optimal pH was determined by assaying the enzyme activity in 20 mM PBS buffer with various pH at 80 °C. The pH stability was investigated by exposing mutant W139F and native GI (inset) in various pHs for 12 h at 60 and 80 °C, respectively. Residual activity was measured under standard conditions (80 °C, pH 6.5). Activity measured with the mutant and native enzyme stored at 4 °C for 12 h (pH 6.5) was set to 100 %, respectively
Effects of metal ions
The effect of various metal ions on the activity of mutant GI from T. saccharolyticum strain B6A was investigated (Table 3). It revealed that both Co2+ and Mg2+ enhance enzyme activity, although the effect of Mg2+ is not obvious as Co2+. Besides, the addition of Co2+ and Mg2+ showed a synergistic activation effect on W139F activity. The presence of Mn2+ inhibited the enzyme activity and Mn2+ decreased the activation effect of Co2+ when they are together. The concentration of 5 mM Mg2+ and 250 μM Co2+ was the best combination which was required to achieve maximum glucose isomerase activity.
The thermal stability of mutant GI was also investigated by measuring residual activity after incubation of the enzyme at 80 °C for 1 h. The results indicated that the glucose isomerase requires Co2+ or Mg2+ for its thermal stability, while Mn2+ has a destructive effect. In addition, the highest thermal stability was achieved in the presence of 5 mM Mg2+ and 250 μM Co2+ due to a synergistic effect.
Estimation of K m and V max
The enzyme reaction was performed in the presence of 5 mM Mg2+ and 250 μM Co2+ after 10 min incubation at pH 6.5 and 80 °C with various concentrations of d-glucose. The Lineweaver–Burk plots of specific activities at various substrate concentrations were performed. The K m and V max at the conditions were estimated and listed in Table 4. For native GI, K m and V max were 149.4 mM and 0.0557 μmol/min, and while for the mutant GI, they were 51.3 mM and 0.0843 μmol/min. The substrate’s affinity preference for mutant GI over native GI was illustrated by a lower K m and higher V max toward substrate glucose, and the mutant GI had a higher catalytic efficiency (K cat./K m) for glucose than the wild-type enzyme of tenfold. This result makes mutant W139F a great candidate for industrial application because the high catalytic efficiency reduces the cost of HFCS production.
Conversion ratio of glucose to fructose
To evaluate the potential industrial application of engineered GI in HFCS production, the conversion ratios of glucose to fructose by the liquid enzyme were investigated in a mimic environment of industrial process.
The substrate was prepared using 50 % (w/v) industrial glucose in pH 8.0 PBS buffer with 5 mM MgCl2 and 250 μM CoCl2. The concentration of GI W139F was 10 mg/ml. First the liquid enzyme was mixed with substrate in a fixed enzyme versus substrate (E/S) ratio of 1:4,000 (w/w) and incubated in water bath of different temperatures for different time. The final conversion ratio of every sample was calculated by fructose quantification. The yellow by-products were estimated with OD425 nm (Table 5).
It seemed that the GI activity was lost entirely within 24 and 12 h because the conversion ratio did not reached the glucose–fructose equilibrium at 75 °C (53.1 %) and 80 °C (53.9 %) [4], respectively. This result is different with previous results that the half-life of W139F was about 20 h at 80 °C in buffer, and was even better in the presence of substrate. The rapid loss of activity may be a result of harmful by-products accumulation in such a sealed system. The higher isomerization temperature is, the more by-products produced. Therefore, the GI was inactivated earlier at 80 °C than 75 °C. In addition, with the increasing time, the conversion ratio remains at a relatively equal level at 75 and 80 °C, and 75 °C was preferred to be the isomerization temperature because of the fewer by-products.
Discussions
Thermostable glucose isomerase from T. saccharolyticum strain B6A and its mutant W139F was purified to electrophoretic homogeneity. Both enzymes were able to recognize glucose (K m 149.4 and 51.3 mM, respectively) as substrate. In both reactions, the GIs required metal ions for thermal stability and catalytic activity, although their requirements were different. The mutant W139F showed a higher catalytic efficiency (K cat./K m) for glucose (Table 4) and a longer half-life (Fig. 4b) than the wild-type GI, and the mutant GI is more thermal stable than native GI. Furthermore, the enzyme activity was stable in the pH range 6.0–8.0 and showed 80 % of its maximum activity at pH 6.0 at 60 °C (Fig. 5b). The mutant derivative W139F is also the most active type ever reported with a maximal activity at 80 °C of 59.58 U/mg on glucose. All of these characterization indicated that this mutant GI is desirable for industrial application of HFCS production.
In conclusion, our research showed that genetic engineering approaches can be utilized to identify amino acid residues responsible for high catalytic activity of glucose isomerase. The W139F mutation significantly enhanced K cat./K m for glucose isomerization to fructose. Genetic engineering altered the important functional amino acid residue to better accommodate glucose as the substrate.
However, except a lower pH optimum, a higher temperature optimum and a higher affinity for glucose the ideal GI should be possessed; the requirement of metal ions for GI activity and stability also should be eliminated. Although introduction of all these properties into a single protein is a herculean task, recent advances in recombinant DNA technology and protein engineering have opened new and encouraging possibilities of combining the desirable properties in a single organism to produce a tailor-made protein.
References
Amore R, Hollenberg C (1989) Xylose isomerase from Adinoplanes missouriensis: primary structure of the gene and the protein. Nucleic Acids Res 17(18):7515. doi:10.1093/nar/17.18.7515
Antrim RL, Colilla W, Schnyder BJ (1979) Glucose isomerase production of high-fructose syrups. Applied biochemistry and bioengineering. Academic Press, London. doi:10.1016/B978-0-12-041102-3.50010-9
Bandlish RK, Michael Hess J, Epting KL, Vieille C, Kelly RM (2002) Glucose-to-fructose conversion at high temperatures with xylose (glucose) isomerases from Streptomyces murinus and two hyperthermophilic Thermotoga species. Biotechnol Bioeng 80(2):185–194. doi:10.1002/bit.10362
Bhosale SH, Rao MB, Deshpande VV (1996) Molecular and industrial aspects of glucose isomerase. Microbiol Rev 60(2):280–300
Bor Y-C, Moraes C, Lee S-P, Crosby WL, Sinskey AJ, Batt CA (1992) Cloning and sequencing the Lactobacillus brevis gene encoding xylose isomerase. Gene 114(1):127–132. doi:10.1016/0378-1119(92)90718-5
Borgi MA, Rhimi M, Bejar S (2007) Involvement of alanine 103 residue in kinetic and physicochemical properties of glucose isomerases from Streptomyces species. Biotechnol J 2(2):254–259. doi:10.1002/biot.200600085
Borgi MA, Srih-Belguith K, Ben Ali M, Mezghani M, Tranier S, Haser R, Bejar S (2004) Glucose isomerase of the Streptomyces sp. SK strain: purification, sequence analysis and implication of alanine 103 residue in the enzyme thermostability and acidotolerance. Biochimie 86 (8):561–568. doi:10.1016/j.biochi.2004.07.003
Brown SH, Sjøholm C, Kelly RM (1993) Purification and characterization of a highly thermostable glucose isomerase produced by the extremely thermophilic eubacterium, Thermotoga maritima. Biotechnol Bioeng 41(9):878–886. doi:10.1002/bit.260410907
Cha J, Batt C (1998) Lowering the pH optimum of d-xylose isomerase: the effect of mutations of the negatively charged residues. Mol Cells 8(4):374
Converti A, Borghi MD (1997) Kinetics of glucose isomerization to fructose by immobilized glucose isomerase in the presence of substrate protection. Bioprocess Eng 18(1):27–33. doi:10.1007/s004490050406
Dekker K, Yamagata H, Sakaguchi K, Udaka S (1991) Xylose (glucose) isomerase gene from the thermophile Clostridium thermohydrosulfuricum; cloning, sequencing, and expression in Escherichia coli. Agric Biol Chem 55(1):221–227. doi:10.1271/bbb1961.55.221
Dische Z, Borenfreund E (1951) A new spectrophotometric method for the detection and determination of keto sugars and trioses. J Biol Chem 192(2):583–587
Hespell RB (1992) Fermentation of xylans by Butyrivibrio fibrisolvens and Thermoanaerobacter strain B6A: utilization of uronic acids and xylanolytic activites. Curr Microbiol 25(4):189–195
Jeltsch A, Lanio T (2002) Site-directed mutagenesis by polymerase chain reaction, vol 182. In vitro mutagenesis protocols. Humana Press, New York. doi:10.1385/1-59259-194-9:085
Jensen VJ, Rugh S (1987) Industrial-scale production and application of immobilized glucose isomerase. Methods Enzymol 136:356–370. doi:10.1016/S0076-6879(87)36035-5
Lama L, Nicolaus B, Calandrelli V, Romano I, Basile R, Gambacorta A (2001) Purification and characterization of thermostable xylose (glucose) isomerase from Bacillus thermoantarcticus. J Ind Microbiol Biotechnol 27(4):234–240. doi:10.1038/sj.jim.7000182
Lee CY, Zeikus JG (1991) Purification and characterization of thermostable glucose isomerase from Clostridium thermosulfurogenes and Thermoanaerobacter strain B6A. Biochem J 273(Pt 3):565–571
Lee Y-E, Ramesh MV, Zeikus JG (1993) Cloning, sequencing and biochemical characterization of xylose isomerase from Thermoanaerobacterium saccharolyticum strain B6A-RI. J Gen Microbiol 139(6):1227–1234. doi:10.1099/00221287-139-6-1227
Meaden PG, Aduse-Opoku J, Reizer J, Reizer A, Lanceman YA, Martin MF, Mitchell WJ (1994) The xylose isomerase-encoding gene (xylA) of Clostridium thermosaccharolyticum: cloning, sequencing and phylogeny of Xy1A enzymes. Gene 141(1):97–101. doi:10.1016/0378-1119(94)90134-1
Meng M, Lee C, Bagdasarian M, Zeikus JG (1991) Switching substrate preference of thermophilic xylose isomerase from d-xylose to d-glucose by redesigning the substrate binding pocket. Proc Natl Acad Sci USA 88(9):4015–4019. doi:10.1073/pnas.88.9.4015
Mu W, Wang X, Xue Q, Jiang B, Zhang T, Miao M (2012) Characterization of a thermostable glucose isomerase with an acidic pH optimum from Acidothermus cellulolyticus. Food Res Int 47(2):364–367. doi:10.1016/j.foodres.2011.09.006
Privalov PL (1990) Cold denaturation of proteins. Crit Rev Biochem Mol Biol 25(4):281–305. doi:10.3109/10409239009090613
Srih-Belghith K, Bejar S (1998) A thermostable glucose isomerase having a relatively low optimum pH: study of activity and molecular cloning of the corresponding gene. Biotechnol Lett 20(6):553–556. doi:10.1023/A:1005393510435
Author information
Authors and Affiliations
Corresponding author
Additional information
H. Xu and D. Shen equally contributed to this study.
Rights and permissions
About this article

Cite this article
Xu, H., Shen, D., Wu, XQ. et al. Characterization of a mutant glucose isomerase from Thermoanaerobacterium saccharolyticum . J Ind Microbiol Biotechnol 41, 1581–1589 (2014). https://doi.org/10.1007/s10295-014-1478-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-014-1478-4