
Abstract
Artemisia annua L. is a medicinal plant valued for its ability to produce artemisinin, a molecule used to treat malaria. Plant nutrients, especially phosphorus (P), can potentially influence plant biomass and secondary metabolite production. Our work aimed to explore the genetic and metabolic response of A. annua to hardly soluble aluminum phosphate (AlPO4, AlP), using soluble monopotassium phosphate (KH2PO4, KP) as a control. Liquid chromatography–mass spectrometry (LC–MS) was used to analyze artemisinin. RNA sequencing, gene ontology (GO), and the Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses were applied to analyze the differentially expressed genes (DEGs) under poor P conditions. Results showed a significant reduction in plant growth parameters, such as plant height, stem diameter, number of leaves, leaf areas, and total biomass of A. annua. Conversely, LC–MS analysis revealed a significant increase in artemisinin concentration under the AlP compared to the KP. Transcriptome analysis revealed 762 differentially expressed genes (DEGs) between the AlP and the KP. GH3, SAUR, CRE1, and PYL, all involved in plant hormone signal transduction, showed differential expression. Furthermore, despite the downregulation of HMGR in the artemisinin biosynthesis pathway, the majority of genes (ACAT, FPS, CYP71AV1, and ALDH1) were upregulated, resulting in increased artemisinin accumulation in the AlP. In addition, 12 transcription factors, including GATA and MYB, were upregulated in response to AlP, confirming their importance in regulating artemisinin biosynthesis. Overall, our findings could contribute to a better understanding the parallel transcriptional regulation of plant hormone transduction and artemisinin biosynthesis in A. annua L. in response to hardly soluble phosphorus fertilizer.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Phosphorus (P) is an essential macronutrient for plant growth and development, involving the composition of membrane phospholipids and nucleic acids and metabolic roles such as energy storage and transfer (Rouached et al. 2010; Veneklaas et al. 2012). However, P exists primarily in soils as hardly soluble phosphate compounds combined with the mineral elements, such as aluminum phosphate (AlPO4, AlP) and the aluminum salt of phosphoric acid in acidic acid soils, and it is difficult for plants to absorb (Pradhan et al. 2017). Therefore, P is a limiting nutrient for plants due to its low availability of P in soils (Augusto et al. 2017). Certainly, in P-poor environments, plants have developed general strategies for obtaining and benefiting from P, including exudation of compounds (Ryan et al. 2001; Shen et al. 2003), root structural specializations (Hu et al. 2010; Williamson et al. 2001), and mycorrhizal symbioses (Smith and Smith 2011; Wan et al.2018), which all can lead to an increase in the bioavailability of AlPO4 in acidic soils to plants. However, the molecular mechanism of plant response to hardly soluble AlPO4 is poorly understood. Various sequencing technologies have recently provided critical information about gene expression changes in some plant species in response to P limitation. Transcriptome analysis of barley revealed that many genes were significantly upregulated or downregulated in response to low P stress. Furthermore, differentially expressed genes (DEGs) were discovered to be primarily involved in P metabolism, sucrose synthesis, phospholipid degradation, hydrolysis of phosphoric enzymes, phosphorylation/dephosphorylation, and post-transcriptional regulation. (Ren et al. 2018). Furthermore, when treated with low phosphorus, DEGs are enriched in carbohydrate metabolic processes, oxidation–reduction processes, biosynthetic processes, and the tricarboxylic acid cycle in oat roots (Chao et al. 2017). To gain a better understanding of these processes, DEGs were studied under low P stress in other crops, including Zea mays L.(Du et al. 2016), Oryza sativa L. (Deng et al. 2018), and Glycine max Linn (Liu et al. 2020), using transcriptome analysis. The above results of studies show that transcriptome can provide more information on the gene regulation related to low P adaptation for plants.
Artemisia annua is an annual herb appreciated for producing artemisinin, a sesquiterpene molecule used to treat fever and malaria (Baraldi et al. 2008; Ma et al. 2007; Wani et al. 2021; Wani et al. 2022). It is widely distributed in most Chinese areas, especially in southwest China (Zhang et al. 2017). It adapts well to different soil types and has no specific nutritional requirements; however, P and potassium (K) supply stimulate its growth even in small quantities (Aftab et al. 2014; Müller and Brandes 1997). For example, Todeschini et al. (2022) showed that P nutrition affected A. annua plant biomass production, and its lowest level led to the highest artemisinin concentration. Therefore, optimizing P supply to A. annua is essential for maximizing dry matter production and/or artemisinin yield (Todeschini et al. 2022). Many studies have explored the ability of crops to acquire P from various hardly soluble forms (Sharma et al. 2013; Giles et al. 2014; Lambers 2022; Lee et al. 2012; Li et al. 2015; Pearse et al. 2007). Currently, minimal effort has been made to understand the adaptive strategies in A. annua against Plimit. A controlled greenhouse experiment was conducted to learn more about the genetic behavior of A. annua in response to the availability of a hardly soluble P source, AlP, to simulate low P availability in acid soil, with the soluble phosphorus form, KP, serving as a control. We then explored the A. annua growth and genetics response to the hardly soluble P source, AlP, via second-generation sequencing analysis. This study could deepen our understanding of the genetic variation of A. annua under low phosphorus availability and suggest strategies to improve its P-use efficiency and the production of biomass and artemisinin with less fertilizer application.
Material and methods
Plant materials
Artemisia annua seeds were collected from the planting area in the Guangxi Medicinal Botanical Garden scientific research base in Nanning, China (108°23′ E, 22°51′ N). The seeds of A. annua were sowed into a plastic container (30 cm × 20 cm × 8 cm, length × width × height, respectively) filled with washed and sterilized river sand and then rinsed with distilled water until the sand was wet, every 2 days during germination. The seedings were supplied with 200 ml of pH 6.5 half-strength Hoagland nutrient solution weekly. All the seedings of similar size with two cotyledons were on standby for the two phosphorus treatments.
Different phosphorus treatments
There were two P sources used in this study: a hardly soluble P source: aluminum phosphate (AlPO4)/(AlP), the water solubility of which is only 1.89 × 10−9 g/100 ml at 20 °C, and a soluble P source: monopotassium phosphate (KH2PO4)/(KP), the water solubility of which is 22.6 g/100 ml at 20 °C. The AlP was the low-P-availability treatment group, and the KP was the control group; each group was replicated five times. River sand was used as a cultivation substrate in this experiment. All the river sand before the experiment was sieved through a 2-mm mesh and cleaned to remove nutrients with running water and then autoclave sterilized for 30 min. The flower pot used for holding the river sand was a height of 12 cm and a diameter of 4 cm. P supplements were added in powder form, 118.06 mg AlPO4 and 131.83 mg KH2PO4, respectively, mixed with the treated river sand, to ensure that each flower pot contained 30 mg P content. After the transplantation of seedlings, all flower pots were put in the greenhouse, with illumination intensity 300 µmol m−2 s−1 during the day, temperature 26 ± 2 °C, and relative humidity 62 ± 2%. All the flower pots’ positions were changed randomly to avoid the influence of environmental differences. Each flowerpot was supplemented with enough distilled water every 2 days, and a 5-ml Hoagland nutrient solution (0.5 × , without P) was added each week. The P treatment was sustained for 3 months (Pearseet al. 2007; Wanet al. 2018).
Plant growth parameters and sample collection
After the different P source treatments, the plant height, stem diameter, leaf number, and leaf area were measured before seedlings were harvested. The leaf area was taken with a digital photo and then calculated with ImageJ software (National Institutes of Health, USA). Three fresh leaves of every flowerpot were collected and wrapped with aluminum foil, immediately frozen in liquid nitrogen for 3–5 min, and then stored in a − 80 ℃ until RNA-Seq and real-time quantitative PCR (RT-qPCR). And then, the final biomass of roots, stems, and leaves were counted after drying at 60 ℃ for 72 h.
Isolation of Artemisinin and Analysis by LC–MS
The leaves were dried at 60 ℃ and prepared for artemisinin determination. Samples of 0.2 g of dried A. annua leaves were extracted using 25 ml petroleum ether (boiling point 30–60) for 40 min with ultrasonic waves, filtrated, transferred to 100 ml evaporating dish, and dried at 40 ℃; then, the evaporating dish was rinsed with methanol repeatedly. All solutions were kept in a volumetric flask at a constant volume of 10 ml (Stringham et al. 2018). The sample extracts were analyzed using an LC–ESI–MS/MS system (HPLC, EXPEC 5210 system1). The analytical conditions were as follows: HPLC column, Waters ACQUITY UPLC BEH C18 (1.7 μm 2.1 × 150 mm); solvent system, water (0.1% acetic acid): acetonitrile; gradient program, 80:20 V/V at 0 min, 5:95 V/V at 4.0 min, 5:95 V/V at 6 min, 80:20 V/V at 6.1 min, 80:20 V/V at 9 min; flow rate, 0.3 ml /min; temperature, 40 ℃; and injection volume, 2 µl. LIT and triple quadrupole (QQQ) scans were acquired on a triple quadrupole linear ion trap mass spectrometer EXPEC 5210 LC/MS/MS system equipped with an ESI. The ESI source operation parameters were as follows: ion source, turbo spray; source temperature, 105 ℃; capillary voltage, (IS) 4800 V; source offset voltage, 700 V; desolvation temperature, 495 ℃; cone gas flow, 72 l/h; and desolvation gas flow300 l/h. Instrument tuning and mass calibration were performed with 10 and 100 µmol/l polypropylene glycol solutions in QQQ and LIT modes, respectively. QQQ scans were acquired as MRM experiments with collision gas (nitrogen) set to 5 psi. DP and CE for individual MRM transitions were done with further DP and CE optimization. A specific set of MRM transitions were monitored for each period according to the metabolites eluted within this period.
RNA extraction and transcriptome sequencing
The RNAprep Pure Plant Kit (TIANGEN, Beijing, China) was used to extract total RNA from frozen samples. The purity of the RNA was determined using the manufacturer’s protocols and a KaiaoK5500 Spectrophotometer (Kaiao, Beijing, China). An RNA Nano 6000 Assay Kit of the Bioanalyzer 2100 system was used to assess RNA concentration and integrity (Agilent Technologies, CA, USA). RNA degradation was monitored on agarose gels. Three replicates of each treatment were deemed high quality and used to build transcriptome libraries. The cDNA construction library refers to the following reagents: oligo (dT) magnetic beads for enriched mRNA from total RNA, divalent cations under elevated temperature in NEB Next First Strand Synthesis Reaction Buffer (5 ×) for fragmentation, random hexamer primer and RNase H for synthesizing the First-strand cDNA, and DNA polymerase I, buffer, dNTPs, and RNase H for synthesizing second-strand cDNA. QiaQuick PCR kits and elution with EB buffer were used to purify the library fragments, and then, the terminal repair, A-tailing, and adapter were implemented. The aimed products were retrieved, PCR was performed, and the library was completed. The library quality was evaluated using the Step One Plus Real-Time PCR Agilent Bioanalyzer 2100 systems. The BioNovo Gene Technology Co., Ltd. (Suzhou, China) sequenced six libraries using an Illumina HiSeq 2500.
qRT-PCR analysis
Total RNA extraction, reverse transcription, and qPCR were operated as described previously (El-Sappah et al. 2021). β-actin mRNA was used as an internal control; all primers, including β-actin as a reference gene, were designed with the Primer 5.0 software (Leišová-Svobodová et al. 2020; Ahmed et al. 2023). The gene-specific primers used for qPCR are listed in Table S1. Gene expression was relatively quantified using the MM Ct method, as described by Livak and Schmittgen (El-Sappah et al. 2023). Each treatment had three replications, and the experiment was performed thrice. The mean and standard errors (SEs) were presented using ORIGIN 8.6 (Singh et al. 2021).
Statistical analysis
The one-way ANOVA was performed using the SPSS Statistics 19.0 software (IBM Corp, Armonk, NY, USA), and the Duncan test was used to determine significant differences (P < 0.05). The data were presented as mean ± SD (standard deviation). The sample was sequenced on the machine, and the software in the sequencing platform was transformed to generate the raw data (RAW Data) of FASTQ, which is the off-machine data. The raw data of each sample was used for statistical analysis. The connector sequence of 3′ ends was removed with Cutadapt, using the HISAT2 software to compare the filtered reads to the reference genome. The read distribution compared to the genome was statistically divided into CDS (coding region), intron (intron), and so on. HTSeq statistics, with three statistical schemes, was used to compare the Read Count value on each gene to the original expression of the gene. RSeQC was used to analyze expression saturation; DESeq was used to analyze the difference in gene expression and to screen the expression of different genes. GO enrichment analysis was performed using top-GO.
Results
Growth parameter assessment and artemisinin concentration
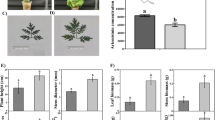
Plant growth parameters, including the plant height, the number of leaves, the stem diameter, and the leaf area, were significantly reduced under the AlP compared to the KP (Fig. 1A-E). Moreover, the leaf biomass and the root biomass had a slight reduction, but the stem biomass recorded a significant reduction under the AlP, compared to the control, KP, which led to a significant reduction in the total biomass of A. annua (Fig. 1F). Conversely, LC–MS analysis revealed not only no decrease but also a slight increase in artemisinin concentration under the AlP, compared to the KP (Fig. 1G).

Plant morphology, growth parameter assessment, and artemisinin concentration in response to the hardly soluble phosphorus source (AlP) compared to control (KP). A Plant height under the AlP, B plant height under the KP, C leaf area under the AlP, D leaf area under the AlP, E morphological traits, F biomass measurements, and G artemisinin concentration (mg kg.−1). Different letters (a and b) indicated significant differences between the AlP and the KP (P < 0.05, Student’s t-test)
Transcriptome sequencing and the DEGs
The cDNA libraries were constructed for generating transcriptome sequences using A. annua leaves of the KPas, a control sample and the AlP as a hardly soluble Psource. Sequence libraries were prepared on the Illumina NextSeq 500 platform from 100 × to 120 × depth. Raw data from paired-end sequencing-by-synthesis generated 44,281,438, and 45,146,916 bp reads from the KP and the AlP, respectively. For all samples, the maximum read length was 6,910,596,600 bp. Following quality control and raw read data processing, 40,888,780 and 41,961,136 reads from the KP and the AlP, respectively, were retained for further assembly. Filtered reads were assembled, and transcripts were generated using Trinity at a hash size of 25. As a result of assembly, 39,274 transcripts of the KP vs the AlP were obtained in treatment comparatives with average transcript lengths of 283.58 bp for the KP vs the AlP (Table S2). The assembled transcript from different replicates showed variation in its numbers, possibly due to variable P absorbed by the plants or noises caused by technology at some point during the sequencing process. The distribution pattern of these transcripts is presented in Fig. 2.

Overview of the RNA-seq data and distribution of DEGs. A Principal component analysis (PCA) of the RNA-seq output. The PCA plot is calculated based on the transcriptome-wide profiles of gene expression. Distances between samples reveal differences in the transcriptome profiles between the samples. B Volcano plots display differentially expressed transcripts. Each dot represented a DEG; dots above the red line displayed the significant DEGs (P < 0.05)
The principal component analysis (PCA) was used to assess variability between RNA-seq experiments. The PCA results revealed a strong correlation between the three replicates at two different treatments (Fig. 2A). A total of 762 DEGs were identified through a comparison of control to treatment (the KP vs the AlP) (323 upregulated, 439 downregulated). To identify common transcripts in the DGE data, transcripts exclusive to the low P availability treatment and the control treatment and downregulated and upregulated transcripts were analyzed for overlap (Fig. 2B).
Identification of the GO and KEGG enrichment analysis
All DEGs were assigned 210 GO terms (P < 0.05), divided into three categories: molecular function, cellular component, and biological process. The top 20 enriched GO terms were shown in Fig. 3 and Fig. S1, with the largest two terms being “protein localization (GO:0,072,662)” and “enzyme inhibitor activity (GO:0,004,857)” from the “biological process” and “molecular function” categories, respectively (Table S3). Furthermore, the MAPK signaling pathway, plant-pathogen interaction, and aminoacyl-tRNA biogenesis subcategories of the “molecular function” category were significantly enriched in more than 50 DEGs.

GO enrichment analysis of all the DEGs between the AlP and the KP. The top 20 enriched GO terms were presented. The horizontal axis represented the rich factor, while the vertical axis represented the GO terms. Number: DEG number; P adjust: adjusted P value
To describe enriched biological pathways, a KEGG pathway enrichment analysis was performed (Fig. 4; Fig. S2; Table S4). The DEGs under the KP, compared to the AlP, were most significantly enriched in “catalytic activity,” “cellular metabolic process,” and “ion binding” categories. Moreover, this comparison was significantly involved in “glyoxylate and dicarboxylate metabolism,” “RNA degradation,” “glycine, serine and threonine metabolism,” and “proteasome” (Fig. 4).

The KEGG enrichment analysis of DEGs between the AlP and the KP. The most enriched KEGG pathways were presented. The horizontal axis represented the rich factor, while the vertical axis represented the pathway names. Number: DEG number; P adjust: adjusted P value
Analysis of DEGs involved in hormone biosynthesis and signal transduction
Several genes involved in phytohormone, tryptophan, carotenoid, and phenylalanine acid signaling were differentially expressed under AlP treatment, according to RNA-seq and RT-PCR analyses (Fig. 5B, C). For example, in the brassinosteroid and carotenoid acid metabolic pathways, DEGs 3 and 2, respectively, were differentially expressed. In the tryptophan biosynthesis pathway, the expression levels of Gretchen Hagen 3 (GH3, CTI12_AA417000) and small auxin-up RNA (SAUR, CTI12_AA600200) were upregulated (Fig. 5B, C).

A Plant hormone signal transduction KEGG pathway in response to the hardly soluble phosphorus source (AlP). The red shapes represented upregulated genes under the AlP; the blue shapes represented downregulated genes under the AlP; the shapes marked with yellow represented both the AlP and the KP upregulated genes. B Validation of RNA sequencing results by quantitative real-time PCR (qRT-PCR) of selected genes. C Heat maps showing variations in the expression of genes involved in hormone signal transduction biosynthesis under the AlP and the KP
The cytokinin response 1 (CRE1, CTI12_AA197330) gene involved in cytokinin (zeatin) signal transduction was significantly downregulated in response to AlP stress.
In brassinosteroid signal transduction, the BRI1-associated receptor kinase 1 BAK1, (CTI12_AA100010), BSK (CTI12_AA091650), and CYCD3 (CTI12_AA210800) were upregulated, whereas BSK (CTI12_AA380960) was downregulated. Only PR1 (CTI12_AA324860) showed upregulation in response to the AlP treatment in phenylalanine metabolism.
Analysis of DEGs involved in the artemisinin biosynthesis pathway
Our transcriptome analysis revealed many DEGs, such as the artemisinin biosynthetic genes. Seven essential artemisinin biosynthesis-related structural genes were examined, and their expression was investigated further using RT-PCR (Fig. 6). There are two independent pathways that lead to isopentenyl diphosphate (IPP) in the synthesis of artemisinin: the mevalonate (MVA) pathway and the methylerythritol phosphate (MEP) pathway in the cytosol and plastid, respectively (Fig. 6A).

A Artemisinin backbone biosynthesis KEGG in response to the hardly soluble phosphorus source (AlP). The red shapes represented upregulated genes under the AlP; the blue shapes represented downregulated genes under the AlP; the shapes marked with yellow represented both the AlP and the KP upregulated genes. B Validation of RNA sequencing results by quantitative real-time PCR (qRT-PCR) of selected genes. C Heat maps showing variations in the expression of genes involved in artemisinin biosynthesis under the AlP and the KP
None of the associated genes in the MEP pathway displayed altered expression in response to the AlP treatment. In the MVA pathway, only the acetyl-CoA acetyltransferase (ACAT, CTI12_AA520360) was upregulated, whereas the 3-hydroxy-3-methylglutaryl-CoAreductase (HMGR) was downregulated. When IPP and dimethylallyl diphosphate (DMAPP) are condensed to farnesyl diphosphate (FPP) via a farnesyl diphosphate synthase (FPS) catalyzed reaction, two distinct pathways are initiated, with the FPS (CTI12_AA302700) demonstrating upregulation. In the steps from farnesyl diphosphate to artemisinin formation, many genes, such as the cytochrome P450 monooxygenase (CYP) gene family, showed differential expression under the AlP treatment. Our transcriptomic findings, consistent with our expression findings, show amorphadiene monooxygenase (CYP71AV1, CTI12 AA566140), which has previously been shown to play a role in the artemisinin synthesis pathway, is being upregulated. The aldehyde dehydrogenase (ALDH, CTI12_AA008900) gene catalyzes dihydroartemisinic aldehyde conversion to dihydroartemisinic acid was upregulated (Fig. 6B, C).
Identification of TFs related to AlP treatment
To better understand A. annua’s transcriptional regulation mechanisms under different P form treatments, 30 TFs changed dramatically in response to the AlP (Fig. 7A). The 17 differentially expressed TF gene families were classified; 12 upregulated TF genes (Fig. 7B; table S5) and 18 downregulated TF genes (Fig. 7C; table S6) were obtained using the Plant TFDB database.

TF family percent distribution.) The upregulated TFs in response to different phosphorus treatments, and B the downregulated TFs in response to different phosphorus treatments
MYB, which contained three members, constituted the most prominent upregulated family among these transcription families. Two families, ERF and WRKY, were the largest among the downregulated TFs, with ten members (Fig. 7).
Discussion
After nitrogen (N), P is the second most crucial nutrient for plants (Amarasinghe et al. 2022; Kvakić et al. 2020). Many biological structures and functions of plants depend on the participation of P, such as the creation of nucleic acids (DNA and RNA), photosynthesis, glycolysis, respiration, membrane formation and stability, and enzyme activation and inactivation (Malhotra et al. 2018; Shen et al. 2011; Todeschini et al.2022; Vance et al.2003). Therefore, the P availability in the growth medium has been shown to have important effects on plant growth and development in numerous studies (Malhotra et al. 2018; Todeschini et al. 2022). P can be found in soil as mineral salts or organic compounds (Cordovil et al. 2020); however, most are hardly soluble (Miller et al. 2010). Therefore, our study compared the effect of AlPO4, as a hardly soluble P source, with the control KH2PO4, as a water-soluble P source. A. annua also showed a significant reduction in growth parameters, such as the plant height, the number of leaves, the stem diameter, and the leaf area (Fig. 1A–E), which also led to a reduction of total biomass under the AlP, compared to the KP (Fig. 1F). Interestingly, the artemisinin concentration was not reduced but significantly increased under the AlP, compared to the KP which was similar to Todeschini et al. (2022) study that showed the inverse relationship between P level and artemisinin concentration. However, plant growth and secondary metabolism responses to P availability are quite complex. For example, optimizing A. annua’s P and boron supply is critical for increasing dry matter production and/or artemisinin yield (Lulie et al. 2017). The yield of faba beans can be increased up to a certain level of P fertilizer application but exceeding that level decreases yield. Furthermore, Kebede et al. (2018) and Singh (2000) found no significant increase in oil content as P concentration increased. Again, limitations in the published data describing A. annua responses to P (Davies et al. 2011; Liu et al. 2003) indicated that artemisinin concentration decreased when P application was greater than 200 mg l−1 (KH2PO4). This decrease in artemisinin production occurred at a P concentration similar to that at which no further increase in plant growth was observed, and the amount of artemisinin per plant decreased dramatically (Liu et al. 2003). Furthermore, the increase in artesiminin concentration under stress, despite the reduction in plant growth parameters, may be due to artesmine’s expected essential role in elevating the harmful effect of stress and participating in plant cell protection. Many previous studies support our hypothesis because they discovered that artemisinin content (except in severe drought) and biosynthetic pathway genes are generally linked (Qureshi, et al. 2005; Yadav, et al. 2017; Vashisth, et al. 2018).
Hence, we performed transcriptomic analysis to gain more knowledge about the genetic behavior under the hardly soluble P source and more knowledge about which genes were responsible for these findings. cDNA libraries were used to generate transcriptome sequences. Using the leaves of the A. annua plant, 762 DEGs were identified in the comparison between the AlP vs. The KP (323 upregulated, 439 downregulated). The results of the GO enrichment analysis revealed that the two most enriched GO terms were “protein localization” and “enzyme inhibitor activity” (Fig. 3). Furthermore, the subcategories “catalytic activity,” “cellular metabolic process,” and “ion binding” were significantly enriched in the 1459 DEGs (Table S2). Based on KEGG pathway enrichment analysis, “glyoxylate and dicarboxylate metabolism,” “RNA degradation,” “glycine, serine and threonine metabolism,” and “proteasome pathways” were significantly enriched under the AlP compared to the KP (Fig. 4; Table S2). These findings suggested that hardly soluble P source could have an adverse effect on the regulation of artemisinin accumulation in A. annua. The AlP treatment also increased the expression of some structural genes involved in artemisinin biosynthesis.
Previous studies have shown that endogenous hormones are necessary for plant growth and development (Pacifici et al. 2015). In RNA sequencing data and RT-PCR analysis (Fig. 5B, C), it was discovered that the AlP treatment varied the expression of many genes involved in tryptophan, carotenoid, and phenylalanine acid pathways. The differentially expressed 3 and 2 DEGs were in the brassinosteroid and carotenoid acid metabolic pathways, respectively. These DEGs are widely thought to control plant growth and stress adaptation (Shi et al. 2020). Tafvizi et al. (2009) and Chen et al. (2007) previously investigated 14 and 10 differentially expressed genes (DEGs) that regulate cotton plant growth in the cytokinin (zeatin) and GA production pathways, respectively. The expression levels of GH3 and SAUR were upregulated in the tryptophan biosynthesis pathway, which promoted cell expansion and might be one of the causes of the maintaining artemisinin concentration under the AlP. According to earlier research, auxin can quickly and briefly increase the expression of three gene families, the SAUR family, the GH3 family, and the Aux/IAA family, which regulate plant development and growth. The auxin response factors (ARFs), controlling most SAUR, GH3 and Aux genes, activate or repress the expression of target genes (Woodward and Bartel 2005). In addition, overexpression of GH3-8 causes abnormal plant morphology as well as slowed growth and development in rice (Ding et al. 2008). On the other hand, the SAURs are the most common family of early auxin response genes, and they play a crucial role in regulating plant growth and development via hormonal and environmental cues (Ren and Gray 2015). Conversely, in our investigation, a factor that decreased cell division and shoot initiation was the downregulation of CRE1. As demonstrated by Laffont et al. (2015) in Medicago truncatula, the cytokinin CRE1 pathway influences root development and tolerance to abiotic and biotic environmental challenges in addition to being necessary for symbiotic nodule organogenesis. In our study, in brassinosteroid signal transduction, both BAK1, BSK (CTI12_AA091650), and CYCD3 were upregulated, whereas the BSK (CTI12_AA380960) was downregulated, which would be expected to have a significant effect on cell division and plant length. Although it has already been established that BSKs and BAK1 are both substrates of the BRI1 kinase, there is evidence to suggest that they have different functions in brassinosteroid signaling (Tang et al. 2008). Moreover, BSK is a crucial family of receptor-like cytoplasmic kinases (RLCK) in the first step of BR signal transduction, activating downstream phosphatase BSU1 (Kim et al. 2009).
Regarding artemisinin biosynthesis, the sesquiterpene route involves numerous enzymatic steps to produce artemisinin (Xie et al. 2016). In the artemisinin syntheses pathway, two distinct mechanisms, the MEP pathway in the plastid and the MVA pathway in the cytosol, are used to generate isopentenyl diphosphate, as shown in Fig. 6A (Vranová et al. 2013). Genes implicated in the MVA and MEP pathways have been described in numerous plants. Our transcriptome analysis uncovered many differentially expressed genes that were identified as producing artemisinin. In our investigation, the MVA pathway had all of the DEGs, whereas the MEP pathway did not exhibit appreciable changes in its gene expression in response to the AlP treatment. In the MVA pathway, the ACAT was upregulated, which produced isopentyl diphosphate (Xieet al.2016), whereas both HMGR and GGPS were downregulated. It showed upregulation of FPS but no change in ADS expression in our study. FPS overexpression increased artemisinin production (Han et al. 2006; Banyai et al. 2010), confirming the role of FPS and substrate availability in the regulation of artemisinin biosynthesis (Ikram and Simonsen 2017; Simonsen et al. 2013).
The CYP71AV1 gene, which is required for the two oxidation steps of artemisinin biosynthesis, amorpha4,11-diene, and artemisinic alcohol, was upregulated (Teoh et al. 2006; Ikram and Simonsen 2017). Furthermore, our study recorded the upregulation of ALDH. ALDH1 is used in yeast and plants for the metabolic engineering of artemisinin precursors (Xie et al.2016). Paddon et al. (2013) successfully produced artemisinic acid on an industrial scale by incorporating ALDH1 into engineered yeast strains. Zhang et al. (2011) found that ALDH1 was overexpressed in tobacco plants. Transgenic plants could synthesize dihydroartemisinic alcohol even if neither artemisinic nor dihydroartemisinic acid was found (Zhanget al.2011). Additionally, according to previous transcriptional investigations, ALDH1 expression in A. annua is directly linked to the generation of artemisinin (Dilshad et al. 2015; Xiang et al. 2015), showing that it participates in the biosynthetic process.
Despite the fact that HMGR was downregulated, artemisinin production increased in the presence of Alp, in contrast to previous studies that showed HMGR upregulation leads to an increase in artemisinin production. Despite the downregulation of HMGR, artemisinin levels increased due to the upregulation of most artemisinin-related genes (ACAT, FPS, CYP71AV1, and ALDH1).
On the contrary, it has been postulated that TFs play significant roles in the transcriptional control of gene expression through their binding to DNA regulatory elements (Hou et al. 2019; Mathelier et al. 2016). In addition, there is strong evidence that TFs have a role in phosphate homeostasis (Castrillo et al. 2013; Nilsson et al. 2007; Secco et al. 2012; Wang et al. 2009). Our study identified 31 TF families, such as GATA, NIN-like, C2H2, GRF, MYB-related, ERF, and HSF, and showed differential expression under the AlP (Fig. 7A, B). These transcription factors, under our treatment, play significant roles in alleviating P starvation, increasing phosphate acquisition, ROS homeostasis, root system establishment, and artemisinin biosynthesis regulation. Many TFs, including one MYB gene (CTI12_AA463820) and one MYB-related gene (CTI12_AA434110), showed upregulated expression after exposure to the AlP. In addition, downregulation was revealed in two MYB-related genes (CTI12_AA271060 and CTI12_AA271110) and two MYB genes (CTI12_AA572640 and CTI12_AA340370), which may be crucial for enhancing phosphate uptake, activating responses to P shortages, and root architecture. An earlier investigation in rice found that the OsMYB2P-1 gene controls downstream genes to repress or activate responses to P shortage and influence root architecture (Dai et al. 2012). Additionally, OsMYB4P overexpression might trigger the expression of several Pht genes and boost phosphate uptake (Yang et al. 2014). Additionally, it has been discovered that the MYB TFs, AaMYB1, AaMIXTA1, and AaTAR2 are crucial for increasing trichome initialization and artemisinin accumulation (Matías-Hernández et al. 2017; Shi et al. 2018; Zhou et al. 2020). Our findings also indicated that two WRKY TF members showed downregulation. Our results demonstrate the anticipated functions of these TF families under phosphate deprivation circumstances. AaWRKY1, the first isolated and characterized A. annua transcription factor, regulates artemisinin biosynthesis (Shen et al. 2016). A previous study (Dai et al. 2016) indicated that WRKY74 modifies rice’s susceptibility to phosphate deprivation. C2H2 displayed differential expression in our work, with just one TF upregulated and one TFs downregulated, both of which may be significantly involved in P starvation. TaZAT8, a C2H2-ZFP-type TF gene in wheat, is crucial in mediating wheat tolerance to a lack of P by controlling P uptake, ROS homeostasis, and the development of the root system (Ding et al. 2016). Additionally, our research showed that BHLH was downregulated, which may impact the final accumulation of artemisinin. Previous research has demonstrated the positive regulation of artemisinin production by AaORA and AabHLH1 (Ji et al. 2014; Lu et al. 2013). Five members of the ERF family were downregulated in response to AlP. As a result, ERF may be crucial in coping with P stress and may result in a large accumulation of artemisinin. ADS and CYP71AV1 were both favorably regulated by AaERF1 and AaERF2 simultaneously, which helped plants produce artemisinin and artemisinic acid (Yu et al. 2012). Overall, our results will help to understand how hardly soluble P fertilizer influences the transcriptional regulation of A. annua L. about artemisinin and plant hormone production in these conditions.
Data availability
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number (s) can be found at GSA, accession number: CRA008375 at the following link: https://bigd.big.ac.cn/gsa/browse/CRA008375.
References
Aftab T, Khan MMA, Ferreira JFS (2014) Effect of mineral nutrition, growth regulators and environmental stresses on biomass production and Artemisinin concentration of Artemisia annua L. In: Aftab T, Ferreira JFS, Khan MMA, Naeem M (eds) Artemisia annua - Pharmacology and Biotechnology. Springer, Berlin Heidelberg Berlin, Heidelberg, pp 157–172
Ahmed S, Chouhan R, Junaid A et al (2023) Transcriptome analysis and differential expression in Arabidopsis thaliana in response to rohitukine (a chromone alkaloid) treatment. Funct Integr Genomics 23:35. https://doi.org/10.1007/s10142-023-00961-0
Amarasinghe T, Madhusha C, Munaweera I, Kottegoda N (2022) Review on mechanisms of phosphate solubilization in rock phosphate fertilizer. Commun Soil Sci Plant Anal 53:1–17. https://doi.org/10.1080/00103624.2022.2034849
Augusto L, Achat DL, Jonard M, Vidal D, Ringeval B (2017) Soil parent material—a major driver of plant nutrient limitations in terrestrial ecosystems. Glob Chang Biol 23:3808–3824. https://doi.org/10.1111/gcb.13691
Banyai W, Kirdmanee C, Mii M, Supaibulwatana K (2010) Overexpression of farnesyl pyrophosphate synthase (FPS) gene affected artemisinin content and growth of Artemisia annua L. Plant Cell Tissue Organ Cult 103:255–265. https://doi.org/10.1007/s11240-010-9775-8
Baraldi R, Isacchi B, Predieri S, Marconi G, Vincieri FF, Bilia AR (2008) Distribution of artemisinin and bioactive flavonoids from Artemisia annua L. during plant growth. Biochem Syst Ecol 36:340–348. https://doi.org/10.1016/j.bse.2007.11.002
Castrillo G, Sánchez-Bermejo E, de Lorenzo L, Crevillén P, Fraile-Escanciano A, Tc M, Mouriz A, Catarecha P, Sobrino-Plata J, Olsson S, Leo Del Puerto Y, Mateos I, Rojo E, Hernández LE, Jarillo JA, Piñeiro M, Paz-Ares J, Leyva A (2013) WRKY6 transcription factor restricts arsenate uptake and transposon activation in Arabidopsis. Plant Cell 25:2944–2957. https://doi.org/10.1105/tpc.113.114009
Chao LM, Liu YQ, Chen DY, Xue XY, Mao YB, Chen XY (2017) Arabidopsis transcription factors SPL1 and SPL12 confer plant thermotolerance at reproductive stage. Mol Plant 10:735–748. https://doi.org/10.1016/j.molp.2017.03.010
Chen Y, Ye G, Zhang L, Wang Y, Zhang X, Chen D (2007) Effect of trans-Bacillus thuringiensis gene on gibberellic acid and zeatin contents and boll development in cotton. Field Crop Res 103:5–10. https://doi.org/10.1016/j.fcr.2007.04.003
Cordovil CMdS, Bittman S, Brito LM, Goss MJ, Hunt D, Serra J, Gourley C, Aarons S, Skiba U, Amon B, Vale MJ, Cruz S, Reis R, Dalgaard T, Hutchings N (2020) Chapter 22 - Climate-resilient and smart agricultural management tools to cope with climate change-induced soil quality decline. In: Prasad MNV, Pietrzykowski M (eds) Climate change and soil interactions. Elsevier, pp 613–662
Dai X, Wang Y, Yang A, Zhang WH (2012) OsMYB2P-1, an R2R3MYB transcription factor, is involved in the regulation of phosphate-starvation responses and root architecture in rice. Plant Physiol 159:169–183. https://doi.org/10.1104/pp.112.194217
Dai X, Wang Y, Zhang WH (2016) OsWRKY74, a WRKY transcription factor, modulates tolerance to phosphate starvation in rice. J Exp Bot 67:947–960. https://doi.org/10.1093/jxb/erv515
Davies MJ, Atkinson CJ, Burns C, Arroo R, Woolley J (2011) Increases in leaf artemisinin concentration in Artemisia annua in response to the application of phosphorus and boron. Ind Crop Prod 34(3):1465–1473. https://doi.org/10.1016/j.indcrop.2011.05.00
Deng QW, Luo XD, Chen YL, Zhou Y, Zhang FT, Hu BL, Xie JK (2018) Transcriptome analysis of phosphorus stress responsiveness in the seedlings of Dongxiang wild rice (Oryza rufipogon Griff.). Biol Res 51:7. https://doi.org/10.1186/s40659-018-0155-x
Dilshad E, Cusido RM, Palazon J, Estrada KR, Bonfill M, Mirza B (2015) Enhanced artemisinin yield by expression of rol genes in Artemisia annua. Malar J 14:424. https://doi.org/10.1186/s12936-015-0951-5
Ding X, Cao Y, Huang L, Zhao J, Xu C, Li X, Wang S (2008) Activation of the indole-3-acetic acid-amido synthetase GH3-8 suppresses expansion expression and promotes salicylate- and jasmonate-independent basal immunity in rice. Plant Cell 20:228–240. https://doi.org/10.1105/tpc.107.055657
Ding W, Wang Y, Fang W, Gao S, Li X, Xiao K (2016) TaZAT8, a C2H2-ZFP type transcription factor gene in wheat, plays critical roles in mediating tolerance to Pi deprivation through regulating P acquisition, ROS homeostasis and root system establishment. Physiol Plant 158:297–311. https://doi.org/10.1111/ppl.12467
Du Q, Wang K, Xu C, Zou C, Xie C, Xu Y, Li WX (2016) Strand-specific RNA-Seq transcriptome analysis of genotypes with and without low-phosphorus tolerance provides novel insights into phosphorus-use efficiency in maize. BMC Plant Biol 16:222. https://doi.org/10.1186/s12870-016-0903-4
El-Sappah AH, Elbaiomy RG, Elrys AS, Wang Y, Zhu Y, Huang Q, Yan K, Xianming Z, Abbas M, El-Tarabily KA, Li J (2021) Genome-wide identification and expression analysis of metal tolerance protein gene family in Medicago truncatula under a broad range of heavy metal stress. Front Genet 12:713224. https://doi.org/10.3389/fgene.2021.713224
El-Sappah AH, Abbas M, Rather SA, Wani SH, Soaud N, Noor Z, Qiulan H, Eldomiaty AS, Mir RR, Li J (2023) Genome-wide identification and expression analysis of metal tolerance protein (MTP) gene family in soybean (Glycine max) under heavy metal stress. Mol Biol Rep 50:2975–2990. https://doi.org/10.1007/s11033-022-08100-x
Giles CD, Hsu P-C, Richardson AE, Hurst MRH, Hill JE (2014) Plant assimilation of phosphorus from an insoluble organic form is improved by addition of an organic anion producing Pseudomonas sp. Soil Biol Biochem 68:263–269. https://doi.org/10.1016/j.soilbio.2013.09.026
Han JL, Liu BY, Ye HC, Wang H, Li ZQ, Li GF (2006) Effects of overexpression of the endogenous farnesyl diphosphate synthase on the artemisinin content in Artemisia annua L. J Integr Plant Biol 48:482–487. https://doi.org/10.1111/j.1744-7909.2006.00208.x
Hou Z, Yin J, Lu Y, Song J, Wang S, Wei S, Liu Z, Zhang Y, Fang Z (2019) Transcriptomic analysis reveals the temporal and spatial changes in physiological process and gene expression in common buckwheat (Fagopyrum esculentum Moench) Grown under Drought Stress. Agronomy 9:569
Hu Y, Ye X, Shi L, Duan H, Xu F (2010) Genotypic differences in root morphology and phosphorus uptake kinetics in brassica napus under low phosphorus supply. J Plant Nutr 33:889–901. https://doi.org/10.1080/01904161003658239
Ikram NK, Simonsen HT (2017) A review of biotechnological artemisinin production in plants. Front Plant Sci 8:1966
Ji Y, Xiao J, Shen Y, Ma D, Li Z, Pu G, Li X, Huang L, Liu B, Ye H, Wang H (2014) Cloning and characterization of AabHLH1, a bHLH transcription factor that positively regulates artemisinin biosynthesis in Artemisia annua. Plant Cell Physiol 55:1592–1604. https://doi.org/10.1093/pcp/pcu090
Kebede B, Nigussie A, Chala M (2018) Response of Artemisia (Artemisia annua L.) to nitrogen and phosphorus fertilizers in Wondo. Acad Res J Agric Sci Res 5(6):407–413. https://doi.org/10.14662/ARJASR2017.051
Kim TW, Guan S, Sun Y, Deng Z, Tang W, Shang JX, Sun Y, Burlingame AL, Wang ZY (2009) Brassinosteroid signal transduction from cell-surface receptor kinases to nuclear transcription factors. Nat Cell Biol 11:1254–1260. https://doi.org/10.1038/ncb1970
Kvakić M, Tzagkarakis G, Pellerin S, Ciais P, Goll D, Mollier A, Ringeval B (2020) Carbon and phosphorus allocation in annual plants: an optimal functioning approach. Front Plant Sci 11:149. https://doi.org/10.3389/fpls.2020.00149
Laffont C, Rey T, André O, Novero M, Kazmierczak T, Debellé F, Bonfante P, Jacquet C, Frugier F (2015) The CRE1 cytokinin pathway is differentially recruited depending on Medicago truncatula root environments and negatively regulates resistance to a pathogen. PLoS ONE 10:e0116819. https://doi.org/10.1371/journal.pone.0116819
Lambers H (2022) Phosphorus acquisition and utilization in plants. Annu Rev Plant Biol 73:17–42. https://doi.org/10.1146/annurev-arplant-102720-125738
Lee K-K, Mok I-K, Yoon M-H, Kim H-J, Chung D (2012) Mechanisms of phosphate solubilization by PSB (phosphate-solubilizing bacteria) in soil. Korean J Soil Sci Fert 45:169–176. https://doi.org/10.7745/KJSSF.2012.45.2.169
Leišová-Svobodová L, Psota V, Stočes Š et al (2020) Comparative de novo transcriptome analysis of barley varieties with different malting qualities. Funct Integr Genomics 20:801–812. https://doi.org/10.1007/s10142-020-00750-z
Li X, Luo L, Yang J, Li B, Yuan H (2015) Mechanisms for solubilization of various insoluble phosphates and activation of immobilized phosphates in different soils by an efficient and salinity-tolerant Aspergillus niger strain An2. Appl Biochem Biotechnol 175:2755–2768. https://doi.org/10.1007/s12010-014-1465-2
Liu C-Z, Guo C, Wang Y, Ouyang F (2003) Factors influencing artemisinin production from shoot cultures of Artemisia annua L. World J Microbiol Biotechnol 19:535–538. https://doi.org/10.1023/A:1025158416832
Liu X, Chu S, Sun C, Xu H, Zhang J, Jiao Y, Zhang D (2020) Genome-wide identification of low phosphorus responsive microRNAs in two soybean genotypes by high-throughput sequencing. Funct Integr Genomics 20:825–838. https://doi.org/10.1007/s10142-020-00754-9
Lu X, Zhang L, Zhang F, Jiang W, Shen Q, Zhang L, Lv Z, Wang G, Tang K (2013) AaORA, a trichome-specific AP2/ERF transcription factor of Artemisia annua, is a positive regulator in the artemisinin biosynthetic pathway and in disease resistance to Botrytis cinerea. New Phytol 198:1191–1202. https://doi.org/10.1111/nph.12207
Lulie B, Nigussie A, Chala M (2017) Response of Artemisia (Artemisia annua L.) to nitrogen and phosphorus fertilizers in wondo response of Artemisia (Artemisia annua L.) to nitrogen and phosphorus fertilizers in Wondo Genet and Koka, Ethiopia. Acad Res J Agric Sci Res 5(6):407–413
Ma C, Wang H, Lu X, Li H, Liu B, Xu G (2007) Analysis of Artemisia annua L. volatile oil by comprehensive two-dimensional gas chromatography time-of-flight mass spectrometry. J Chromatogr A 1150:50–53. https://doi.org/10.1016/j.chroma.2006.08.080
Malhotra H, Vandana, Sharma S, Pandey R (2018) Phosphorus nutrition: plant growth in response to deficiency and excess. In: Hasanuzzaman M, Fujita M, Oku H, Nahar K, Hawrylak-Nowak B (eds) Plant nutrients and abiotic stress tolerance. Springer, Singapore. https://doi.org/10.1007/978-981-10-9044-8_7
Mathelier A, Fornes O, Arenillas DJ, Chen CY, Denay G, Lee J, Shi W, Shyr C, Tan G, Worsley-Hunt R, Zhang AW, Parcy F, Lenhard B, Sandelin A, Wasserman WW (2016) JASPAR 2016: a major expansion and update of the open-access database of transcription factor binding profiles. Nucleic Acids Res 44:D110-115. https://doi.org/10.1093/nar/gkv1176
Matías-Hernández L, Jiang W, Yang K, Tang K, Brodelius PE, Pelaz S (2017) AaMYB1 and its orthologue AtMYB61 affect terpene metabolism and trichome development in Artemisia annua and Arabidopsis thaliana. Plant J 90:520–534. https://doi.org/10.1111/tpj.13509
Miller SH, Browne P, Prigent-Combaret C, Combes-Meynet E, Morrissey JP, O’Gara F (2010) Biochemical and genomic comparison of inorganic phosphate solubilization in Pseudomonas species. Environmental Microbiol Reports 2:403–411. https://doi.org/10.1111/j.1758-2229.2009.00105.x
Müller M, Brandes D (1997) Growth and development of Artemisia annua L. on different soil types. Verhandlungen der Gesellschaft für Ökologie 27:435–460. https://doi.org/10.24355/dbbs.084-200511080100-270
Nilsson L, Müller R, Nielsen TH (2007) Increased expression of the MYB-related transcription factor, PHR1, leads to enhanced phosphate uptake in Arabidopsis thaliana. Plant Cell Environ 30:1499–1512. https://doi.org/10.1111/j.1365-3040.2007.01734.x
Pacifici E, Polverari L, Sabatini S (2015) Plant hormone cross-talk: the pivot of root growth. J Exp Bot 66:1113–1121. https://doi.org/10.1093/jxb/eru534
Paddon CJ, Westfall PJ, Pitera DJ, Benjamin K, Fisher K, McPhee D, Leavell MD, Tai A, Main A, Eng D, Polichuk DR, Teoh KH, Reed DW, Treynor T, Lenihan J, Fleck M, Bajad S, Dang G, Dengrove D, Diola D, Dorin G, Ellens KW, Fickes S, Galazzo J, Gaucher SP, Geistlinger T, Henry R, Hepp M, Horning T, Iqbal T, Jiang H, Kizer L, Lieu B, Melis D, Moss N, Regentin R, Secrest S, Tsuruta H, Vazquez R, Westblade LF, Xu L, Yu M, Zhang Y, Zhao L, Lievense J, Covello PS, Keasling JD, Reiling KK, Renninger NS, Newman JD (2013) High-level semi-synthetic production of the potent antimalarial artemisinin. Nature 496:528–532. https://doi.org/10.1038/nature12051
Pearse SJ, Veneklaas EJ, Cawthray G, Bolland MD, Lambers H (2007) Carboxylate composition of root exudates does not relate consistently to a crop species’ ability to use phosphorus from aluminium, iron or calcium phosphate sources. New Phytol 173:181–190. https://doi.org/10.1111/j.1469-8137.2006.01897.x
Pradhan M, Sahoo RK, Pradhan C, Tuteja N, Mohanty S (2017) Contribution of native phosphorous-solubilizing bacteria of acid soils on phosphorous acquisition in peanut (Arachis hypogaea L.). Protoplasma 254:2225–2236. https://doi.org/10.1007/s00709-017-1112-1
Qureshi MI, Israr M, Abdin M, Iqbal M (2005) Responses of Artemisia annua L. to lead and salt-induced oxidative stress. Environ Exp Bot 53:185–193
Ren H, Gray WM (2015) SAUR proteins as effectors of hormonal and environmental signals in plant growth. Mol Plant 8:1153–1164. https://doi.org/10.1016/j.molp.2015.05.003
Ren P, Meng Y, Li B, Ma X, Si E, Lai Y, Wang J, Yao L, Yang K, Shang X, Wang H (2018) Molecular mechanisms of acclimatization to phosphorus starvation and recovery underlying full-length transcriptome profiling in barley (Hordeum vulgare L.). Front Plant Sci 9:500. https://doi.org/10.3389/fpls.2018.00500
Rouached H, Arpat AB, Poirier Y (2010) Regulation of phosphate starvation responses in plants: signaling players and cross-talks. Mol Plant 3:288–299. https://doi.org/10.1093/mp/ssp120
Ryan P, Delhaize E, Jones D (2001) Function and mechanism of organic anion exudation from plant roots. Annu Rev Plant Physiol Plant Mol Biol 52:527–560. https://doi.org/10.1146/annurev.arplant.52.1.527
Secco D, Wang C, Arpat BA, Wang Z, Poirier Y, Tyerman SD, Wu P, Shou H, Whelan J (2012) The emerging importance of the SPX domain-containing proteins in phosphate homeostasis. New Phytol 193:842–851. https://doi.org/10.1111/j.1469-8137.2011.04002.x
Sharma SB, Sayyed RZ, Trivedi MH, Gobi TA (2013) Phosphate solubilizing microbes: sustainable approach for managing phosphorus deficiency in agricultural soils. SpringerPlus 2:1–14
Shen J, Rengel Z, Tang C, Zhang F (2003) Role of phosphorus nutrition in development of cluster roots and release of carboxylates in soil-grown Lupinus albus. Plant Soil 248:199–206. https://doi.org/10.1023/A:1022375229625
Shen J, Yuan L, Zhang J, Li H, Bai Z, Chen X, Zhang W, Zhang F (2011) Phosphorus dynamics: from soil to plant. Plant Physiol 156:997–1005. https://doi.org/10.1104/pp.111.175232
Shen Q, Yan T, Fu X, Tang K (2016) Transcriptional regulation of artemisinin biosynthesis in Artemisia annua L. Science Bulletin 61:18–25. https://doi.org/10.1007/s11434-015-0983-9
Shi P, Fu X, Shen Q, Liu M, Pan Q, Tang Y, Jiang W, Lv Z, Yan T, Ma Y, Chen M, Hao X, Liu P, Li L, Sun X, Tang K (2018) The roles of AaMIXTA1 in regulating the initiation of glandular trichomes and cuticle biosynthesis in Artemisia annua. New Phytol 217:261–276. https://doi.org/10.1111/nph.14789
Shi J, Wang N, Zhou H, Xu Q, Yan G (2020) Transcriptome analyses provide insights into the homeostatic regulation of axillary buds in upland cotton (Gossypium hirsutum L). BMC Plant Biol 20:228. https://doi.org/10.1186/s12870-020-02436-x
Simonsen HT, Weitzel C, Christensen SB (2013) Guaianolide sesquiterpenoids: pharmacology and biosynthesis. In: Ramawat K, Mérillon JM (eds) Natural products. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-22144-6_134
Singh M (2000) Effect of nitrogen, phosphorus and potassium nutrition on herb, oil and artemisinin yield of Artemisia annua under semi-arid tropical condition. J Med Arom Plant Sci 22:368–369
Singh D, Singh CK, Taunk J et al (2021) Transcriptome skimming of lentil (Lens culinaris Medikus) cultivars with contrast reaction to salt stress. Funct Integr Genomics 21:139–156. https://doi.org/10.1007/s10142-020-00766-5
Smith SE, Smith FA (2011) Roles of arbuscular mycorrhizas in plant nutrition and growth: new paradigms from cellular to ecosystem scales. Annu Rev Plant Biol 62:227–250. https://doi.org/10.1146/annurev-arplant-042110-103846
Stringham RW, Moore GL, Teager DS, Yue TY (2018) Analysis and isolation of potential artemisinin precursors from waste streams of Artemisia Annua extraction. ACS Omega 3:7803–7808. https://doi.org/10.1021/acsomega.8b00974
Tafvizi F, Farahani F, Sheidai M, Nejadsattari T (2009) Effects of zeatin and activated charcoal in proliferation of shoots and direct regeneration in cotton (Gossypium hirsutum L.). Afr J Biotechnol 8(22):6220–6227. https://doi.org/10.5897/AJB09.1211
Tang W, Kim TW, Oses-Prieto JA, Sun Y, Deng Z, Zhu S, Wang R, Burlingame AL, Wang ZY (2008) BSKs mediate signal transduction from the receptor kinase BRI1 in Arabidopsis. Science 321:557–560. https://doi.org/10.1126/science.1156973
Teoh KH, Polichuk DR, Reed DW, Nowak G, Covello PS (2006) Artemisia annua L. (Asteraceae) trichome-specific cDNAs reveal CYP71AV1, a cytochrome P450 with a key role in the biosynthesis of the antimalarial sesquiterpene lactone artemisinin. FEBS Lett 580:1411–1416. https://doi.org/10.1016/j.febslet.2006.01.065
Todeschini V, Anastasia F, Massa N, Marsano F, Cesaro P, Bona E, Gamalero E, Oddi L, Lingua G (2022) Impact of phosphatic nutrition on growth parameters and artemisinin production in Artemisia annua plants inoculated or not with Funneliformis mosseae. Life 12(4):497. https://doi.org/10.3390/life12040497
Vance CP, Uhde-Stone C, Allan DL (2003) Phosphorus acquisition and use: critical adaptations by plants for securing a nonrenewable resource. New Phytol 157:423–447. https://doi.org/10.1046/j.1469-8137.2003.00695.x
Vashisth D, Kumar R, Rastogi S, Patel VK, Kalra A, Gupta MM, Gupta AK, Shasany AK (2018) Transcriptome changes induced by abiotic stresses in Artemisia annua. Sci Rep 8:3423. https://doi.org/10.1038/s41598-018-21598-1
Veneklaas EJ, Lambers H, Bragg J, Finnegan PM, Lovelock CE, Plaxton WC, Price CA, Scheible WR, Shane MW, White PJ, Raven JA (2012) Opportunities for improving phosphorus-use efficiency in crop plants. New Phytol 195:306–320. https://doi.org/10.1111/j.1469-8137.2012.04190.x
Vranová E, Coman D, Gruissem W (2013) Network analysis of the MVA and MEP pathways for isoprenoid synthesis. Annu Rev Plant Biol 64:665–700. https://doi.org/10.1146/annurev-arplant-050312-120116
Wan L-Y, Qi S-S, Dai Z-C, Zou CB, Song Y-G, Hu Z-Y, Zhu B, Du D-L (2018) Growth responses of Canada goldenrod (Solidago canadensis L.) to increased nitrogen supply correlate with bioavailability of insoluble phosphorus source. Ecol Res 33:261–269. https://doi.org/10.1007/s11284-017-1552-2
Wang C, Ying S, Huang H, Li K, Wu P, Shou H (2009) Involvement of OsSPX1 in phosphate homeostasis in rice. The Plant j: Cell Molecular Biol 57:895–904. https://doi.org/10.1111/j.1365-313X.2008.03734.x
Wani KI, Choudhary S, Zehra A, Naeem M, Weathers P, Aftab T (2021) Enhancing artemisinin content in and delivery from Artemisia annua: a review of alternative, classical, and transgenic approaches. Planta 254:1–15
Wani KI, Zehra A, Choudhary S, Naeem M, Khan MMA, Khan R, Aftab T (2022) Exogenous strigolactone (GR24) positively regulates growth, photosynthesis, and improves glandular trichome attributes for enhanced artemisinin production in Artemisia annua. J Plant Growth Regul. https://doi.org/10.1007/s00344-022-10654-w
Williamson LC, Ribrioux SPCP, Fitter AH, Leyser HMO (2001) Phosphate availability regulates root system architecture in Arabidopsis. Plant Physiol 126:875–882. https://doi.org/10.1104/pp.126.2.875
Woodward AW, Bartel B (2005) Auxin: regulation, action, and interaction. Ann Bot 95:707–735. https://doi.org/10.1093/aob/mci083
Xiang L, Zhu S, Zhao T, Zhang M, Liu W, Chen M, Lan X, Liao Z (2015) Enhancement of artemisinin content and relative expression of genes of artemisinin biosynthesis in Artemisia annua by exogenous MeJA treatment. Plant Growth Regul 75:435–441. https://doi.org/10.1007/s10725-014-0004-z
Xie D-Y, Ma D-M, Judd R, Jones AL (2016) Artemisinin biosynthesis in Artemisia annua and metabolic engineering: questions, challenges, and perspectives. Phytochem Rev 15:1093–1114. https://doi.org/10.1007/s11101-016-9480-2
Yadav RK, Sangwan RS, Srivastava AK, Sangwan NS (2017) Prolonged exposure to salt stress affects specialized metabolites-artemisinin and essential oil accumulation in Artemisia annua L.: metabolic acclimation in preferential favour of enhanced terpenoid accumulation accompanying vegetative to reproductive phase transition. Protoplasma 254:505–522. https://doi.org/10.1007/s00709-016-0971-1
Yang WT, Baek D, Yun DJ, Hwang WH, Park DS, Nam MH, Chung ES, Chung YS, Yi YB, Kim DH (2014) Overexpression of OsMYB4P, an R2R3-type MYB transcriptional activator, increases phosphate acquisition in rice. Plant Physiol Biochem: PPB 80:259–267. https://doi.org/10.1016/j.plaphy.2014.02.024
Yu ZX, Li JX, Yang CQ, Hu WL, Wang LJ, Chen XY (2012) The jasmonate-responsive AP2/ERF transcription factors AaERF1 and AaERF2 positively regulate artemisinin biosynthesis in Artemisia annua L. Mol Plant 5:353–365. https://doi.org/10.1093/mp/ssr087
Zhang Y, Nowak G, Reed DW, Covello PS (2011) The production of artemisinin precursors in tobacco. Plant Biotechnol J 9:445–454. https://doi.org/10.1111/j.1467-7652.2010.00556.x
Zhang XB, Guo LP, Qiu ZD, Qu XB, Wang H, Jing ZX, Huang LQ (2017) Analysis of spatial distribution of artemisinin in Artemisia annua in China. China Journal of Chinese Materia Medica 42(22):4341–4345. https://doi.org/10.19540/j.cnki.cjcmm.2017.0168
Zhou Z, Tan H, Li Q, Li Q, Wang Y, Bu Q, Li Y, Wu Y, Chen W, Zhang L (2020) Trichome and artemisinin regulator 2 positively regulates trichome development and artemisinin biosynthesis in Artemisia annua. New Phytol 228:932–945. https://doi.org/10.1111/nph.16777
Funding
This research was funded by the National Natural Science Foundation of China (81903752), Guangxi Major Science and Technology Project of China (GuikeAA22096021), Natural Science Foundation of Guangxi Province (2019GXNSFBA18502), Scientific Research Funding Project of Guangxi Botanical Garden of Medicinal Plants (GYJ202013), Research and Innovation Team Building Project of Guangxi Botanical Garden of Medicinal Plants (GYCH2019008), National Natural Science Foundation of China (81560623), Key Laboratory Construction Program of Guangxi Health commission (ZJC2020003), and Hainan Natural Science Foundation (No. 2019RC316).
Author information
Authors and Affiliations
Contributions
Conceptualization, L. W., Q. H., Z. Z., S. W., J. F., and A. H. El-S.; methodology, L. W., Q. H., A. H. El-S., and S. W.; software, L. W., Q. H., S. W., and A. H. El-S.; validation, L. P. and J. F.; formal analysis, L. W., Q. H., S. W., and A. H. El-S.; investigation, R. G. E., A. H. El-S., and Z. Z.; resources, W. L.; writing—original draft preparation, L. W., Q. H., S. W., and A. H. El-S.; writing—review and editing, L. W., Q. H., A. S. E., M. M. A. E., G. J., S. W., L. G., and A. H. El-S.; visualization, X. J., L. S., Z. Z.; project administration, L. W.; funding acquisition, L. W., S. W., Z. Z., and J. F. J. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Wan, L., Huang, Q., Ji, X. et al. RNA sequencing in Artemisia annua L explored the genetic and metabolic responses to hardly soluble aluminum phosphate treatment. Funct Integr Genomics 23, 141 (2023). https://doi.org/10.1007/s10142-023-01067-3
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10142-023-01067-3