
Abstract
The medicinal herb Artemisia annua L. is prized for its capacity to generate artemisinin, which is used to cure malaria. Potentially influencing the biomass and secondary metabolite synthesis of A. annua is plant nutrition, particularly phosphorus (P). However, most soil P exist as insoluble inorganic and organic phosphates, which results to low P availability limiting plant growth and development. Although plants have developed several adaptation strategies to low P levels, genetics and metabolic responses to P status remain largely unknown. In a controlled greenhouse experiment, the sparingly soluble P form, hydroxyapatite (Ca5OH(PO4)3/CaP) was used to simulate calcareous soils with low P availability. In contrast, the soluble P form KH2PO4/KP was used as a control. A. annua’s morphological traits, growth, and artemisinin concentration were determined, and RNA sequencing was used to identify the differentially expressed genes (DEGs) under two different P forms. Total biomass, plant height, leaf number, and stem diameter, as well as leaf area, decreased by 64.83%, 27.49%, 30.47%, 38.70%, and 54.64% in CaP compared to KP; however, LC–MS tests showed an outstanding 37.97% rise in artemisinin content per unit biomass in CaP contrary to KP. Transcriptome analysis showed 2015 DEGs (1084 up-regulated and 931 down-regulated) between two P forms, including 39 transcription factor (TF) families. Further analysis showed that DEGs were mainly enriched in carbohydrate metabolism, secondary metabolites biosynthesis, enzyme catalytic activity, signal transduction, and so on, such as tricarboxylic acid (TCA) cycle, glycolysis, starch and sucrose metabolism, flavonoid biosynthesis, P metabolism, and plant hormone signal transduction. Meanwhile, several artemisinin biosynthesis genes were up-regulated, including DXS, GPPS, GGPS, MVD, and ALDH, potentially increasing artemisinin accumulation. Furthermore, 21 TF families, including WRKY, MYB, bHLH, and ERF, were up-regulated in reaction to CaP, confirming their importance in P absorption, internal P cycling, and artemisinin biosynthesis regulation. Our results will enable us to comprehend how low P availability impacts the parallel transcriptional control of plant development, growth, and artemisinin production in A. annua. This study could lay the groundwork for future research into the molecular mechanisms underlying A. annua’s low P adaptation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Recent attempts to integrate phosphorus (P) in terrestrial biosphere models (Wang et al. 2010; Thum et al. 2019) show that P is crucial for the Earth’s biogeochemical cycles, particularly the carbon (C) cycle. P is a component of membrane phospholipids and RNA, and its metabolic roles include the transfer and storage of energy and energizing binding sites for metabolic turnover (Rouached et al. 2010; Veneklaas et al. 2012). P impacts the organisms at all levels of the organization, from the individual to the community (Vitousek et al. 2010). Despite being plentiful in soil, phosphate (P) mainly occurs as sparsely soluble phosphate compounds via absorbing aluminum (Al) and iron (Fe) exposed to the surfaces of soil elements in acidic soils or to calcium (Ca) in calcareous soils (Wan et al. 2018). Plant bioavailability of these insoluble phosphates is often poor (Behera et al. 2014). P, a crucial macronutrient for plant growth and development, is usually a limiting issue for plants due to its inadequate availability in natural contexts (Augusto et al. 2017), which is being acknowledged more and more (Vitousek et al. 2010; Wieder et al. 2015; Vicca et al. 2018). Inorganic soluble P fertilizer is typically the primary choice to remedy soil P shortage to achieve high crop yields and high-quality products (Jalali and Jalali 2020).
Certainly, a variety of adaptive strategies to cope with P limitation have developed in plants, including root structural specializations (Williamson et al. 2001; Hu et al. 2010) exudation of compounds (Ryan et al. 2001; Shen et al. 2003), mycorrhizal symbioses (Smith and Smith 2011; Wan et al. 2018), modulation of C metabolism bypassing steps that require P, perfusion of organic acids, increasing production and secretion of phosphatase, and enhancing Pi transporters expression (Raghothama 1999; Vance et al. 2003; Plaxton 2004), which leads to an increase in the bioavailability of insoluble phosphate to plants. In addition, plant bioavailability of insoluble phosphate can also be affected by soil environmental changes, such as changes in pH (Hinsinger 2001). However, little is known about genetics and metabolic mechanisms for low P adaptation in plants.
In recent years, different sequencing technologies supported us with essential knowledge about P starvation and its responsive genes expression globally in several plant species and organs, including Arabidopsis thaliana leaves, shoots, and roots (Hammond et al. 2003; Wu et al. 2003; Misson et al. 2005), Lupinus albus proteoid roots (Uhde-Stone et al. 2003), and Oryza sativa roots and leaves (Wasaki et al. 2003a, 2006). According to transcriptome analysis of barley, many genes were dramatically up- or down-regulated by low P stress. The differentially expressed genes (DEGs) were mainly involved in P metabolisms like phosphorylation/dephosphorylation, post-transcriptional regulation, sucrose synthesis, hydrolysis of phosphoric enzymes, and phospholipid degradation (Ren et al. 2018). Furthermore, under low-P treatment, DEGs were used in the tricarboxylic acid cycle, oxidation–reduction process, biosynthetic process, and the carbohydrate metabolic process of oat roots (Chao et al. 2017). Transcriptome analysis was used to study DEGs under low-P stress in different crops, such as soybean (Liu et al. 2020), rice (Ding et al. 2016), and maize (Du et al. 2016) to better understand these processes. According to the studies’ findings above, transcriptome analysis can reveal additional details about the gene regulation behind plants’ low P adaption.
Artemisia annua, also known as sweet wormwood, sweet annie, sweet sagewort, annual mugwort, or annual wormwood, is an annual herb that produces artemisinin, a sesquiterpene molecule used to treat fever and malaria (Ma et al. 2007; Baraldi et al. 2008). It is prevalent throughout much of China, particularly in the southwest (Zhang et al. 2017). It has no particular nutritional needs and adapts well to many soil types. However, even in small amounts, potassium (K) and P supplies inhibited development (Müller and Brandes 1997; Aftab et al. 2013). Optimizing P to A. annua is important for increasing the output of artemisinin and/or dry matter (Todeschini et al. 2022). Numerous research studies have investigated how plants can absorb P from different forms that are sparingly soluble (Pearse et al. 2007; Lee et al. 2012; Giles et al. 2014; Li et al. 2015; Bhattacharya 2019; Lambers 2022). Little effort is being made to comprehend how plants adapt to decreasing P supplies. Todeschini et al. (2022) demonstrated that P nutrition influences the biomass production of A. annua and that the lowest amount of P results in the maximum artemisinin concentration. For more knowledge about the genetic behavior of our plant in response to low P availability, a controlled greenhouse experiment was conducted using sparingly soluble P form, calcium phosphate hydroxyl apatite Ca5(OH, F)(PO4)3/CaP to simulate the calcareous soils with low P availability compared to the soluble P form KH2PO4/KP as a control. Ca5(OH, F)(PO4)3 is by far the most abundant phosphate mineral in nature (Kumari and Phogat 2008). As the rock phosphate (Ca5(OH, F)(PO4)3) is a relatively sparingly soluble (insoluble) material, its geometric surface area has a significant bearing on its rate of dissolution in soil (Kumari and Phogat 2008). Then, we explored the A. annua genetics and metabolic response to the sparingly soluble P forms Ca5OH(PO4)3 via second-generation sequencing analysis. This research can help us better understand the genetic variety of A. annua in low-phosphorus environments and offer ideas for how to increase their P usage efficiency and produce more artemisinin and biomass with less fertilizer.
Material and method
Plant materials
The Artemisia annua seeds were gathered from the region used for planting in the Guangxi Medicinal Botanical Garden scientific research center in Nanning, China (108°23′ E, 22°51′ N). A. annua seeds were planted in an acrylic container (30 cm length × 20 cm width × 8 cm height) filled with thoroughly cleaned driving sand. The sand was watered every two days with distilled water to keep it moist during germination at Guangxi Botanical Garden of Medicinal Plants. Weekly seedlings were cultivated using 200 mL of pH 6.5 half-strength Hoagland nutrition solution. The two cotyledon seedlings were all of a similar size and awaited two phosphorus treatments.
Different phosphorus treatments
In this investigation, two P sources were employed; an insoluble inorganic P source, calcium phosphate hydroxyapatite (Ca5OH(PO4)3/CaP), in which water solubility is only 0.3 mg/mL at 20 ℃, and a soluble inorganic P source: monopotassium phosphate (KH2PO4/KP), in which water solubility is 222 mg/mL at 20 ℃.The resulting two treatments were replicated five times. The CaP was treated with decreased P availability, while the KP served as the control group: five copies of each group were made. In this experiment, rivers were employed as cultivation substrates. Before the treatment experiment, the river sand was sieved through a 2-mm screen, rinsed with running water to remove nutrients, and then autoclaved for 30 min to sterilize it. The flower pot, which was intended to store river sand, had a diameter of 4 cm and a height of 12 cm. 162.19 mg Ca5OH(PO4)3 and 131.83 mg KH2PO4 were added in powder form and appropriately blended with the cleaned river sand to make sure each flower pot contained 30 mg P content. Following transplanting, every flower pot was placed in a greenhouse with a day time light intensity of 300 mol m−2 s−1, a temperature of 26 °C, and a relative humidity of 62%. To counteract the effects of environmental variations, all of the flower pots had their placements altered at random. Every two days, adequate distilled water was provided to each flower pot, and every week, 5 mL of Hoagland nutritional solution (0.5x, without P) was added. Three months of P therapy was continued (Pearse et al. 2007; Wan et al. 2018).
Plant characteristic analysis and sample collection
The height, diameter of the stem, number of leaves, and area of the leaves of each plant were all measured before the seedlings were harvested after 90 days of two P source treatments. The leaf area was calculated using ImageJ software from the National Institutes of Health in the USA on a digital picture. Each flower pot had three fresh leaves that were taken out, wrapped in aluminum foil, promptly frozen in liquid nitrogen for 3 to 5 min, and then stored at − 80 °C until they could be utilized for RNA-Seq and real-time quantitative PCR (RT-qPCR). Following a 72-h drying period at 60 °C, the ultimate biomass of the roots, stems, and leaves was calculated.
Isolation of artemisinin and analysis by LC–MS
The leaves were dried at 60 °C and prepared for artemisinin analysis. Samples of 0.2 g dried A. annua leaves were ultrasonically extracted with 25 mL petroleum ether (boiling point 30–60) for 40 min, filtered, transferred to a 100-mL evaporation plate, and dried at 40 °C. Finally, we cover with methanol again.
Store all solutions in volumetric flasks and measure up to a fixed volume of 10 mL (Stringham et al. 2018). Samples were fixed using an Agilent Eclipse system equipped with an Acquity UPLC® XDB C18 column (5 μm, 4.6 × 250 mm). The mobile phase is 70% methyl (A), 30% water, and 0.1% formic acid (B). The following eluent values were used at a 1.0 mL/min flow rate.
RNA extraction and transcriptome sequencing
Following the manufacturer’s instructions, total RNA was isolated from frozen materials using the RNA prep Pure Plant Kit (TIANGEN, Beijing, China). The purity of the RNA was evaluated using a Kaiao K5500® Spectrophotometer (Kaiao, Beijing, China). RNA integrity and concentration were assessed using the RNA Nano 6000 Assay Kit of the Bioanalyzer 2100 system (Agilent Technologies, CA, USA). RNA degradation was observed (El-Sappah et al. 2023).
Enrich mRNA was obtained from total RNA using oligo (dT) magnetic beads to construct the cDNA libraries. The fragmented mRNA was subjected to first-strand synthesis using a random hexamer primer and RNase H. After purification of the library fragments using QiaQuick PCR kits and elution with EB buffer, terminal repair, A-tailing, and adapter ligation were performed. Subsequently, second-strand cDNA was synthesized using DNA polymerase I, buffer, dNTPs, and RNase H. The resulting library was amplified using PCR and assessed for quality using an Agilent Bioanalyzer 2100 instrument. Six libraries were sequenced by BioNovoGene Technology Co., Ltd. (Suzhou, China) using an Illumina HiSeq 2500.
Quantitative real-time PCR (qRT-PCR) analysis
EASYspin Plus Plant RNA Kit was used for total RNA extraction from leaf samples, following the manufacturer’s protocols. Then, 500 ng of total RNA, a gDNA eraser, and a PrimeScript RT kit were used for synthesized cDNA, diluted tenfold, and used as a qRT-PCR template (El-Sappah et al. 2021a). All primers, including β-actin as reference genes, were designed with Primer Premier 5.0. Following the manufacturer’s protocols, the qRT-PCR was performed using TB Green (TaKaRa) on a CFX 96 real-time PCR system (BioRad, Hercules, CA, USA). A 20-L volume was used for PCR amplification, which included 100 ng cDNA, 0.6 L of each primer with a total of 10 mol, and a 10 L PCR mix (2). The following conditions for all reactions: 30 s/95 °C, 40 cycles of 10 s/95 °C, and 30 s/55 °C, and the melting curve were generated to confirm the PCR specificity (El-Sappah et al. 2021b).The non-treated control treatment was used to standardize all samples, and the relative expression levels were calculated using the 2 −ΔΔCt method (Livak and Schmittgen 2001).
Statistical analysis
The one-way ANOVA was performed using the SPSS Statistics 19.0 software (IBM Corp, Armonk, NY, USA), and the Duncan test was used to determine significant differences (P < 0.05). Data were presented as mean ± SD (standard deviation). The sample was sequenced on the machine, and the sequencing platform’s software generated the raw data (RAW Data) of FASTQ, which is the off-machine data. The raw data from each sample was used for statistical analysis. The 3′ end connector sequence was removed with Cutadapt, and the filtered reads were compared to the reference genome using the HISAT2 software. The read distribution was statistically divided into CDS (coding region), intron (intron), and soon. The Read Count value on each gene was compared to the gene’s original expression using HTSeq statistics, which included three statistical schemes. RSeQC was used to analyze expression saturation, while DESeq was used to analyze gene expression differences and screen for various genes. GO enrichment analysis was performed using top-GO.
Results
Growth parameter assessment and artemisinin concentration
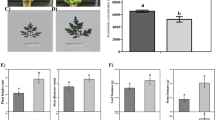
Plant growth factors such as plant height, leaf number, leaf area, and stem diameter were severely reduced by 27.49%, 30.47%, 38.70%, and 54.64%, respectively, under CaP treatment compared to KP (Fig. 1A–E). Additionally, CaP resulted in significantly lower leaf, stem, and root biomass compared to KP, leading to an overall reduction of 64.83% in A. annua biomass (Fig. 1F). Interestingly, in contrast to KP, the artemisinin content per unit biomass showed a significant increase of 37.97% under CaP treatment (Fig. 1G).

Pant morphology and growth parameter assessment in response to the insoluble phosphorus resource (CaP) compared to KP as a soluble resource. A Plant morphology showed plant height under CaP fertilizer. Plant morphology showed the B height of plant under KP fertilizer, C leaf size area of CaP′ plants, D leaf area of KP′ plant, E morphological traits, and F biomass measurements. G The artemisinin concentration (mg kg.−1) in response to the insoluble phosphorus resource (CaP) compared to the soluble resource (KP)
Sequencing and differentially expressed gene (DEG) analysis of transcriptomes
The A. annua leaves from the KP treatment, which served as the control, and the CaP treatment, which resulted in low P availability, were used to build the cDNA libraries for creating the transcriptome sequences. From the KP and the CaP, raw data from paired-end sequencing-by-synthesis produced 44, 281, 438, 47, 866, and 365 reads, respectively. The most excellent read length for all samples was 7, 396, 967, and 700 bp. For subsequent assembly, 40,888,780 and 44,252,874 of the KP and the CaP, were kept after quality check and raw reading data processing. Using Trinity with a hash size of 25, filtered readings were combined, and transcripts were produced. The assembly acquired 39,065 transcripts for the KP vs. the CaP, with the average transcript length for the KP vs. the CaP being 291.31 bp (Table S1). The assembled transcript from different replicates had varying numbers, which could be attributed to variable P absorbed by the plants or noises caused by technology at some stage during the sequencing process. Figure 2 shows the distribution pattern of these transcripts. RNA-seq experiment variability was evaluated using the principal component analysis (PCA).

RNA-seq and DEG distribution overview. A The RNA-seq output PCA based on the transcriptome-wide gene expression profiles. Distances between samples reflect differences in transcriptome profiles. B Volcano plots depicting differentially expressed transcripts. Each dot represents a DEG; dots above the red line reflect significant DEGs (P < 0.05)
According to the PCA results (Fig. 2A), PC1 accounted for 28.1% of the apparent transcript expression variance across the three samples at the two treatments. Comparing the KP and CaP yielded a total of 2015 DEGs (1084 up-regulated and 931 down-regulated), which were then identified. Transcripts exclusive to the control treatment and the low P availability therapy, as well as those that are down-regulated and up-regulated, were analyzed for overlapping to find common transcripts in DGE data (Fig. 2B).
Gene function annotation and classification
Using the COG databases, all DEGs were functionally classified. Based on sequence homology, 1884 DEGs were divided into 25 groups (Fig. 3). Among these categories, the largest group was function unknown (457 DEGs, 20.99%), followed by general function prediction only (419 DEGs, 19.25%), signal transduction mechanisms (225 DEGs, 10.34%), posttranslational modification, protein turnover, chaperones (168 DEGs, 7.72%), transcription (129 DEGs, 5.93%), carbohydrate transport and metabolism (96 DEGs, 4.41%), and secondary metabolite biosynthesis, transport, and catabolism (92 DEGs, 4.23%).

COG function classification analysis of the metabolic pathway
GO enrichment analysis showed that 1044 of 2015 DEGs were assigned to 110 GO terms (P value < 0.05), divided into three categories: only 11 GO terms belonging to cellular components, 47 to molecular function, and 52 to biological process. The top 20 enriched GO terms are shown in (Fig. 4), with the most significant GO term being catalytic activity (GO: 003824) and the second GO term being ion binding (GO: 0043167) from the molecular function category (Table S2).

GO enrichment study of all DEGs associated with the CaP phosphorus resource. The top twenty enhanced GO words are displayed. The horizontal axis represents the rich factor, while the vertical axis represents the GO words. Number: the number of DEGS; Padjust: the corrected P value
Furthermore, the three GO terms within the biological process were L-glutamate transport (GO:0015813), glucosinolate metabolic process (GO:0019760), and glucose transmembrane transport (GO:1,904,659). A study of KEGG pathway enrichment was done to describe enriched biological pathways (Fig. 5, Table S3).

KEGG enrichment analysis of DEGs under CaP phosphorus resource. The most enriched KEGG pathways at KP_vs_CaP are presented. The horizontal axis represents the rich factor, while the vertical axis represents the pathway names. Number: DEGs number; Padjust: adjusted P value
In the KEGG annotated results, out of 2015 DEGs, 802 were annotated into 114 different biological pathways, categorized into five main categories and 19 subcategories. The majority of the subcategories were related to carbohydrate metabolism (12.28%) and lipid metabolism (12.28%), followed by amino acid metabolism (9.65%), metabolism of cofactors and vitamins (7.89%), and biosynthesis of other secondary metabolites (6.14%). Figure 5 displays the top 20 abundant biochemical pathways with the respective numbers of assigned DEGs. Among the DEGs showing significant differential expression, several were involved in pathways like glyoxylate and dicarboxylate metabolism (ko00630), circadian rhythm-plant (ko04712), and flavonoid biosynthesis (ko00941).
The results of COG, GO, and KEGG enrichment analyses suggested that the DEGs between the CaP and the KP are mainly related to genes involved in carbohydrate metabolism, secondary metabolites biosynthesis, enzyme catalytic activity, circadian rhythm-plant, and signal transduction, and so on.
Analysis of DEGs involved in carbohydrate metabolism
The functions of genes differentially expressed between the CaP and the KP were centered around carbohydrate metabolic networks (Fig. 6).

Carbohydrate metabolism. The red shapes represent experimental up-regulation genes while the blue shapes represent experimental down-regulation genes
Compared to the KP, 18 genes were significantly up-regulated in the CaP, including four BGL genes (beta-glucosidase), one GUN gene (endoglucanase), one INV gene (fructan 6-exohydrolase), two glgC genes (glucose-1-phosphate adenylyltransferase), one gpmI gene (2,3-bisphosphoglycerate-independent phosphoglycerate mutase), one PGAM gene (2,3-bisphosphoglycerate-dependent phosphoglycerate mutase), one ENO gene (enolase), one DLD gene (dihydrolipoamide dehydrogenase), one DLAT gene (dihydrolipoamide acetyltransferase), one CS gene (citrate synthase), three ACO genes (aconitate hydratase), and one SUCB gene (succinate-CoA ligase). Moreover, six genes were significantly down-regulated in the CaP, such as two BAM genes (beta-amylase), one PYG gene (glucan phosphorylase), one pfp gene (pyrophosphate-fructose 6-phosphate 1-phosphotransferase), one ALDO gene (fructose-bisphosphate aldolase), and one PK gene (pyruvate kinase).
Analysis of DEGs involved in flavonoid biosynthesis
The transcript levels of flavonoid biosynthesis genes, including one C4H gene (cinnamate-4-hydroxylase), two CHS genes (chalcone synthase), and two CCOAOMT genes (caffeoyl-CoA O-methyl transferase) were much higher in the CaP than in the KP. Moreover, one PAL gene (phenylalanine ammonia-lyase) was down-regulated in the CaP. However, five HCT genes (shikimate O-hydroxycinnamoyl transferase) were significantly up-regulated or down-regulated in the CaP and the KP (Fig. 7).

Flavonoid biosynthesis. The red shapes represent experimental up-regulation genes, the blue shapes represent experimental down-regulation genes, and the yellow forms represent both experimental and control up-regulation genes
Analysis of DEGs involved in P metabolism
Four genes involved in P transportation, one PHT gene (phosphate transporter), one SLC25A44 gene (solute carrier family 25, member 44), and two SPX genes (SPX domain protein) were significantly up-regulated in the CaP. In addition, nine genes encoding phosphomonoesterases, including five PPA genes (purple acid phosphatase), two PLA genes (phospholipase A), and two PLD genes (phospholipase D) were all significantly up-regulated in the CaP.
Analysis of DEGs involved in hormone biosynthesis and signal transduction
After CaP treatment, both RNA-Seq data and qRT-PCR analysis revealed differential expression of several genes involved in phytohormone, carotenoid, tryptophan, and linolenic acid signaling (Fig. 8B, C).

A The KEGG pathway of plant hormone signal transduction in response to an insoluble phosphorus supply (CaP). The red shapes represent experimental up-regulation genes, the blue shapes represent experimental down-regulation genes, and the yellow forms represent both experimental and control up-regulation genes. B Results of RNA sequencing validation of identified genes using qRT-PCR. C Heatmaps showing variations in gene expression in hormone signal transduction biosynthesis under CaP and KP treatments
DEGs 5, 7, and 4 exhibited differential expression in the carotenoid, tryptophan, and linolenic acid metabolic pathways. Within the tryptophan biosynthesis pathway, the expression levels of YUCCA (CTI12_AA189550 and CTI12_AA526120) were up-regulated. Additionally, the expression of AUX (CTI12_AA007170), involved in auxin signal transduction, showed a significant increase in plant cells. Furthermore, two SAUR (CTI12_AA189550 and CTI12_AA526120) were up-regulated, while the three genes (CTI12_AA421350, CTI12_AA448510, and CTI12_AA404350) were down-regulated (Fig. 6B, C). For multiple type-B response regulators family (ARR-B) genes involved in cytokinin (zeatin) signal transduction, one ARR-B (CTI12_AA069810) was significantly down-regulated in response to CaP stress. Also, BSK (CTI12_AA380960), which is involved in brassinosteroid signal transduction and mediates the signal transduction from the receptor kinase BRI1, was down-regulated. Furthermore, the three abscisic acid-related genes PYL, PP2C, and ABF, which are involved in signal transduction; COI and JAZ, which are associated with jasmonic acid; and NPR1, which is related to salicylic acid, all showed differential expression. Two PYL (CTI12_AA441590 and CTI12_AA064610), two PP2C (CTI12_AA621200 and CTI12_AA166960), one ABF (CTI12_AA367130), two JAZ (CTI12_AA385850 and CTI12_AA276470), and one NPR1 (CTI12_AA562790) gene were significantly up-regulated in response to CaP stress. At the same time, one PYL (CTI12_AA335910) and one COI (CTI12_AA380950) genes were down-regulated in response to CaP stress. Finally, at diterpenoid biosynthesis, three genes (CTI12_AA491890, CTI12_AA083800, and CTI12_AA510120) showed high expression with CaP treatments (Fig. 8).
Analysis of DEGs involved in the artemisinin biosynthesis pathway
Our transcriptome analysis identified several differentially expressed genes (DEGs), including those involved in artemisinin biosynthesis. Among the 31 artemisinin biosynthesis-related structural genes analyzed, their expression profiles were further investigated using qRT-PCR (Fig. 9).

A Artemisinin backbone biosynthesis KEGG in response to the insoluble phosphorus resource (CaP). B Validation of RNA sequencing results by qRT-PCR of selected genes. C Heatmaps showing variations in the expression of genes involved in artemisinin biosynthesis under CaP and KP treatments
Artemisinin biosynthesis occurs through two distinct pathways: the mevalonate (MVA) pathway in the cytosol and the methylerythritol phosphate (MEP) pathway in the plastid (Fig. 9A). In the MEP pathway, the deoxy-D-xylulose-5-phosphate synthase (DXS) (CTI12_AA379900) and geranyl diphosphate synthase (GPPS) (CTI12_AA281200) genes were significantly up-regulated. Conversely, the 4-hydroxy-3-methylbut-2-en-1-yl diphosphate reductase (HMDR) showed down-regulation in response to CaP treatments. In the MVA pathway, the diphosphomevalonate decarboxylase (MVD) (CTI12_AA318610) and geranylgeranyl diphosphate synthase (GGPS) (CTI12_AA137060) exhibited up-regulation. Both pathways converge when isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP) combine to form farnesyl diphosphate (FPP). Several genes, including those from the cytochrome P450 monooxygenase (CYP) family, displayed differential expression under CaP treatment in the processes leading from farnesyl diphosphate to artemisinin synthesis. Eleven of CYP family (CTI12_AA566140, CTI12_AA083800, CTI12_AA110770, CTI12_AA329710, CTI12_AA078680, CTI12_AA111420, CTI12_AA493230, CTI12_AA416010, CTI12_AA510120, CTI12_AA491890, and CTI12_AA024600) were significantly up-regulated while 14 of CYP family such as CTI12_AA210550 and CTI12_AA005010 were down-regulated. The aldehyde dehydrogenase (ALDH) (CTI12_AA521530) enzyme catalyzes the conversion from dihydroartemisinic aldehyde to dihydroartemisinic acid was up-regulated (Fig. 9B, C).
Identification of TFs related to CaP treatment
There were 72 TFs that altered significantly in response to CaP in order to better understand the mechanism governing transcriptional regulation in A. annua under various phosphorus form treatments (Fig. 10A).

TF family distribution. A The total distribution of TFs in A. annua plant, where the horizontal axis represents the names of all 57 annotated TF families, while the vertical axis represents the number of gene members, B The up-regulated TFs in response to different phosphorus (Table S4), and C the down-regulated TFs in response to different phosphorus treatments (Table S5)
Using the Plant TFDB database, the 39 differentially expressed TF families were divided into 18 down-regulated TF families (Fig. 10C; Table S5) and 21 up-regulated TF families (Fig. 10B; Table S4). Among these six-membered transcription families, C3H and WRKY were the most notable up-regulated families. The two families with the most members among the down-regulated TFs were MYB and WRKY, which each had ten members (Fig. 10).
Discussion
In our study, the P source used in the CaP was calcium phosphate hydroxyl apatite (Ca5OH(PO4)3, which its water solubility is only 0.3 mg/mL at 20 ℃, while the P source used in the KP was monopotassium phosphate KH2PO4, which its water solubility is 222 mg/mL at 20 ℃. Thus, the P availability of the CaP is significantly lower than that of the KP. Therefore, we also found that there were significant decreases in morphological characteristics and growth responses of A. annus grown in the CaP in contrast to the KP (Fig. 1). The significant effects of P availability on plant growth and development have been shown in a variety of research (Malhotra et al. 2018; Todeschini et al. 2022) For instance, the study of Todeschini et al. (2022) showed the significant effect of different P nutrition in A. annua biomass production, which is similar to our findings. A particular amount of P fertilizer treatment can also boost faba bean output; however, going beyond that amount causes yield reductions. Therefore, we conclude that differences in P availability between the CaP and the KP are an essential factor determining the growth and development of A. annus.
It has been well reported that P is one of the most important nutrient elements involved in many biological processes during plant growth and development (Kvakić et al. 2020; Amarasinghe et al. 2022), such as the creation of nucleic acids (DNA and RNA), glycolysis, photosynthesis, enzyme activation and inactivation, cellular signaling, and carbohydrate metabolism (Vance et al. 2003; Shen et al. 2011; Malhotra et al. 2018; Todeschini et al. 2022). However, very little is known about the availability of various P and the genes that control them in A. annua. Therefore, identifying the genes under various P circumstances is extremely important. Therefore, we conducted a transcriptome study to learn more about the genetic behavior under the hardly soluble P resource and identify the genes that are in charge of these results. Then, 2015 DEGs in total (1084 up-regulated and 931 down-regulated) were found in our study when the CaP and KP were compared. The DEGs between the CaP and the KP are primarily associated with genes involved in glucose metabolism, enzyme catalytic activity, circadian rhythm-plant, secondary metabolites biosynthesis, and signal transduction, according to our COG, GO, and KEGG enrichment analyses.
Three primary mechanisms are involved in carbohydrate metabolism: the tricarboxylic acid (TCA) cycle, glycolysis, and starch and sucrose metabolism. Several enzyme genes engaged in starch synthesis play crucial roles in starch and sucrose metabolism. Up-regulation of glgC, a major enzyme in starch synthesis, and down-regulation of PYG and BAM indicate starch buildup in leaves under low P availability conditions. Research studies on rice and other plants’ chloroplasts have demonstrated starch accumulation under low P availability conditions (Fredeen et al. 1990; Qiu and Israel 1992; Usuda and Shimogawara 1992; Ciereszko et al. 2001). It has been proposed that starch accumulation in leaves under low P availability is caused by the disruption of triose phosphate export from the stroma due to decreased P concentration (Natr 1992). Natr (1992) also observed P release through enhanced starch synthesis. Given that starch synthesis and P-utilizing enzyme induction are synchronized, it is reasonable to speculate that starch accumulation in leaves with low P availability results from maintaining internal P concentration. Moreover, several up-regulated enzyme genes, such as BGL, GUN, and INV, are involved in converting polysaccharides (glucoside, cellulose, and sucrose) to glucose, which accelerates carbohydrate metabolism, leading to decreased carbohydrate content and increased glucose content. Similar up-regulation of enzyme genes associated with polysaccharide conversion was observed in Scenedesmus sp. and Phaeodactylum tricornutum under low P availability. Zhang et al. (2014) also reported increased starch levels and decreased sucrose levels in response to P deficit. These findings suggest that adaptations allowing leaves to sustain Pi levels under low phosphorus stress include storing starch in maize chloroplasts and limiting sucrose synthesis.
The glycolysis process can transform glucose into acetyl-CoA. As a result, we discovered that the expression of genes encoding pfp and ALDO reduced in the glycolysis pathway, which may have indicated that the conversion of glyceraldehyde-3P to carbohydrates was repressed. While the expression level of genes encoding gpmI, PGAM, ENO, DLAT, and DLD increased, catalyzing acetyl-CoA formation. These findings indicate that carbohydrate conversion to acetyl-CoA might be promoted under conditions of low P availability. While acetyl-CoA is the starting point for the TCA cycle, it follows that the accumulation of acetyl-CoA in the glycolysis pathway provides a substrate for the TCA cycle. We also found that all enzyme genes in the TCA cycle were up-regulated, including one CS gene, three ACO genes, and one SUCB gene. Citrate synthase is the rate-limiting enzyme for the condensation reaction of oxaloacetate and acetyl-CoA (Fig. 7). This result is similar to previous results, that is, low P availability induces the gene expression of key enzymes in the TCA cycle (Wasaki et al. 2003b; Wang et al. 2012; Yang et al. 2019). The up-regulated expression of these enzyme genes involved in starch and sucrose metabolism, glycolysis, and TCA cycle could lead to the control of energy metabolism, which is well known to produce energy and NADPH. Therefore, our results indicate that under low P availability conditions, controlling energy metabolism in A. annua is likely to be very important by increasing the supply of ATP, NADH, and NADPH.
Our study conducted an enrichment analysis, revealing that flavonoid biosynthesis was a significantly enriched pathway in both CaP and KP treatments (Fig. 7). In response to low P availability conditions, the transcription levels of flavonol biosynthesis genes, including C4H, CHS, HCT, and CCOAOMT, increased in the CaP treatment. This likely led to an increase in flavonoid content in A. annua. Previous research has also shown that P stress affects the expression of genes related to flavonoid biosynthesis in apple trees. Flavonoids are low molecular weight secondary metabolites widespread in the plant community. Flavonols, a type of flavonoid, play crucial roles in various biological processes in plants, such as UV protection, resistance to pathogens, regulation of plant growth, and protection against oxidative stresses induced by drought (Nakabayashi et al. 2014), temperature (Catalá et al. 2011), and changes in nitrogen, P, or carbon nutrition (Fei et al. 2014; Shi and Xie 2014). These studies imply that flavonols are key regulators of plant biofunction under stress conditions (Xu et al. 2014; Martinez et al. 2016). Thus, A. annua responds to low P availability by regulating the expression of genes and metabolites in the flavonoid pathway to cope with the stress.
To cope with low P availability, plants can enhance P absorption and internal P cycling by regulating the expression of genes and enzymes involved in P metabolism pathways, such as phosphorus transporter, purple acid phosphatase, phospholipid degradation, phosphorylation/dephosphorylation, hydrolysis of phosphoric enzymes, and posttranscriptional regulation (Ren et al. 2018). Phosphorus transport into and within plants is facilitated by phosphorus transporter (PHT), which acts as a high-affinity P transporter essential for maintaining P levels under low P availability (Muneer and Jeong 2015; Del-Saz et al. 2018). As a cell surface transporter, the SLC family plays a fundamental role in P transport, including P absorption from interstitial fluid (Ravera et al. 2007). Our transcriptomic analysis revealed that a PHT gene and an SLC25A44 gene were up-regulated in response to low P availability. This expression pattern is consistent with PHT genes identified in Arabidopsis and rice (Mudge et al. 2002; Paszkowski et al. 2002).For example, in rice, the expression of OsPT2 and OsPT6 increases in leaves under Pi starvation. Additionally, we observed significant up-regulation of SPX domain protein transcripts under low P availability. SPX domain proteins are involved in fine-tuning P transport and starvation signaling in plants (Duan et al. 2008; Hürlimann et al. 2009). Examples include Pho87, Pho90, and Pho91, which are part of the regulon and mediate Pi translocation under P-deficient conditions (Chiou and Lin 2011). Moreover, the mRNA levels of OsSPX-MFS1, which contains an SPX domain, are induced by Pi deficiency and function as a P transporter in leaf P reallocation (Wu et al. 2013). These observations suggest the involvement of P transporter genes in regulating P in A. annua under conditions of low P availability.
Purple acid phosphatase (PAP) and phospholipase (e.g., PLA and PLD) enzymes can regulate internal P cycling by utilizing various organic P forms and facilitating intertissue transport in plants (Tran et al. 2010). For instance, AtPAP10, AtPAP12, and AtPAP26 in Arabidopsis release P from glycerol 3-phosphate, ADP, or DNA, respectively (Wang et al. 2014). Our study observed significant up-regulation of five PAPs, two PLD, and two PLA genes under low P availability, which aligns with findings in Arabidopsis (Suen et al. 2015). The increased expression levels of PAP, PLD, and PLA genes in A. annua suggest a physiological response aimed at synthesizing phosphatase and phospholipase enzymes to activate P recovery and intertissue transport under P deficiency.
The GO enrichment analysis revealed that ‘Monooxygenase activity’ and ‘Oxidation–reduction process’ were the most highly enriched GO keywords (Fig. 4). Additionally, several subcategories such as ‘Organic substance metabolic process (GO:0071704),’ ‘Primary metabolic process (GO:0044238),’ ‘Cellular metabolic process (GO:0009812),’ ‘Organic cyclic compound binding (GO:0097159),’ and ‘Heterocyclic compound binding (GO:1,901,363)’ showed significant enrichment in the 1692 DEGs (Table S2). KEGG pathway enrichment analysis indicated that ‘Plant hormone signal transduction’ and ‘Flavonoid biosynthesis’ pathways were notably enriched when comparing CaP to KP. These results suggest that the regulation of artemisinin accumulation in A. annua may be adversely affected by the limited use of phosphorus fertilizer. Moreover, some structural genes involved in artemisinin production exhibited higher expression levels under CaP treatment.
Previous studies have shown that endogenous hormones play a crucial role in plant growth and development (Pacifici et al. 2015). Treatment with CaP resulted in changes in the expression of several genes involved in the tryptophan, carotenoid, and linolenic acid pathways, as observed through RNA sequencing data and RT-PCR analyses. Specifically, genes related to tryptophan, carotenoid, and linolenic acid metabolic pathways were differentially expressed (5, 7, and 4 DEGs, respectively). These DEGs are known to be involved in plant development and stress tolerance, as noted by Shi et al. (2020). Earlier research by Tafvizi et al. (2009) and Chen et al. (2007) investigated differentially expressed genes (DEGs) in the cytokinin (zeatin) and GA synthesis pathways, respectively, affecting cotton plant development. In the tryptophan biosynthesis pathway, YUCCA, AUX, and SAUR expression levels were up-regulated, likely promoting cell expansion and contributing to the increase in artemisinin concentration. YUCCA proteins, a class of plant flavin mono-oxygenases, oxidize tryptophan (Trp) to form IPyA, which is further oxidized by YUCCA proteins to produce IAA (Dai et al. 2013; Nishimura et al. 2014). YUC genes have also been implicated in IAA biosynthesis in monocots (Yamamoto et al. 2007; Fujino et al. 2008; Gallavotti et al. 2008). Additionally, AUX has been proposed to stimulate root growth and lateral root proliferation in the presence of P deficiency (López-Bucio et al. 2002). SAURs are the most common family of early auxin response genes, playing a key role in the regulation of plant growth and development in response to hormones and environmental cues (Ren and Gray 2015). Regarding gibberellin production, GID2 expression was increased. The F-box protein gibberellin-insensitive dwarf2 (GID2) from rice is crucial for GA-mediated degradation of the DELLA protein (Gomi et al. 2004). Our study, on the other hand, discovered the down-regulation of brassinosteroid-signaling kinase (BSK), which is involved in brassinosteroid biosynthesis and may have a negative impact on plant development. The BSK family of receptor-like cytoplasmic kinases (RLCK) plays an important role in the early stages of BR signal transduction by activating downstream phosphatase BSU1 (Kim et al. 2009).
As mentioned by Xie et al. (2016), the biosynthesis of artemisinin involves several enzymatic steps in the sesquiterpene pathway. The biosynthesis of artemisinin is supplied through two separate pathways: the methylerythritol phosphate (MEP) pathway in the plastid and the mevalonate (MVA) pathway in the cytosol, as illustrated in Fig. 9A (Vranová et al. 2013). These pathways, along with genes associated with MVA and MEP pathways, have been extensively studied in numerous plants (Chang et al. 2013; Vranová et al. 2013; Rodríguez-Concepción and Boronat 2015). In our transcriptome analysis, we identified several genes that were differentially expressed and implicated in artemisinin biosynthesis. Notably, the expression of DXS and GPPS genes was significantly up-regulated in the MEP pathway. Deoxy-D-xylulose-5-phosphate synthase (DXS) is the first enzyme in the mevalonate-independent route responsible for producing 1-deoxy-D-xylulose-5-phosphate in plastids. DXS catalyzes the condensation of pyruvate and D-glyceraldehyde-3-phosphate (Weathers et al. 2006).
When exposed to light stress, it has been noted that root cultures of A. annua grown in light as opposed to dark significantly enhanced DXS transcript accumulation (Souret et al. 2003). To create geranyl diphosphate, GPPS catalyzes the condensation of dimethylallyl diphosphate and isopentenyl diphosphate (Gilg et al. 2005). In the MVA pathway, the MVD and GGPS were up-regulated and produced isopentyl diphosphate and geranylgeranyl pyrophosphate, respectively (Xie et al. 2016). Furthermore, the up-regulation of ALDH was observed in our study. ALDH1 was employed to design artemisinin precursors in yeast and plants metabolically (Xie et al. 2016). By including ALDH1 in modified yeast strains, Paddon et al. (2013) successfully synthesized artemisinic acid on an industrial scale. ALDH1 was discovered to be overexpressed in tobacco plants by Zhang et al. (2011). Even when no artemisinic acid nor dihydroartemisinic acid was present, transgenic plants were still able to produce dihydroartemisinic alcohol (Zhang et al. 2011). Additionally, earlier transcriptional studies demonstrate a clear correlation between the expression of ALDH1 and the synthesis of artemisinin in A. annua, indicating that ALDH1 participates in the biosynthetic process (Dilshad et al. 2015; Xiang et al. 2015).
TFs (transcription factors) have been proposed to play a crucial role in the regulation of gene expression by binding to DNA regulatory elements (Mathelier et al. 2016; Hou et al. 2019). Several studies (Nilsson et al. 2007; Wang et al. 2009; Secco et al. 2012; Castrillo et al. 2013) have provided substantial evidence supporting the involvement of TFs in phosphate homeostasis. Our investigation identified 39 TF families, including WRKYs, MYBs, GATA, bZIPs, CO-like, NACs, FAR1, and C2H2 zinc finger proteins, which displayed differential expression when exposed to CaP (Fig. 10A–C). These transcription factors were affected by our treatment and played critical roles in enhancing responses to P starvation, promoting phosphate acquisition, maintaining ROS homeostasis and root system, and regulating the synthesis of artemisinin. In response to CaP treatment, several TFs, including MYB (CTI12 AA068790) and two MYB-related genes (CTI12 AA469700 and CTI12 AA366500), showed elevated expression levels. In contrast, five MYB genes (CTI12 AA135050, CTI12 AA280420, CTI12 AA321750, and CTI12 AA340370) and two MYB-related genes (CTI12 AA398370 and CTI12 AA535620) displayed down-regulation. These TFs are likely essential for enhancing phosphate uptake, activating responses to P shortages, and regulating root architecture. Prior research on rice revealed that OsMYB2P-1 modulates downstream genes to suppress or activate responses to P shortage and control root architecture (Dai et al. 2012). Additionally, overexpression of OsMYB4P may promote phosphate absorption by increasing the expression of several Pht genes (Yang et al. 2014).
Our findings also indicated that WRKY TF members expressed differently in response to CaP, with 5 WRKY (CTI12 AA191430, CTI12 AA367540, CTI12 AA449960, CTI12 AA505740, and CTI12 AA575820) down-regulated and 3 WRKY (CTI12 AA402370, CTI12 AA424020, and CTI12 AA498820) up-regulated. Our findings show that these TF families perform as expected when faced with phosphate deficiency.
Artemisinin biosynthesis in A. annua is regulated by AaWRKY1, which is the first transcription factor identified and characterized in this plant (Shen et al. 2016). Additionally, MYB TFs AaMYB1, AaMIXTA1, and AaTAR2 have been found to be crucial for enhancing trichome initialization and artemisinin accumulation, as highlighted by research conducted by Matías-Hernández et al. (2017), Shi et al. (2018), and Zhou et al. (2020). In a previous study of Dai et al. (2016), it was revealed that WRKY74 plays a role in altering rice’s susceptibility to phosphate shortage. In our study, one C2H2 TF was up-regulated, and five were down-regulated. These C2H2 TFs might be essential in responding to P starvation. Wheat’s TaZAT8, a C2H2-ZFP-type TF gene, has been demonstrated to mediate wheat tolerance to phosphorus shortage by regulating phosphorus absorption, ROS homeostasis, and root system development (Ding et al. 2016). Furthermore, our research indicated the overexpression of BHLH (CTI12 AA394200) and the down-regulation of other BHLH (CTI12 AA385960). Prior studies have shown that AaORA and AabHLH1 positively regulate artemisinin synthesis (Lu et al. 2013; Ji et al. 2014).
In our study, we observed diverse expression patterns within the ERF family, with one member showing up-regulation and four showing down-regulation. In response to phosphorus (P) stress, one member of the bZIP family was up-regulated, while the other two were down-regulated. Consequently, the ERF and BZIP families may play significant roles in coping with P stress, potentially leading to a substantial accumulation of artemisinin. AaERF1 and AaERF2 were found to concurrently positively regulate ADS and CYP71AV1, enabling the synthesis of artemisinin and artemisinic acid in plants (Yu et al. 2012). However, another study by Tan et al. (2015) identified TAR1 as a crucial regulator of artemisinin biosynthesis, essential for the growth of trichomes in A. annua plants. Additionally, transgenic A. annua plants overexpressing AabZIP1 displayed elevated levels of ADS and CYP71AV1 expression, leading to enhanced artemisinin biosynthesis (Zhang et al. 2015). Overall, our findings contribute to a better understanding of how sparingly soluble phosphorus fertilizer influences the simultaneous transcriptional regulation of artemisinin and plant hormone transduction production in A. annua L.
Conclusion
The medicinal herb Artemisia annua L. is valued for its ability to produce artemisinin, which is used to treat malaria. Plant nutrition, specifically phosphorus (P), can influence A. annua biomass and secondary metabolite synthesis. However, most soil phosphates exist as insoluble inorganic and organic phosphates, and low P availability in soils is always a major abiotic factor limiting plant growth and development. Although plants have evolved several adaptation strategies to low P levels, the genetics and metabolic responses to P status are largely unknown. In a controlled greenhouse experiment, the sparingly soluble P form calcium hydroxide phosphate (Ca5OH(PO4)3/CaP) simulated calcareous soils with limited P availability. In contrast, the soluble P form KH2PO4/KP was a control. A. annua’s morphological traits, growth, and artemisinin concentration were determined, and RNA sequencing, clusters of orthologous genes (COG), Kyoto Encyclopedia and Gene Ontology (GO) of genomes (KEGG), and gene enrichment evaluations were used to identify genes that are expressed differently (DEGs) under two different P forms. Total biomass, plant height, leaf number, and stem diameter, as well as leaf area, decreased by 64.83%, 27.49%, 30.47%, 38.70%, and 54.64% in CaP compared to KP, respectively; however, LC–MS tests revealed a remarkable 37.97% increase in artemisinin content per unit biomass in CaP versus KP. Transcriptome analysis revealed 2015 DEGs (1084 up-regulated and 931 down-regulated) between two P forms, representing 39 TF families.
Further analysis revealed that DEGs were primarily involved in carbohydrate metabolism, secondary metabolite biosynthesis, enzyme catalytic activity, signal transduction, etc. These pathways were primarily associated with genes that encode key enzymes/proteins involved in the tricarboxylic acid (TCA) cycle, glycolysis, starch and sucrose metabolism, flavonoid biosynthesis, P metabolism, and plant hormone signal transduction. Meanwhile, several artemisinin biosynthesis genes were up-regulated, including DXS, GPPS, GGPS, MVD, and ALDH, which could increase artemisinin accumulation. Furthermore, 21 TF families, including WRKY, MYB, bHLH, and ERF, were up-regulated in response to CaP, demonstrating their importance in P absorption, internal P cycling, and artemisinin biosynthesis regulation. Overall, our study could pave the way for future research into the molecular mechanisms that underpin A. annua’s low P adaptation mechanism.
Data availability
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at BIG Sub, accession number: PRJAC012205.
References
Aftab T, Khan MMA, Ferreira JFS (2014) Effect of Mineral Nutrition, Growth Regulators and Environmental Stresses on Biomass Production and Artemisinin Concentration of Artemisia annua L. In: Aftab T, Ferreira J, Khan M, Naeem M (eds) Artemisia annua - Pharmacology and Biotechnology. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-41027-7_10
Amarasinghe T, Madhusha C, Munaweera I, Kottegoda N (2022) Review on mechanisms of phosphate solubilization in rock phosphate fertilizer. Commun Soil Sci Plant Anal 53(8):944–960
Augusto L, Achat DL, Jonard M, Vidal D, Ringeval B (2017) Soil parent material—a major driver of plant nutrient limitations in terrestrial ecosystems. Glob Change Biol 23(9):3808–3824
Baraldi R, Isacchi B, Predieri S, Marconi G, Vincieri FF, Bilia AR (2008) Distribution of artemisinin and bioactive flavonoids from Artemisia annua L. during plant growth. Biochem Syst Ecol 36(5–6):340–348
Behera B, Singdevsachan SK, Mishra R, Dutta S, Thatoi H (2014) Diversity, mechanism and biotechnology of phosphate solubilising microorganism in mangrove—a review. Biocatal Agric Biotechnol 3(2):97–110
Bhattacharya A (2019) Changing environmental condition and phosphorus-use efficiency in plants. Changing climate and resource use efficiency in plants, 241–305. https://doi.org/10.1016/C2017-0-04681-5
Castrillo G, Sánchez-Bermejo E, de Lorenzo L, Crevillén P, Fraile-Escanciano A, Tc M et al (2013) WRKY6 transcription factor restricts arsenate uptake and transposon activation in Arabidopsis. Plant Cell 25(8):2944–2957
Catalá R, Medina J, Salinas J (2011) Integration of low temperature and light signaling during cold acclimation response in Arabidopsis. Proc Natl Acad Sci 108(39):16475–16480
Chang W-C, Song H, Liu H-W, Liu P (2013) Current development in isoprenoid precursor biosynthesis and regulation. Curr Opin Chem Biol 17(4):571–579
Chao L-M, Liu Y-Q, Chen D-Y, Xue X-Y, Mao Y-B, Chen X-Y (2017) Arabidopsis transcription factors SPL1 and SPL12 confer plant thermotolerance at reproductive stage. Mol Plant 10(5):735–748
Chen Y, Ye G, Zhang L, Wang Y, Zhang X, Chen D (2007) Effect of trans-Bacillus thuringiensis gene on gibberellic acid and zeatin contents and boll development in cotton. Field Crop Res 103(1):5–10
Chiou T-J, Lin S-I (2011) Signaling network in sensing phosphate availability in plants. Annu Rev Plant Biol 62:185–206
Ciereszko I, Johansson H, Hurry V, Kleczkowski LA (2001) Phosphate status affects the gene expression, protein content and enzymatic activity of UDP-glucose pyrophosphorylase in wild-type and pho mutants of Arabidopsis. Planta 212:598–605
Dai X, Wang Y, Yang A, Zhang W-H (2012) OsMYB2P-1, an R2R3 MYB transcription factor, is involved in the regulation of phosphate-starvation responses and root architecture in rice. Plant Physiol 159(1):169–183
Dai X, Mashiguchi K, Chen Q, Kasahara H, Kamiya Y, Ojha S et al (2013) The biochemical mechanism of auxin biosynthesis by an Arabidopsis YUCCA flavin-containing monooxygenase. J Biol Chem 288(3):1448–1457
Dai X, Wang Y, Zhang W-H (2016) OsWRKY74, a WRKY transcription factor, modulates tolerance to phosphate starvation in rice. J Exp Bot 67(3):947–960
Del-Saz NF, Romero-Munar A, Cawthray GR, Palma F, Aroca R, Baraza E et al (2018) Phosphorus concentration coordinates a respiratory bypass, synthesis and exudation of citrate, and the expression of high-affinity phosphorus transporters in Solanum lycopersicum. Plant, Cell Environ 41(4):865–875
Dilshad E, Cusido RM, Palazon J, Estrada KR, Bonfill M, Mirza B (2015) Enhanced artemisinin yield by expression of rol genes in Artemisia annua. Malar J 14(1):1–10
Ding W, Wang Y, Fang W, Gao S, Li X, Xiao K (2016) TaZAT8, a C2H2-ZFP type transcription factor gene in wheat, plays critical roles in mediating tolerance to Pi deprivation through regulating P acquisition, ROS homeostasis and root system establishment. Physiol Plant 158:297–311
Du Q, Wang K, Xu C, Zou C, Xie C, Xu Y, Li WX (2016) Strand-specific RNA-Seq transcriptome analysis of genotypes with and without low-phosphorus tolerance provides novel insights into phosphorus-use efficiency in maize. BMC Plant Biol 16:222
Duan K, Yi K, Dang L, Huang H, Wu W, Wu P (2008) Characterization of a sub-family of Arabidopsis genes with the SPX domain reveals their diverse functions in plant tolerance to phosphorus starvation. Plant J 54(6):965–975
El-Sappah AH, Elrys AS, Desoky E-SM, Zhao X, Bingwen W, El-Sappah HH et al (2021b) Comprehensive genome wide identification and expression analysis of MTP gene family in tomato (Solanum lycopersicum) under multiple heavy metal stress. Saudi J Biol Sci 28(12):6946–6956
El-Sappah AH, Abbas M, Rather SA, Wani SH, Soaud N, Noor Z et al (2023) Genome-wide identification and expression analysis of metal tolerance protein (MTP) gene family in soybean (Glycine max) under heavy metal stress. Mol Biol Rep 50(4):2975–2990. https://doi.org/10.1007/s11033-022-08100-x
El-Sappah AH, Elbaiomy RG, Elrys AS, Wang Y, Zhu Y, Huang Q, Yan K, Xianming Z, Abbas M, El-Tarabily KA, Li J (2021a). Genome-wide identification and expression analysis of metal tolerance protein gene family in Medicago truncatula under a broad range of heavy metal stress. Front Genet 12:713224. https://doi.org/10.3389/fgene.2021.713224
Fei H, Ellis BE, Vessey JK (2014) Carbon partitioning in tissues of a gain-of-function mutant (MYB75/PAP1-D) and a loss-of-function mutant (myb75-1) in Arabidopsis thaliana. Botany 92(2):93–99
Fredeen AL, Raab TK, Rao IM, Terry N (1990) Effects of phosphorus nutrition on photosynthesis in Glycine max (L.) Merr. Planta 181:399–405
Fujino K, Matsuda Y, Ozawa K, Nishimura T, Koshiba T, Fraaije MW et al (2008) NARROW LEAF 7 controls leaf shape mediated by auxin in rice. Mol Genet Genomics 279:499–507
Gallavotti A, Barazesh S, Malcomber S, Hall D, Jackson D, Schmidt RJ et al (2008) sparse inflorescence1 encodes a monocot-specific YUCCA-like gene required for vegetative and reproductive development in maize. Proc Natl Acad Sci 105(39):15196–15201
Giles CD, Hsu P-CL, Richardson AE, Hurst MR, Hill JE (2014) Plant assimilation of phosphorus from an insoluble organic form is improved by addition of an organic anion producing Pseudomonas sp. Soil Biol Biochem 68:263–269
Gilg AB, Bearfield JC, Tittiger C, Welch WH, Blomquist GJ (2005) Isolation and functional expression of an animal geranyl diphosphate synthase and its role in bark beetle pheromone biosynthesis. Proc Natl Acad Sci U S A 102(28):9760–9765. https://doi.org/10.1073/pnas.0503277102
Gomi K, Sasaki A, Itoh H, Ueguchi-Tanaka M, Ashikari M, Kitano H et al (2004) GID2, an F-box subunit of the SCF E3 complex, specifically interacts with phosphorylated SLR1 protein and regulates the gibberellin-dependent degradation of SLR1 in rice. Plant J 37(4):626–634
Hammond JP, Bennett MJ, Bowen HC, Broadley MR, Eastwood DC, May ST et al (2003) Changes in gene expression in Arabidopsis shoots during phosphate starvation and the potential for developing smart plants. Plant Physiol 132(2):578–596
Hinsinger P (2001) Bioavailability of soil inorganic P in the rhizosphere as affected by root-induced chemical changes: a review. Plant Soil 237(2):173–195
Hou Z, Yin J, Lu Y, Song J, Wang S, Wei S et al (2019) Transcriptomic analysis reveals the temporal and spatial changes in physiological process and gene expression in common buckwheat (Fagopyrum esculentum Moench) grown under drought stress. Agronomy 9(10):569
Hu Y, Ye X, Shi L, Duan H, Xu F (2010) Genotypic differences in root morphology and phosphorus uptake kinetics in Brassica napus under low phosphorus supply. J Plant Nutr 33(6):889–901
Hürlimann HC, Pinson B, Stadler-Waibel M, Zeeman SC, Freimoser FM (2009) The SPX domain of the yeast low-affinity phosphate transporter Pho90 regulates transport activity. EMBO Rep 10(9):1003–1008
Jalali M, Jalali M (2020) Effect of organic and inorganic phosphorus fertilizers on phosphorus availability and its leaching over incubation time. Environ Sci Pollut Res 27(35):44045–44058
Ji Y, Xiao J, Shen Y, Ma D, Li Z, Pu G et al (2014) Cloning and characterization of AabHLH1, a bHLH transcription factor that positively regulates artemisinin biosynthesis in Artemisia annua. Plant Cell Physiol 55(9):1592–1604
Kim T-W, Guan S, Sun Y, Deng Z, Tang W, Shang J-X et al (2009) Brassinosteroid signal transduction from cell-surface receptor kinases to nuclear transcription factors. Nat Cell Biol 11(10):1254–1260
Kumari K, Phogat VK (2008) Rock phosphate: its availability and solubilization in the soil – a review. Agric Rev 29:108–116
Kvakić M, Tzagkarakis G, Pellerin S, Ciais P, Goll D, Mollier A et al (2020) Carbon and phosphorus allocation in annual plants: an optimal functioning approach. Front Plant Sci 11:149
Lambers H (2022) Phosphorus acquisition and utilization in plants. Annu Rev Plant Biol 73:17–42
Lee K-K, Mok I-K, Yoon M-H, Kim H-J, Chung D-Y (2012) Mechanisms of phosphate solubilization by PSB (phosphate-solubilizing bacteria) in soil. Korean J Soil Sci Fert 45(2):169–176
Li X, Luo L, Yang J, Li B, Yuan H (2015) Mechanisms for solubilization of various insoluble phosphates and activation of immobilized phosphates in different soils by an efficient and salinity-tolerant Aspergillus niger strain An2. Appl Biochem Biotechnol 175:2755–2768
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2− ΔΔCT method. Methods 25(4):402–408
López-Bucio J, Hernández-Abreu E, Sánchez-Calderón L, Nieto-Jacobo MF, Simpson J, Herrera-Estrella L (2002) Phosphate availability alters architecture and causes changes in hormone sensitivity in the Arabidopsis root system. Plant Physiol 129(1):244–256
Lu X, Zhang L, Zhang F, Jiang W, Shen Q, Zhang L et al (2013) AaORA, a trichome-specific AP 2/ERF transcription factor of Artemisia annua, is a positive regulator in the artemisinin biosynthetic pathway and in disease resistance to Botrytis cinerea. New Phytol 198(4):1191–1202
Liu X, Chu S, Sun C, Xu H, Zhang J, Jiao Y, Zhang D (2020) Genome-wide identification of low phosphorus responsive microRNAs in two soybean genotypes by high-throughput sequencing. Funct Integr Genomics 20:825–838
Ma C, Wang H, Lu X, Li H, Liu B, Xu G (2007) Analysis of Artemisia annua L. volatile oil by comprehensive two-dimensional gas chromatography time-of-flight mass spectrometry. J Chromatogr A 1150(1–2):50–53
Malhotra H, Vandana, Sharma S, Pandey R (2018) Phosphorus nutrition: plant growth in response to deficiency and excess. In: Hasanuzzaman M, Fujita M, Oku H, Nahar K, Hawrylak-Nowak B (eds) Plant Nutrients and Abiotic Stress Tolerance. Springer, Singapore. https://doi.org/10.1007/978-981-10-9044-8_7
Martinez V, Mestre TC, Rubio F, Girones-Vilaplana A, Moreno DA, Mittler R, Rivero RM (2016) Accumulation of flavonols over hydroxycinnamic acids favors oxidative damage protection under abiotic stress. Front Plant Sci 7:838. https://doi.org/10.3389/fpls.2016.00838
Mathelier A, Fornes O, Arenillas DJ, Chen C-Y, Denay G, Lee J et al (2016) JASPAR 2016: a major expansion and update of the open-access database of transcription factor binding profiles. Nucleic Acids Res 44(D1):D110–D115
Matías-Hernández L, Jiang W, Yang K, Tang K, Brodelius PE, Pelaz S (2017) AaMYB 1 and its orthologue AtMYB61 affect terpene metabolism and trichome development in Artemisia annua and Arabidopsis thaliana. Plant J 90(3):520–534
Misson J, Raghothama KG, Jain A, Jouhet J, Block MA, Bligny R et al (2005) A genome-wide transcriptional analysis using Arabidopsis thaliana Affymetrix gene chips determined plant responses to phosphate deprivation. Proc Natl Acad Sci 102(33):11934–11939
Mudge SR, Rae AL, Diatloff E, Smith FW (2002) Expression analysis suggests novel roles for members of the Pht1 family of phosphate transporters in Arabidopsis. Plant J 31(3):341–353
Müller M, Brandes D (1997) Growth and development of Artemisia annua L. on different soil types. Verh-Ges Fur Okologie 27:453–460
Muneer S, Jeong BR (2015) Proteomic analysis provides new insights in phosphorus homeostasis subjected to Pi (inorganic phosphate) starvation in tomato plants (Solanum lycopersicum L.). PLoS One 10(7):e0134103
Nakabayashi R, Yonekura-Sakakibara K, Urano K, Suzuki M, Yamada Y, Nishizawa T et al (2014) Enhancement of oxidative and drought tolerance in Arabidopsis by overaccumulation of antioxidant flavonoids. Plant J 77(3):367–379
Natr L (1992) Mineral nutrients-a ubiquitous stress factor for photosynthesis. Photosynthetica (Czech Republic) 27:3
Nilsson L, Müller R, Nielsen TH (2007) Increased expression of the MYB-related transcription factor, PHR1, leads to enhanced phosphate uptake in Arabidopsis thaliana. Plant, Cell Environ 30(12):1499–1512
Nishimura T, Hayashi Ki, Suzuki H, Gyohda A, Takaoka C, Sakaguchi Y et al (2014) Yucasin is a potent inhibitor of YUCCA, a key enzyme in auxin biosynthesis. Plant J 77(3):352–366
Pacifici E, Polverari L, Sabatini S (2015) Plant hormone cross-talk: the pivot of root growth. J Exp Bot 66(4):1113–1121
Paddon CJ, Westfall PJ, Pitera DJ, Benjamin K, Fisher K, McPhee D et al (2013) High-level semi-synthetic production of the potent antimalarial artemisinin. Nature 496(7446):528–532
Paszkowski U, Kroken S, Roux C, Briggs SP (2002) Rice phosphate transporters include an evolutionarily divergent gene specifically activated in arbuscular mycorrhizal symbiosis. Proc Natl Acad Sci 99(20):13324–13329
Pearse SJ, Veneklaas EJ, Cawthray G, Bolland MD, Lambers H (2007) Carboxylate composition of root exudates does not relate consistently to a crop species’ ability to use phosphorus from aluminium, iron or calcium phosphate sources. New Phytol 173(1):181–190
Plaxton WC (2004) Plant response to stress: biochemical adaptations to phosphate deficiency. Encyclopedia of plant and crop science. Marcel Dekker, New York, pp 976–980
Qiu J, Israel DW (1992) Diurnal starch accumulation and utilization in phosphorus-deficient soybean plants. Plant Physiol 98(1):316–323
Raghothama K (1999) Phosphate acquisition. Annu Rev Plant Biol 50(1):665–693
Ravera S, Virkki LV, Murer H, Forster IC (2007) Deciphering PiT transport kinetics and substrate specificity using electrophysiology and flux measurements. Am J Physiol Cell Physiol 293(2):C606–C620
Ren H, Gray WM (2015) SAUR proteins as effectors of hormonal and environmental signals in plant growth. Mol Plant 8(8):1153–1164
Ren P, Meng Y, Li B, Ma X, Si E, Lai Y et al (2018) Molecular mechanisms of acclimatization to phosphorus starvation and recovery underlying full-length transcriptome profiling in barley (Hordeum vulgare L.). Front Plant Sci 9:500
Rodríguez-Concepción M, Boronat A (2015) Breaking new ground in the regulation of the early steps of plant isoprenoid biosynthesis. Curr Opin Plant Biol 25:17–22
Rouached H, Arpat AB, Poirier Y (2010) Regulation of phosphate starvation responses in plants: signaling players and cross-talks. Mol Plant 3(2):288–299
Ryan P, Delhaize E, Jones D (2001) Function and mechanism of organic anion exudation from plant roots. Annu Rev Plant Biol 52(1):527–560
Secco D, Wang C, Arpat BA, Wang Z, Poirier Y, Tyerman SD et al (2012) The emerging importance of the SPX domain-containing proteins in phosphate homeostasis. New Phytol 193(4):842–851
Shen J, Rengel Z, Tang C, Zhang F (2003) Role of phosphorus nutrition in development of cluster roots and release of carboxylates in soil-grown Lupinus albus. Plant Soil 248:199–206
Shen J, Yuan L, Zhang J, Li H, Bai Z, Chen X et al (2011) Focus issue on phosphorus plant physiology: phosphorus dynamics: from soil to plant. Plant Physiol 156(3):997
Shen Q, Yan T, Fu X, Tang K (2016) Transcriptional regulation of artemisinin biosynthesis in Artemisia annua L. Sci Bull 61:18–25
Shi M-Z, Xie D-Y (2014) Biosynthesis and metabolic engineering of anthocyanins in Arabidopsis thaliana. Recent Pat Biotechnol 8(1):47–60
Shi P, Fu X, Shen Q, Liu M, Pan Q, Tang Y et al (2018) The roles of Aa MIXTA 1 in regulating the initiation of glandular trichomes and cuticle biosynthesis in Artemisia annua. New Phytol 217(1):261–276
Shi J, Wang N, Zhou H, Xu Q, Yan G (2020) Transcriptome analyses provide insights into the homeostatic regulation of axillary buds in upland cotton (G. hirsutum L.). BMC Plant Biol 20:1–14
Smith SE, Smith FA (2011) Roles of arbuscular mycorrhizas in plant nutrition and growth: new paradigms from cellular to ecosystem scales. Annu Rev Plant Biol 62:227–250
Souret FF, Kim Y, Wyslouzil BE, Wobbe KK, Weathers PJ (2003) Scale-up of Artemisia annua L. hairy root cultures produces complex patterns of terpenoid gene expression. Biotechnol Bioeng 83(6):653–667
Stringham RW, Moore GL, Teager DS, Yue TY (2018) Analysis and isolation of potential artemisinin precursors from Waste streams of Artemisia annua extraction. ACS omega 3(7):7803–7808
Suen PK, Zhang S, Sun SS-M (2015) Molecular characterization of a tomato purple acid phosphatase during seed germination and seedling growth under phosphate stress. Plant Cell Rep 34:981–992
Tafvizi F, Farahanei F, Sheidai M, Nejadsattari T (2009) Effects of zeatin and activated charcoal in proliferation of shoots and direct regeneration in cotton (Gossypium hirsutum L.). Afr J Biotechnol 8(22):6220
Tan H, Xiao L, Gao S, Li Q, Chen J, Xiao Y et al (2015) Trichome and artemisinin regulator 1 is required for trichome development and artemisinin biosynthesis in Artemisia annua. Mol Plant 8(9):1396–1411
Thum T, Caldararu S, Engel J, Kern M, Pallandt M, Schnur R et al (2019) A new terrestrial biosphere model with coupled carbon, nitrogen, and phosphorus cycles (QUINCY v1. 0; revision 1772). Geosci Model Dev Discuss 2019:1–38
Todeschini V, Anastasia F, Massa N, Marsano F, Cesaro P, Bona E et al (2022) Impact of phosphatic nutrition on growth parameters and artemisinin production in Artemisia annua plants inoculated or not with Funneliformis mosseae. Life 12(4):497
Tran HT, Hurley BA, Plaxton WC (2010) Feeding hungry plants: the role of purple acid phosphatases in phosphate nutrition. Plant Sci 179(1–2):14–27
Uhde-Stone C, Zinn KE, Ramirez-Yáñez M, Li A, Vance CP, Allan DL (2003) Nylon filter arrays reveal differential gene expression in proteoid roots of white lupin in response to phosphorus deficiency. Plant Physiol 131(3):1064–1079. https://doi.org/10.1104/pp.102.016881
Usuda H, Shimogawara K (1992) Phosphate deficiency in maize: III. Changes in enzyme activities during the course of phosphate deprivation. Plant Physiol 99(4):1680
Vance CP, Uhde-Stone C, Allan DL (2003) Phosphorus acquisition and use: critical adaptations by plants for securing a nonrenewable resource. New Phytol 157(3):423–447
Veneklaas EJ, Lambers H, Bragg J, Finnegan PM, Lovelock CE, Plaxton WC et al (2012) Opportunities for improving phosphorus-use efficiency in crop plants. New Phytol 195(2):306–320
Vicca S, Stocker BD, Reed S, Wieder WR, Bahn M, Fay PA et al (2018) Using research networks to create the comprehensive datasets needed to assess nutrient availability as a key determinant of terrestrial carbon cycling. Environ Res Lett 13(12):125006
Vitousek PM, Porder S, Houlton BZ, Chadwick OA (2010) Terrestrial phosphorus limitation: mechanisms, implications, and nitrogen–phosphorus interactions. Ecol Appl 20(1):5–15
Vranová E, Coman D, Gruissem W (2013) Network analysis of the MVA and MEP pathways for isoprenoid synthesis. Annu Rev Plant Biol 64:665–700
Wan L-Y, Qi S-S, Dai Z-C, Zou CB, Song Y-G, Hu Z-Y et al (2018) Growth responses of Canada goldenrod (Solidago canadensis L.) to increased nitrogen supply correlate with bioavailability of insoluble phosphorus source. Ecol Res 33:261–269
Wang C, Ying S, Huang H, Li K, Wu P, Shou H (2009) Involvement of OsSPX1 in phosphate homeostasis in rice. Plant J 57(5):895–904
Wang Y, Law R, Pak B (2010) A global model of carbon, nitrogen and phosphorus cycles for the terrestrial biosphere. Biogeosciences 7(7):2261–2282
Wang L, Lu S, Zhang Y, Li Z, Du X, Liu D (2014) Comparative genetic analysis of Arabidopsis purple acid phosphatases AtPAP10, AtPAP12, and AtPAP26 provides new insights into their roles in plant adaptation to phosphate deprivation. J Integr Plant Biol 56(3):299–314. https://doi.org/10.1111/jipb.12184
Wang C, Huang W, Ying Y, Li S, Secco D, Tyerman S et al (2012) Functional characterization of the rice SPX-MFS family reveals a key role of OsSPX-MFS1 in controlling phosphate homeostasis in leaves. New Phytol 196(1):139–148
Wasaki J, Yamamura T, Shinano T, Osaki M (2003a) Secreted acid phosphatase is expressed in cluster roots of lupin in response to phosphorus deficiency. Plant Soil 248:129–136
Wasaki J, Yonetani R, Kuroda S, Shinano T, Yazaki J, Fujii F et al (2003b) Transcriptomic analysis of metabolic changes by phosphorus stress in rice plant roots. Plant, Cell Environ 26(9):1515–1523
Wasaki J, Shinano T, Onishi K, Yonetani R, Yazaki J, Fujii F et al (2006) Transcriptomic analysis indicates putative metabolic changes caused by manipulation of phosphorus availability in rice leaves. J Exp Bot 57(9):2049–2059
Weathers PJ, Elkholy S, Wobbe KK (2006) Artemisinin: the biosynthetic pathway and its regulation in Artemisia annua, a terpenoid-rich species. In Vitro Cell Dev Biol-Plant 42:309–317
Wieder WR, Cleveland CC, Smith WK, Todd-Brown K (2015) Future productivity and carbon storage limited by terrestrial nutrient availability. Nat Geosci 8(6):441–444
Williamson LC, Ribrioux SP, Fitter AH, Leyser HO (2001) Phosphate availability regulates root system architecture in Arabidopsis. Plant Physiol 126(2):875–882
Wu P, Ma L, Hou X, Wang M, Wu Y, Liu F et al (2003) Phosphate starvation triggers distinct alterations of genome expression in Arabidopsis roots and leaves. Plant Physiol 132(3):1260–1271
Wu P, Shou H, Xu G, Lian X (2013) Improvement of phosphorus efficiency in rice on the basis of understanding phosphate signaling and homeostasis. Curr Opin Plant Biol 16(2):205–212
Xiang L, Zhu S, Zhao T, Zhang M, Liu W, Chen M et al (2015) Enhancement of artemisinin content and relative expression of genes of artemisinin biosynthesis in Artemisia annua by exogenous MeJA treatment. Plant Growth Regul 75:435–441
Xie D-Y, Ma D-M, Judd R, Jones AL (2016) Artemisinin biosynthesis in Artemisia annua and metabolic engineering: questions, challenges, and perspectives. Phytochem Rev 15:1093–1114
Xu Q, Yin XR, Zeng JK, Ge H, Song M, Xu CJ, Li X, Ferguson IB, Chen KS (2014) Activator- and repressor-type MYB transcription factors are involved in chilling injury induced flesh lignification in loquat via their interactions with the phenylpropanoid pathway. J Exp Bot 65:4349–4359. https://doi.org/10.1093/jxb/eru208
Yamamoto Y, Kamiya N, Morinaka Y, Matsuoka M, Sazuka T (2007) Auxin biosynthesis by the YUCCA genes in rice. Plant Physiol 143(3):1362–1371
Yang WT, Baek D, Yun D-J, Hwang WH, Park DS, Nam MH et al (2014) Overexpression of OsMYB4P, an R2R3-type MYB transcriptional activator, increases phosphate acquisition in rice. Plant Physiol Biochem 80:259–267
Yang J, Xie M-Y, Yang X-L, Liu B-H, Lin H-H (2019) Phosphoproteomic profiling reveals the importance of CK2, MAPKs and CDPKs in response to phosphate starvation in rice. Plant Cell Physiol 60(12):2785–2796
Yu Z-X, Li J-X, Yang C-Q, Hu W-L, Wang L-J, Chen X-Y (2012) The jasmonate-responsive AP2/ERF transcription factors AaERF1 and AaERF2 positively regulate artemisinin biosynthesis in Artemisia annua L. Mol Plant 5(2):353–365
Zhang Y, Nowak G, Reed DW, Covello PS (2011) The production of artemisinin precursors in tobacco. Plant Biotechnol J 9(4):445–454
Zhang K, Liu H, Tao P, Chen H (2014) Comparative proteomic analyses provide new insights into low phosphorus stress responses in maize leaves. PLoS ONE 9(5):e98215
Zhang F, Fu X, Lv Z, Lu X, Shen Q, Zhang L et al (2015) A basic leucine zipper transcription factor, AabZIP1, connects abscisic acid signaling with artemisinin biosynthesis in Artemisia annua. Mol Plant 8(1):163–175
Zhang X-B, Guo L-P, Qiu Z-D, Qu X-B, Wang H, Jing Z-X et al (2017) Analysis of spatial distribution of artemisinin in Artemisia annua in China Zhongguo Zhong yao za zhi= Zhongguo Zhongyao Zazhi= China. J Chin Mater Med 42(22):4277–4281
Zhou Z, Tan H, Li Q, Li Q, Wang Y, Bu Q et al (2020) Trichome and artemisinin regulator 2 positively regulates trichome development and artemisinin biosynthesis in Artemisia annua. New Phytol 228(3):932–945
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 82360753), the Guangxi Major Science and Technology Project of China (Grant No. GuikeAA22096021), the Natural Science Foundation of Guangxi Province (Grant No. 2023GXNSFAA026330), the Innovative Team for Traditional Chinese Medicinal Materials Quality of Guangxi (Grant No. GZKJ2305), the Scientific Research Funding Project of Guangxi Botanical Garden of Medicinal Plants (Grant No. GYJ202013), the Research and Innovation Team Building Project of Guangxi Botanical Garden of Medicinal Plants (Grant No. GYCH2019008), National Natural Science Foundation of China (Grant No. 81560623), the Key Laboratory Construction Program of Guangxi Health commission (Grant No. ZJC2020003), and the Key Techniques Research and Promotion of Guangxi Medicinal Materials Varieties (Grant No. GZKJ2314).
Author information
Authors and Affiliations
Contributions
Conceptualization: LW, JH, WS, QH, SW, and AE-S. Designed the experiments: LW, IJ, and SW. Performed the experiments: LR, JH, WH, and LD. Analyzed the results: LW, JH, QH, and AE-S. Draw the figures: AE-S, LW, and QH. Contributed reagents/materials: LW, LS, JH, ZZ, LP, and JF. Contributed to writing the original manuscript draft: LW, QH, and AE-S. Review and editing of the manuscript: AE-S, HQ, RE, SS, RMYH, SAS, MAAE, and MA. Writing final copy: AE-S. All authors contributed to the article and approved the submitted version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Wan, L., Huo, J., Huang, Q. et al. Genetics and metabolic responses of Artemisia annua L to the lake of phosphorus under the sparingly soluble phosphorus fertilizer: evidence from transcriptomics analysis. Funct Integr Genomics 24, 26 (2024). https://doi.org/10.1007/s10142-024-01301-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10142-024-01301-6