
Abstract
The extreme process condition of high temperature and high alkali limits the applications of most of natural xylanases in pulp and paper industry. Recently, various methods of protein engineering have been used to improve the thermal and alkalic tolerance of xylanases. In this work, directed evolution and site-directed mutagenesis were performed to obtain a mutant xylanase improved both on alkali stability and thermostability from the native Paenibacillus campinasensis Family-11 xylanase (XynG1-1). Mutant XynG1-1B43 (V90R/P172H) with two units increased in the optimum pH (pH 7.0–pH 9.0) and significant improvement on alkali stability was selected from the second round of epPCR library. And the further thermoduric mutant XynG1-1B43cc16 (V90R/P172H/T84C-T182C/D16Y) with 10 °C increased in the optimum temperature (60–70 °C) was then obtained by introducing a disulfide bridge (T84C-T182C) and a single amino acid substitution (D16Y) to XynG1-1B43 using site-directed mutagenesis. XynG1-1B43cc16 also showed higher thermostability and catalytic efficiency (k cat /K m ) than that of wild-type (XynG1-1) and XynG1-1B43. The attractive improved properties make XynG1-1B43cc16 more suitable for bioleaching of cotton stalk pulp under the extreme process condition of high temperature (70 °C) and high alkali (pH 9.0).
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Xylanases (endo-1, 4-β-d-xylanohydrolase; E.C. 3.2.1.8) are important enzymes for the degradation of the major hemicellulosic polysaccharide of hardwood, since they hydrolyze xylopyranosyl linkages of β-1, 4-xylan [6]. As the key xylan-degrading enzymes, xylanases have attracted considerable attention because of their application potential in various industrial processes, especially in paper industry such as bio-pulping and bio-bleaching [4, 5]. The problem faced by the xylanases using in pulp and paper industry, however, is retaining active under high temperature and high alkali conditions. Since the availability of natural xylanases is limited, improvements in the thermal and alkali tolerance of xylanases have been attempted by mutational methods using rational approaches based on three-dimensional structures or non-rational approaches based on directed screening [22, 26].
According to the similarity of amino acid sequences of their catalytic domain, xylanases have been grouped commonly into glycoside hydrolase (GH) families 10 and 11 [3]. Among the highly conserved amino acids in the family 11 xylanases, there are two conserved glutamic acids (Glu93 and Glu182 in a Bacillus pumilus xylanase) located on either side of the cleft [9]. These catalytic residues participate together in the retention of anomeric configuration of the glycosidic oxygen involving a double displacement catalytic mechanism [6]. Besides, a mass of protein engineering work had been devoted to improving the stability of xylanases such as the thermostability and operative pH range. Despite increasing knowledge of microbial xylanolytic systems recognized through previous research, further studies are still required to achieve better understanding of the thermoduric mechanism and pH-tolerant mechanism of xylanases.
Recently, P. campinasensis G1-1 (CGMCC NO.5,023) was newly isolated from a Chinese paper mill that exhibits a strong capacity to degrade xylan and produces a family 11 xylanase (XynG1-1) with the optimal activity at 60 °C and pH 7.0 [28]. Then the recombinant xylanase (XynG1-1R) expressed in Bacillus megaterium MS941 was purified and used for biobleaching of cotton stalk pulp and saccharification of recycled paper sludge. The results indicated that XynG1-1R can reduce chlorine consumption by 50 % in biobleaching of cotton stalk pulp and increase saccharification efficiency of recycled paper sludge by 10 % at its optimum condition [27]. However, due to the high temperature and high alkali process condition of paper industry, the xylanase used in this process need to be more thermostable and alkali-stable. Therefore, in this study, directed evolution in combination with site-directed mutagenesis were employed to enhance the alkali and thermal tolerance of the wild-type xylanase from P. campinasensis G1-1. A series of mutants with the improvements on alkali tolerance or thermal tolerance or both were compared based on their characterizations and kinetics. Furthermore, the effect of bio-bleaching of cotton stalk pulp using the mutant xylanase under the high-temperature and high-alkali-process condition was evaluated.
Materials and methods
Gene, vector, strains, and chemicals
The wild-type xylanase gene was amplified from Paenibacillus campinasensis G1-1 (CGMCC NO.5,023) isolated by our laboratory [28]. pET-XynG1-1 was constructed by inserting the wild-type gene (1,017 bp) to the expression vector pET22b(+) [28]. E. coli DH5α (preserved in our laboratory) and E. coli BL21 (DE3) (preserved in our laboratory) were used for gene cloning, sequencing, and expression, respectively. The recombinant E. coli BL21 harboring pET-XynG1-1 was regarded as the wild-type strain. Birchwood xylan was purchased from Sigma Chemical Co. (St. Louis, MO, USA). Pyrobest polymerase, Taq polymerase, dATP, dGTP, dTTP, and dCTP were purchased from Fermentas China Co. Ltd. All other chemicals were of analytical reagent-grade purity and obtained from commercial sources.
Construction of error-prone PCR libraries
To generate a library of XynG1-1 variants, mutations were introduced using two-round error-prone PCR (epPCR). For the first round of epPCR, plasmid pET-XynG1-1 encoding wild-type XynG1-1 was used as the template. For the second round of epPCR, plasmids harboring all the beneficial mutations from the first round screening were pooled and used as templates. The detailed gene operation was performed as reported in [28].
Screening for alkali-stable variants
Because the recombinant xylanase expressed in E. coli BL21 (DE3) was mostly secreted outside the cell [28], the mutant recombinant xylanases could be screened on the plates via hydrolysis zone and be determined extracellular enzyme activity straightly. All the positive mutants were detected by their ability to produce a hydrolysis zone on xylan plates (LB-Agar plate added 1 % xylan, 100 μg/ml ampicillin, and 1 mM IPTG, pH 7.0). Then the positive mutants were transferred to alkali xylan plates (pH 10.0 for the mutants of the first ground epPCR and pH 11.0 for the mutants of the second ground epPCR) for further screening. The recombinant E. coli BL21 (pET-XynG1-1) was as a control. Single colonies from the mutant library of the second round epPCR were picked with sterile toothpicks and cultured in 200 μl of LB medium containing ampicillin (100 μg/ml) and 1 mM IPTG in sterile 96-well microplates and incubated overnight at 37 °C with shaking at 180 rpm for 24 h. For keeping strains, half of the content of each well was transferred into a fresh microplate adding sterile glycerin at a final concentration of 15 %. The rest of plates were subjected to centrifugation at 4,000 rpm for 20 min. Enzymatic assay was then carried out at 60 °C for 10 min after the addition of 5 μl of supernatants and 5 μl of 1 % (w/v) of xylan prepared in 20 mM Tris–HCl buffer (pH 9.0) in a fresh microplate. The reactions were quenched by the addition of 30 μl of DNS reagent [2] and heated at 105 °C for 10 min to determine the amount of reducing sugar. After heating, 210 μl of deionized water was added into the reactions and absorbance was measured at 540 nm. The clones revealing higher xylanase activity than that of E. coli BL21 (pET-XynG1-1) were collected for further enzymatic properties studying and gene sequencing.
Design and construction of site-directed mutants
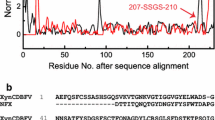
To further improve thermostability of the alkali-stable mutant (XynG1-1B43) of XynG1-1, similarity analysis using the protein sequence of XynG1-1B43 (GenBank: AGG82433.1) was performed online with the NCBI BLAST software (http://blast.ncbi.nlm.nih.gov). Four homologous sequences of family-11 xylanases with the optimum temperature more than 70 °C from thermophilic bacteria were selected. The xylanase sequences shared 62–79 % sequence similarities with XynG1-1B43 (Fig. 1). Mutant site D16Y and a disulfide bridge (C84–C182) were proposed based on the comparison of the XynG1-1B43 sequence with the consensus sequence (Fig. 1). Site-directed mutagenesis was performed using an overlapping PCR technique [17]. The sequences of oligonucleotides containing the appropriate base changes are listed in Table 1. All mutants with single substitutions were constructed with plasmid pET-XynG1-1B43 as the template. The amplified fragments were digested with HindIII and XhoI and ligated into the pET-22b(+) vector previously digested with the same enzymes, and then the recombinant plasmids were transformed into E. coli BL21. The successful introductions of the desired mutations were confirmed by sequencing at Beijing Genomic Institute. The formations of disulfide bridges in proteins were demonstrated as previously described by treating with 1–5 mM dithiothreitol (DTT) prior to SDS-PAGE [23, 24].

Amino acid sequence alignment of XynG1-1B43 and four thermophile xylanases obtained from GenBank. The numbers on the left are the GenBank accession numbers of the sequences. gb|AAB95327.1| is xylanase from Caldicellulosiruptor sp. Rt69B.1; gb|AAC46361.1| is xylanase 229B from Dictyoglomus thermophilum; gi|11,513,709| is xylanase XynB from Dictyoglomus thermophilum Rt46b.1; gi|206,900,912| is xylanase A from Dictyoglomus thermophilum H-6-12 [14, 15]. The marks under the sequences are as follows: the asterisk indicates positions that have fully conserved residues; the dot indicates conservation within the same type of amino acids. The mutation sites are highlighted in the boxes
Expression and purification of wild-type and mutant enzymes
Each colony of E. coli BL21 (harboring encoding gene of wild-type and mutant xylanases) was inoculated into 5 ml of LB medium (100 μg/ml Amp) and was cultured overnight at 37 °C in a rotary shaker. The purification of the extracellular xylanases of wild type and mutants were performed as reported in [27].
Analysis of the enzyme activity
Xylanase activity was measured by determining the amount of reducing sugars (xylose) liberated from birchwood xylan (Sigma) using 3,5-dinitrosalicylic acid under the optimum condition of each enzyme [13]. The method was described previously by Zheng et al. [27]. One unit of xylanase activity was defined as amount enzyme that liberated 1 μmol of reducing sugars per minute.
Properties of wild-type and mutant enzymes
The optimum temperature and pH of the purified recombinant xylanases were determined as reported in [27] with modifications. The temperature range was 40–90 °C and the pH range was 4.0–11.0.
Under the condition of pH 11.0, the alkali stability was determined by incubating purified enzymes in a water bath at 60 °C for 30, 60, 90, 120, 150, and 180 min, respectively, and then the residual enzyme activity was shown to analyze the alkali stability. Thermostability of XynG1-1 and mutant xylanases were performed by maintaining the purified enzyme solution in a water bath at 90 °C for 20, 40, 60, 80, 100, and 120 min, respectively, and then running the activity assay.
Kinetic properties of wild-type and mutant enzymes
The kinetic parameters (K m , V max and k cat ) of wild-type and mutant xylanases were estimated as reported in [28].
Biobleaching of cotton stalk pulp with wild-type XynG1-1 and mutant XynG1-1B43cc
The optimum dosage of xylanase was 15 IU/g dry pulp determined in the previous experiment [27]. The process of pulp bleaching and determination of paper parameters were consistent with the previous experiment [27], except that the enzymatic bleaching was performed at 70 °C and pH 9.0 for 3 h and then the chemical bleaching (D0 ED1) with 2.5 % of chlorine dioxide in D1.
Computer modeling methods
To obtain the theoretical structure of mutant and native xylanases, models were built on a public website, Swiss-Model [1, 7, 18], using the coordinates of xylanase J from Bacillus sp. strain 41 M-1 (2dcjB pdb entry) as a template, which was received from the Protein Databank [25]. The three-dimensional structure of wild-type XynG1-1 and mutants were viewed and analyzed by the Swiss PDB Viewer and PyMOL molecular graphics system.
Nucleotide sequence accession numbers
The nucleotide sequences of mutant xylanase genes, XynG1-1B43 and XynG1-1B43cc16, have been submitted in GenBank with the accession numbers as JX908142.1 and JX908143.1, respectively.
Results and discussion
Screening mutants with enhanced alkali tolerance from epPCR library
Randomly picked clones from the first-round epPCR library showed 2.66 base substitutions/kb and 2,850 colonies displayed xylanase activity on xylan plates (pH 10.0). The xylanase activities of 86 clones produced larger zones of hydrolysis than XynG1-1 from E. coli BL21 (pET-XynG1-1) were assayed for xylanase activity at pH 9.0. Six clones showing higher xylanase activity than XynG1-1 at pH 9.0 and 60 °C were subjected to the second round of mutagenesis (data not shown).
For the second round of mutagenesis, 950 clones displayed hydrolysis zones on the xylan plates (pH 11.0) were evaluated for enzyme activity and alkali stability. Five of the most alkali-stable mutants (XynG1-1B05, XynG1-1B22, XynG1-1B43, XynG1-1B56, and XynG1-1B72) were selected for further evaluation (Table 2). A total of three amino acid substitutions from the mutants, V90R, P172H, and I124T, were found to be possibly beneficial for the enhanced alkali tolerance of XynG1-1 (Table 2).
Characterization of wild-type XynG1-1 and mutants from epPCR library
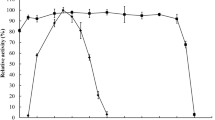
Further enzymatic characterizations were performed for purified XynG1-1 and purified mutants. The assays for xylanase activity were performed at various pHs (pH 4.0–11.0). The results revealed marked changes of the optimum pH due to amino acid substitutions (Fig. 2a). XynG1-1 revealed the maximum activity at pH 7.0. XynG1-1B05 and XynG1-1B22 had the maximum activities at pH 8.0, XynG1-1B43 and XynG1-1B56 showed the maximum activities at pH 9.0. However, XynG1-1B72 showed the same optimum pH compared with XynG1-1 (Fig. 2a). The alkali stability of wild-type XynG1-1 and the mutants were determined by measuring their residual activities after incubating the purified enzymes for 180 min at pH 11.0. Compared with XynG1-1, all of the mutants exhibited increased alkali stability at pH 11.0 (Fig. 2b). XynG1-1B43 retained the highest residual activity (42 %), which was 1.5 times higher than that of XynG1-1 after incubation at pH 11.0 for 180 min (Fig. 2b). According to the results described above, the amino acid substitutions of V90R and P172H showed an obvious effect on enhancing the alkali stability of XynG1-1. However, in order to determine the effect of the amino acid substitution of I124T, site-directed mutagenesis for I124T was performed. The mutant (I124T) showed no significant improvement on the properties of XynG1-1 (data not shown). Besides, the influence of temperature on XynG1-1 and the mutants were studied in order to determine the thermostability of the enzymes. Wild-type XynG1-1 and the mutants showed similar properties and tendency of thermal inactivation at high temperature, indicating that there was no change in the thermostable property after mutation (data not shown).

The optimum pH (a) and alkali stability (b) of wild-type XynG1-1 and epPCR mutants. a The activities were assayed under various pH levels (pH 4.0–11.0) and 60 °C. b The residual activities of the enzymes were determined after incubating at 60 °C and pH 11.0 for different interval times. The initial activity of each enzyme was defined as 100 %. Data are presented as mean ± SD (n = 3)
Val90 with high hydrophobic nature is the last residue of β-sheet seven on the outside of the catalytic cleft domain, and its hydrophobic side chain pointed towards the solvent (Fig. 3). It is generally assumed that in proteins hydrophobic residues are not favorable at solvent-exposed sites for protein stability [10]. So Arg with a hydrophilic side chain was found to stabilize the protein in replacement of Val90. The hydrophilic nature of the surface was increased when compared with Val90. Meanwhile, Arg90 at this position was involved in hydrogen bonding with Asp91, which would further enhance the stability of the region according to the theoretical structure (Fig. 4b). Many previous reports also confirmed that the increase in the number of arginine on the protein surface could enhance the alkali adaptation of enzymes. Shirai et al. [20], via analyzing the relationship of structure and function of M-protease, suggested that high-alkali adaptation would be accompanied by an increase in arginine and neutral hydrophilic amino acids (histidine, asparagine, and glutamine) residues and a decrease in the number of negatively charged amino acids (aspartic acid and glutamic acid) and lysine residues. Some mutant enzymes shifted the activity profile to the alkalic region by 0.5–1.0 pH units were prepared from Trichoderma reesei xylanase II by inducing excess Arg residues on its protein surface [21]. The optimum pH of xylanase J from Bacillus sp. 41 M-1 was increased from 8.5 to 9.5 by introducing excess Arg residues on the protein surface and reinforcing the characteristic salt bridge in the catalytic cleft. In this study, the coincident conclusion was obtained that the optimum pH of XynG1-1 was increased from 7.0 to 8.0 by the amino acid substitution of V90R (Fig. 2a). Moreover, Pro172 as a circular amino acid residue was very close to the alpha-helix on the protein surface (Fig. 3). However, proline in the structure of protein is generally to form cis peptide bonds that go against the alpha helix formation [12]. So the His-to-Pro substitution was supposed to be beneficial for stability of the alpha-helix of XynG1-1, then reinforcing the capacity of resistance at extreme environment. In addition, His172 as a polar residue is involved in a hydrogen bonding network: His172↔Gly170 and His172↔Tyr144 according to the theoretical structure (Fig. 4d). However, Pro172 only provides a hydrogen bond to Tyr144. As suggested by Shirai et al. [20], neutral hydrophilic amino acid (histidine, asparagine, and glutamine) residues on the protein surface were beneficial for high-alkali adaptation—the same conclusion was obtained in this study with the results that the mutant XynG1-1B72 (P172H) showed higher residual activity than that of XynG1-1 after incubation at pH 11.0 for 180 min (Fig. 2b).

Three-dimensional structures of wild-type XynG1-1 (a) and mutant B43cc16 (b). The models were obtained from a public Web site, Swiss-Model (http://www.expasy.ch/swissmod/SWISS-MODEL.htmL) [1, 7, 18], using the coordinate of 2dcj.pdb entry as template [25], and analysis by PyMOL molecular graphics system. It was shown that XynG1-1 consisted of two domains: a family 11 catalytic domain (green) and a non-catalytic xylan-binding domain (blue). According to the retaining catalytic mechanism of family 11 xylanases, Glu95 and Glu184 (highlighted in red) located on either side of the cleft were supposed to be nucleophile and proton donor in the catalytic reaction, respectively. The amino acid residues of mutant sites are highlighted in yellow. There was no extensive conformational change of mutant XynG1-1B43cc16 structure by the amino acid replacements compared with XynG1-1

Stereo views of the amino acid residues and their surroundings at the mutant sites before (a, c) and after (b, d) epPCR mutation. The residues at the mutant sites are shown in yellow and the surrounding residues are shown in red. The green dashed lines indicate hydrogen bondings
The combination of the two substitutions of V90R and P172H resulted in a cumulative effect observed in the double-mutated XynG1-1B43 (V90R/P172H) showing the highest activity of the mutants at pH 9.0 with 2.5 times higher than that of XynG1-1. The cumulative effect of multiple-positions mutant was usually observed in mutant xylanases using directed evolution or site-directed mutagenesis [11, 16, 22]. On the basis of study on the XynG1-1 mutants, it was concluded that Arg residues and His residues on the protein surface were beneficial for alkali adaptation of XynG1-1.
Characterization of wild-type XynG1-1, epPCR mutants, and site-directed mutants
XynG1-1B43 with the maximum enhancement of alkali stability of XynG1-1 was selected as a template for further improving thermostability by site-directed mutagenesis. A disulfide bridge mutant XynG1-1B43cc (C84–C182) and a single-mutant XynG1-1B4316 (D16Y) as well as the corresponding double-mutant XynG1-1B43cc16 (T84C-T182C/D16Y) with excellent thermostability were purified from the recombinant E. coli BL21, and their properties were compared with those of XynG1-1 and XynG1-1B43 produced and analyzed under the same conditions (Fig. 5). The results showed that the optimum temperature of XynG1-1B43cc and XynG1-1B43cc16 were 70 °C compared to 60 °C of XynG1-1 and XynG1-1B43 (Fig. 5a). Meanwhile, the thermostability of the three site-directed mutants dramatically increased after incubation at 90 °C (Fig. 5a). The site-directed mutants retained at least 24.5 % of its activity after incubation at 90 °C for 120 min, while XynG1-1 and XynG1-1B43 were completely inactivated (Fig. 5b). The double site-directed mutant XynG1-1B43cc16 revealed the most remarkable improvement in both the optimum temperature and thermostability (Fig. 5), and no change in the temperature properties (data not shown) compared with that of XynG1-1B43.

The optimum temperature (a) and thermostability (b) of wild-type XynG1-1, mutant XynG1-1B43 and site-directed mutants. a The activities were assayed under various temperatures (40–90 °C) and pH 9.0. b The residual activities of the enzymes were determined after incubating at 90 °C and pH 9.0 for different interval times. The initial activity of each enzyme was defined as 100 %. Data are presented as mean ± SD (n = 3)
XynG1-1B43cc and XynG1-1B43cc16 with various concentration of DTT treatment (1–5 mM) resulted in more migratory bands than that of XynG1-1B43 in SDS-PAGE, which indicates the presence of disulfide bridges in XynG1-1B43cc and XynG1-1B43cc16 (data not shown). The new disulfide bridge between β-sheet 7 and β-sheet 13 inside the catalytic cleft of the mutant XynG1-1B43 was constructed by the substitutions of both Thr84Cys and Thr182Cys (Fig. 3). The new disulfide bridge was designed to stabilize the relative position of the two catalytic sites (E95 and E184) for improvement of temperature resistance according to the corresponding disulfide bridge existing in nature of a family 11 xylanase (GenBank: gb|AAB95327.1|) with the optimal temperature of 75 °C from the extreme thermophile Caldicellulosiruptor strain Rt69B.1 [15] and a xylanase XynB (GenBank: gi|11,513,709|) with the optimal temperature of 85 °C from Dictyoglomus thermophilum Rt46B.1 [14] (Fig. 1). The wild-type xylanase XynG1-1 and alkali mutant XynG1-1B43 have a hydrogen bonding network: Thr84↔Val85, Thr8↔Ile98, and Thr182↔Ala180 according to the theoretical structure (Fig. 6a). However, a covalent disulfide bond is more stable than hydrogen bonds, which is more readily disrupted by heating up compared with disulfide bonds [19]. A new hydrogen bond that formed between C84 and Met181 was further consolidating the structure around the disulfide bond according to the theoretical structure (Fig. 6b). Therefore, the mutant XynG1-1B43cc showed more thermostability than XynG1-1 and XynG1-1B43 (Fig. 5). In addition, the new disulfide bridge inside the catalytic cleft of the xylanase was supposed to dramatically influence the properties of the catalytic sites (E95 and E184) resulting in the optimum temperature of mutant XynG1-1B43 increasing from 60 to 70 °C. Moreover, the substitution of Asp to Tyr at position 16 of the N-terminal of XynG1-1B43cc was designed to further improve its thermostability based on the formation of the hydrophobic domain (Y15–Y17), which was conserved in thermophilic xylanases such as xylanase XynB (GenBank: gi|11,513,709|) from Dictyoglomus thermophilum Rt46B.1 [14] and a family 11 xylanase (GenBank: gb|AAB95327.1|) from Caldicellulosiruptor strain [15] (Fig. 1). The substitution of Asp to Tyr resulted in breakages of two hydrogen bonds of Asp16 ↔ Thr11 and Asp16 ↔ Lys54 according to the theoretical structure (Fig. 6 c, d), which indicated that the hydrophobic interactions among the three tyrosines are more significant for the thermostability of XynG1-1 than hydrogen bonds. Besides, clusters or “sticky patches” formed by aromatic residues on the surface of the enzyme are responsible for the tendency of the protein to aggregate at high concentrations in the absence of reagents and their hydrophobic contacts may also contribute to the thermostability of the enzyme [8]. However, the combination of aromatic residues on the protein surface could not influence the optimum temperature of XynG1-1.

Stereo views of the amino acid residues and their surroundings at the mutant sites before (a, c) and after (b, d) site-directed mutation. The residues at the mutant sites were shown in yellow; the residues forming hydrogen bonds with the residues at the mutant sites were shown in red and the residues have hydrophobic interaction with the residues at the mutant sites were shown in blue; The green dashed lines indicated hydrogen bondings
The synergism of the formation of aromatic residues cluster and introduction of a disulfide bridge was responsible for increasing the thermostability of XynG1-1B43. The final mutant XynG1-1B43cc16 possessed the optimum temperature of 70 °C, which was 10 °C higher than that of XynG1-1and XynG1-1B43 and showed the highest thermostability among all the mutants and wild-type XynG1-1.
Kinetic parameters of wild-type XynG1-1, mutant XynG1-1B43, and XynG1-1B43cc16
A comparison of the kinetic parameters of mutant XynG1-1B43 and XynG1-1B43cc16 with XynG1-1 at a condition of pH 9.0 and 70 °C was carried out using birchwood xylan and oat spelt xylan as substrates. As shown in Table 3, both mutants XynG1-1B43 and XynG1-1B43cc16 had greater affinity for birchwood xylan than oat spelt xylan, which was consistent with XynG1-1. At the condition of pH 9.0 and 70 °C, XynG1-1B43cc16 showed a significant decrease in K m and increase in k cat , which resulted in catalytic efficiency (k cat /K m ) increasing compared with XynG1-1. The k cat /K m of XynG1-1B43cc16 were about two times higher than that of XynG1-1 at pH 9.0 and 70 °C using birchwood xylan as a substrate (Table 3). However, XynG1-1B43cc16 and XynG1-1B43 showed equivalent values of K m (5.55 mg/ml), which were lower than that (7.06 mg/ml) of XynG1-1.
Biobleaching of cotton stalk pulp with wild-type XynG1-1 and mutant XynG1-1B43cc
Fifteen IU/g dry pulp of XynG1-1 and XynG1-1B43cc were used to pretreat the cotton stalk pulp at the condition of 70 °C and pH 9.0, and followed by chemical bleaching (D0ED1) with 2.5 % of chlorine dioxide in D1. The results in Table 4 show that both XynG1-1 and XynG1-1B43cc pretreatment indicated positive effects in the bleaching of cotton stalk pulp. XynG1-1 and XynG1-1B43cc pretreatment gained brightness levels of 73.39 and 78.95 %, however, the brightness of the control with no xylanase pretreatment was 69.66 % (Table 4). XynG1-1B43cc pretreatment resulted in a lower kappa number of 2.3 compared with that (4.21) of XynG1-1 pretreatment (Table 4). The paper strength parameters were similar after pretreating by the two xylanases (Table 4). Besides, XynG1-1B43cc pretreatment at 70 °C and pH 9.0 resulted in similar brightness, kappa number, and paper strength parameters to those of pulps pretreated by XynG1-1R at 60 °C and pH 8.0 [27]. However, the wild-type XynG1-1 pretreatment resulted in lower brightness and higher kappa number at 70 °C and pH 9.0 compared with those of pulps pretreated by XynG1-1B43cc (Table 4). All of the results indicated that the mutant xylanase XynG1-1B43cc is appropriate for biobleaching in the paper industry.
Conclusion
Protein engineering of xylanase XynG1-1 from P. campinasensis G1-1 using directed evolution in combination with site-directed mutagenesis revealed a series of mutants with enhanced alkali or thermal tolerance. The mutant XynG1-1B43cc16 with the optimum pH at 9.0 and the optimum temperature at 70 °C is potentially useful for the paper industry. Moreover, the mutants generated in this study will be instrumental for further research on the relationship between the structure and function of xylanases.
References
Arnold K, Bordoli L, Kopp J, Schwede T (2006) The SWISS-MODEL workspace: a Web-based environment for protein structure homology modelling. Bioinformatics 22(2):195–201
Bailey MJ, Biely P, Poutanen K (1992) Interlaboratory testing of methods for assay of xylanase activity. J Biotechnol 23(3):257–270
Balaa BA, Brijs K, Gebruers K, Vandenhaute J, Wouters J, Housen I (2009) Xylanase XYL1p from Scytalidium acidophilum: site-directed mutagenesis and acidophilic adaptation. Bioresour Technol 100(24):6465–6471
Beg QK, Kapoor M, Mahajan L, Hoondal GS (2001) Microbial xylanases and their industrial applications: a review. Appl Microbiol Biotechnol 56(3–4):326–338
Birijlall N, Manimaran A, Santhosh Kumar K, Permaul K, Singh S (2011) High-level expression of a recombinant xylanase by Pichia pastoris NC38 in a 5L fermenter and its efficiency in biobleaching of bagasse pulp. Bioresour Technol 102(20):9723–9729
Collins T, Gerday C, Feller G (2005) Xylanases, xylanase families and extremophilic xylanases. FEMS Microbiol Rev 29(1):3–23
Guex N, Peitsch MC (1997) SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18(15):2714–2723
Harris GW, Pickersgill RW, Connerton I, Debeire P, Touzel JP, Breton C, Perez S (1997) Structural basis of the properties of an industrially relevant thermophilic xylanase. Proteins 29(1):77–86
Ko EP, Akatsuka H, Moriyama H, Shinmyo A, Hata Y, Katsube Y, Urabe I, Okada H (1992) Site-directed mutagenesis at aspartate and glutamate residues of xylanase from Bacillus pumilus. Biochem J 288(Pt 1):117–121
Liu YH, Lu FP, Li Y, Wang JL, Gao C (2008) Acid stabilization of Bacillus licheniformis alpha amylase through introduction of mutations. Appl Microbiol Biotechnol 80(5):795–803
Liu YH, Lu FP, Li Y, Yin XB, Wang Y, Gao C (2008) Characterisation of mutagenised acid-resistant alpha-amylase expressed in Bacillus subtilis WB600. Appl Microbiol Biotechnol 78(1):85–94
Matthews BW (1993) Structural and genetic analysis of protein stability. Annu Rev Biochem 62:139–160
Miller GL (1959) Use of dinitrosalicylic acid reagent for determination of reducing sugar. Anal Chem 13:426–428
Morris DD, Gibbs MD, Chin CW, Koh MH, Wong KK, Allison RW, Nelson PJ, Bergquist PL (1998) Cloning of the xynB gene from Dictyoglomus thermophilum Rt46B.1 and action of the gene product on kraft pulp. Appl Environ Microbiol 5(64):1759–1765
Morris DD, Gibbs MD, Ford M, Thomas J, Bergquist PL (1999) Family 10 and 11 xylanase genes from Caldicellulosiruptor sp. strain Rt69B.1. Extremophiles 3(2):103–111
Nakamura S, Nakai R, Namba K, Kubo T, Wakabayashi K, Aono R, Horikoshi K (1995) Structure-function relationship of the xylanase from alkaliphilic Bacillus sp. strain 41 M-1. Nucleic Acids Symp Ser 34:99–100
Nishimoto M, Kitaoka M, Hayashi K (2002) Employing chimeric xylanases to identify regions of an alkaline xylanase participating in enzyme activity at basic pH. J Biosci Bioeng 94(5):395–400
Schwede T, Kopp J, Guex N, Peitsch MC (2003) SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res 31(13):3381–3385
Shao Q, Gao YQ (2011) The relative helix and hydrogen bond stability in the B domain of protein A as revealed by integrated tempering sampling molecular dynamics simulation. J Chem Phys 135(13):135102
Shirai T, Suzuki A, Yamane T, Ashida T, Kobayashi T, Hitomi J, Ito S (1997) High-resolution crystal structure of M-protease: phylogeny aided analysis of the high-alkaline adaptation mechanism. Protein Eng 10(6):627–634
Turunen O, Vuorio M, Fenel F, Leisola M (2002) Engineering of multiple arginines into the Ser/Thr surface of Trichoderma reesei endo-1,4-beta-xylanase II increases the thermotolerance and shifts the pH optimum towards alkaline pH. Protein Eng 15(2):141–145
Umemoto H, Ihsanawati Inami M, Yatsunami R, Fukui T, Kumasaka T, Tanaka N, Nakamura S (2009) Improvement of alkaliphily of Bacillus alkaline xylanase by introducing amino acid substitutions both on catalytic cleft and protein surface. Biosci Biotechnol Biochem 73(4):965–967
Wakarchuk W, Sung W, Campbell R, Cunningham A, Watson D, Yaguchi M (1994) Thermostabilization of the Bacillus circulans xylanase by the introduction of disulfide bonds. Protein Eng 7:1379–1386
Yang HM, Yao B, Meng K, Wang YR, Bai YG, Wu NF (2007) Introduction of a disulfide bridge enhances the thermostability of a Streptomyces olivaceoviridis xylanase mutant. J Ind Microbiol Biotechnol 34(3):213–218
Yazawa R, Takakura J, Sakata T, Ihsanawati, Yatsunami R, Fukui T, Kumasaka T, Tanaka N, Nakamura S (2011) A calcium-dependent xylan-binding domain of alkaline xylanase from alkaliphilic Bacillus sp. strain 41 M-1. Biosci Biotechnol Biochem 75(2):379–381
Zhang Z-G, Yi Z-L, Pei X-Q, Wu Z-L (2010) Improving the thermostability of Geobacillus stearothermophilus xylanase XT6 by directed evolution and site-directed mutagenesis. Bioresour Technol 101(23):9272–9278
Zheng H, liu Y, Liu X, Han Y, Wang J, Lu F (2012) Overexpression of a Paenibacillus campinasensis xylanase in Bacillus megaterium and its applications to biobleaching of cotton stalk pulp and saccharification of recycled paper sludge. Bioresour Technol 125:182–187
Zheng H, Liu Y, Liu X, Wang J, Han Y, Lu F (2012) Isolation, purification and characterization of a thermostable xylanase from a novel strain Paenibacillus campinasensis G1-1. J Microbiol Biotechnol 22(7):930–958
Acknowledgments
Financial support from the National High Technology Research and Development Program of China (Grant 2013AA102106), Program for Changjiang Scholars and Innovative Research Team in University (Grant IRT1166), the National Natural Science Fund (Grant 31,101,219) and China Postdoctoral Science Fund (Grant 2013M540076) are gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Additional information
H. Zheng and Y. Liu, who contributed equally to this work, are both nominated as the first author.
Rights and permissions
About this article
Cite this article
Zheng, H., Liu, Y., Sun, M. et al. Improvement of alkali stability and thermostability of Paenibacillus campinasensis Family-11 xylanase by directed evolution and site-directed mutagenesis. J Ind Microbiol Biotechnol 41, 153–162 (2014). https://doi.org/10.1007/s10295-013-1363-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-013-1363-6