
Abstract
The study of the food microbiome has gained considerable interest in recent years, mainly due to the wide range of applications that can be derived from the analysis of metagenomes. Among these applications, it is worth mentioning the possibility of using metagenomic analyses to determine food authenticity, to assess the microbiological safety of foods thanks to the detection and tracking of pathogens, antibiotic resistance genes and other undesirable traits, as well to identify the microorganisms responsible for food processing defects. Metataxonomics and metagenomics are currently the gold standard methodologies to explore the full potential of metagenomes in the food industry. However, there are still a number of challenges that must be solved in order to implement these methods routinely in food chain monitoring, and for the regulatory agencies to take them into account in their opinions. These challenges include the difficulties of analysing foods and food-related environments with a low microbial load, the lack of validated bioinformatics pipelines adapted to food microbiomes and the difficulty of assessing the viability of the detected microorganisms. This review summarizes the methods of microbiome analysis that have been used, so far, in foods and food-related environments, with a specific focus on those involving Next-Generation Sequencing technologies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
A microbiome is a dynamic ecosystem involving an interacting microbial community (the microbiota) and the surrounding environment, in which the microbes display different activities that shape a specific biological niche. Foods and feeds, basically made from animal and plant origin, provide the essential macro and micronutrients to sustain the life of other organisms, constituting an ecological niche for the proliferation of a large microbial diversity. Specific microbial consortia are associated not only with the raw materials and foods themselves, but also with the processing environments, being a kind of “feedback system” that must be balanced in order to obtain safe products with high quality standards (De Filippis et al. 2021). Almost every food has an associated microbiome and/or microbiome-related DNA (the metagenome). The food microbiota is considered pivotal in the imprinting of distinct and specific organoleptic features, mainly in fermented foods. Traditionally, this microbiota has been studied using culture-dependent techniques. Today, these techniques are still the most common to characterize microorganisms from food, to establish the methodology needed to guarantee the microbiological safety of the products and to determine the risk assessment in the agri-food industry. The main limitation of culture-dependent methods is that they only allow the study of culturable microorganisms; therefore, there is a risk of not having an accurate view of the biodiversity of the microbial population present in the food.
It was at the beginning of this century when the so-called culture-independent techniques began to be applied in food microbiology. These methods are based on the analysis of macromolecules (mainly DNA), which allows the identification of the members of the micro-ecosystem to be determined without the need to culture them (Cocolin et al. 2013). The first methods developed were based on the direct analysis of the DNA extracted from the sample, followed by PCR amplification of DNA fragments and a further separation of the amplicons using electrophoretic or chromatographic techniques, or using DNA-DNA hybridization, giving rise to methods such as PCR-DGGE (PCR-Denaturing Gradient Gel Electrophoresis), T-RFLP (Terminal Restriction Fragment Length Polymorphism) or FISH (Fluorescence in situ Hybridization) (Pogacic et al. 2010). However, in recent years, methods based on Next Generation Sequencing (NGS) methodologies have revolutionized how we study the food microbiome. Thanks to these techniques, we are able to extract the information encrypted in the microbiome and unmask desirable and undesirable members of the community, as well as their potential metabolic functions. This information gives novel opportunities to explore the diversity and functionality of microbes in foods, as well as in food processing and producing environments (De Filippis et al. 2021).
Among the NGS-based approaches most commonly used at present, it is important to highlight that metataxonomic approaches, which are based on sequencing 16S rRNA as a marker gene for taxonomic composition characterization, should be clearly differentiated from metagenomic techniques, which involve untargeted sequencing of all the genetic material retrieved from microbial community samples. Microbial fingerprinting using shotgun or gene-targeted NGS analyses could be a valuable extension of current microbiological methods for the detection and tracking of pathogens in the food industry, to support the authenticity and/or geographical origin of foods, as well as a powerful tool to analyse the spread of antibiotic resistance genes, among other applications. In this review, we intend to give an insight into how the introduction of novel methods of microbiome analysis have impacted on food microbiology, focusing on those methods that use NGS technologies for the study of metagenomes, and the potential applications derived from these analyses.
Sequencing methodologies: short-reads vs long-reads
From the first DNA sequencing methodologies developed in the mid-1970s to date, steady advances have dramatically increased the power, resolution and speed of analysis offered by the different generations of sequencing platforms. This revolution has been accompanied by a dramatic reduction in sequencing costs and has posited DNA sequencing as a preeminent tool in several biology fields. In the last few decades, it has also found application in deciphering and monitoring the microbial communities associated with foodstuffs, food production processes, and food processing environments, known to represent key determinants of food quality and safety.
The first appearance of the so-called second generation of sequencing platforms has largely increased sequencers performance by enabling the sequencing in parallel of millions of DNA molecules in a single experiment (Heather and Chain 2016). These include pyrosequencing platforms such as the one developed by Roche which, although it is not in use any longer, was the first technology from these second generation of platforms broadly offered commercially. This technology was rapidly followed by other sequencing platforms such as those offered by Illumina or Solexa which are still widely employed. These second generation platforms are characterized for generating millions of short DNA sequencing reads, usually ranging from 150 bp up to 500 bp in length, in a single experiment. Besides, they have enabled scientists to multiplex or parallelize several samples to be sequenced simultaneously in a single run. The constant upgrades of second-generation sequencing platforms released to the market have continuously offered impressive advances, particularly concerning ease of library preparation and sequencer manipulation, increase in the amount of data generated and enhanced accuracy, while reducing processing time. These platforms have revolutionized how we tackle the study of complex microbial communities and have made possible the study of the implication of a broad range of microorganisms in diverse biological processes and to discern their role in complex microbial ecosystems, including those related to food production. Versatility, affordances, and performance of these platforms have promoted their application for fungal profiling in complex microbial communities, based on Internal Transcribed Spacer (ITS) and/or 18S rRNA gene sequencing; bacterial profiling based on16S rRNA gene sequencing; and functional analyses based on shotgun metagenomics or other marker gene sequencing approaches. These methods provide a better characterization of the microbial diversity in a given sample as compared to traditional microbial community analyses, such as those relying on DGGE-electrophoresis or culture dependent analyses. Besides, whole genome sequencing-based typing approaches allow the most comprehensive comparison possible of individual strains as compared to traditional single-nucleotide polymorphism (SNP) and multilocus sequence typing (MLST) analyses, thus NGS technologies have revolutionized both microbial community and individual strain profiling. Specifically, some of these second-generation sequencing platforms have seen exceptional applications of relevance in food production, from farm to fork, as a means of rapidly tracking the origin of outbreaks (Koutsoumanis et al. 2019), to the surveillance of foodborne viruses or microorganisms that could impose health threats (Desdouits et al. 2020); to identify microbial populations responsible for food processing defects (Quigley et al. 2016; Ritschard et al. 2018; Xue et al. 2021a), or quality attributes in fermented foods (Dertli and Çon 2017; Walsh et al. 2016), or even to assess food authenticity, in terms of geographic origin or production processes, based on microbial fingerprints (Anagnostopoulos et al. 2019; Haynes et al. 2019; Kamilari et al. 2019; Mezzasalma et al. 2017). The application of such tools to identify and monitor the appearance of undesirable traits among the bacterial communities inhabiting food processing environments such as antibiotic resistance, traits related to microbial persistence in food processing environments or those involved in food spoilage, quality defects or safety issues, is more recent, but shows promise that they would be able to anticipate undesirable events (Stasiewicz et al. 2015).
Third-generation sequencing instruments have come to light in the last decade further stepping ahead NGS-based applications to tackle research and monitoring of dynamic processes within food manufacturing processes and environments. These are mainly represented by Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT). The main merits of these third-generation sequencing platforms is that they are able to conduct long read sequencing, theoretically as long as the DNA molecules present in the sample, and usually well above 5–10 kb. They also conduct single DNA molecule sequencing, thus avoiding the need to conduct PCR amplification steps during library production; and for these reasons, and the chemistry involved in the sequencing reaction, they are able to detect physical–chemical modifications, such as epigenetic marks, including acetylation or methylation, in the nucleotides sequenced (Munroe and Harris 2010). Moreover, the existence among these platforms of miniaturized sequencing devices, that can sequence DNA in real time in the food industry, has appeared as a promising tool which could be implemented as a routine monitoring of microbial dynamics in real settings, without the need to conduct the analyses in highly specialized laboratories. Besides, these platforms enable (i) a much finer resolution in taxonomic assignments as, for instance, they are able to sequence the full 16S rRNA in microbial communities; (ii) to sequence whole bacterial genomes with a lower amount of reads; and (iii) to discern molecular modifications in the DNA. All these advantages, in addition to the availability of miniaturized sequencing devices, make long-read platforms appealing to conduct sequencing-based monitoring of food processing environments to guarantee foodstuffs quality and safety. Long-read platforms have also been demonstrated to be valuable in tracking mobile genetic elements and their spread through microbial communities, as the DNA methylation patterns in these usually resemble those encountered in the chromosomal genomes of the strains they originated from (Carr et al. 2021). In this way, monitoring how new methylation patterns appear within a given mobile genetic element can provide real time information to decipher its spread across members in the communities, and its correlation with desirable or undesirable traits, such as microbial persistence within the food industry. Despite the many advantages and promises offered by long-read sequencing platforms, it is worth remarking that they do also generally need larger DNA quantities, which can be a limiting factor for certain applications or sample types, such as those obtained from food processing environments (McHugh et al. 2021). In addition, long-read sequencing instruments have far higher error rates leading to 88–94% and 85–87% accuracy values for Nanopore and PacBio instruments, respectively (Pearman et al. 2020). In contrast, error rates yielded by short-read sequencing instruments like Illumina platforms are in the range of 0.1 to 0.6%. Therefore, the lower accuracy of long-read sequencing instruments may affect the success of current taxonomic classification methods. Moreover, long-read sequencing instruments are not optimal for single nucleotide variation detection analyses because insertions and deletions may be included in the errors (Kono and Arakawa, 2019). Nevertheless, the diverse NGS-based approaches currently available offer numerous possibilities to design tailored-made applications which might be of interest within the food industry, yet the design of specific and standardized sample collection, DNA extraction, bioinformatic pipelines, and proper databases is essential so as to take advantage of the full potential that microbiome sequencing can deliver in food settings.
Data analysis of metataxonomic and metagenomic data
NGS technologies generate large volumes of data comprising entire communities of microorganisms. Although most bioinformatics pipelines to date have been developed in the frame of human gut microbiome research, and both pipelines and databases might need to be adapted to food and food processing environments, they can serve as a reference guide to tackle food metagenomic studies. A glossary of terms defining most technical terms used in this section is presented in Table 1.
Metataxonomic analysis based on bacterial 16S rRNA gene has been widely used for decades (Johnson et al. 2019), although sequencing of 18S gene (Karst et al. 2018), as well as ITS region (Rui et al. 2019), is also employed for taxonomic identification of eukaryotic microorganisms including fungal communities. Targeting of 16S variable regions with short-read sequencing platforms, such as Illumina, is commonly reported for taxonomic annotation of microorganisms, providing reliable identification at genus level. It is worth remarking that short-read metataxonomic analyses should not be used for strain- or species-specific monitoring purposes as they cannot provide reliable taxonomic assignments to describe microbial diversity at those levels. Besides, these short-read metataxonomic approaches have some other limitations including bias for taxonomic assignment of sequences depending on the variable region chosen for the analysis (Santos et al. 2020). Long-read sequencing platforms like ONT and PacBio, where the entire 16S gene is sequenced, can achieve better resolution and have the potential to characterize microbial communities at species level (Johnson et al. 2019).
Most computational pipelines developed for metataxonomic data analysis have been designed for short 16S rRNA gene fragments sequenced as paired-end reads on an Illumina platform, even though other genes and sequencing platforms could be used (Hall and Beiko 2018). The Quantitative Insights Into Microbial Ecology (QIIME) versions 1 and 2 is one popular software suite for microbial marker-gene analysis that generates microbial community descriptors by a series of computational transformations of the original sequence data (Bolyen et al. 2019). Some of these transformations include sequence quality filtering, sequence alignments, phylogeny building, taxonomic classification and microbial diversity analysis (Bolyen et al. 2019; Hall and Beiko 2018). A typical QIIME2 workflow involves the following steps (Hall and Beiko 2018): (1) importing sequences and sample metadata, (2) assessing sequence quality depending on sequencing platform and target gene, (3) removing primers to prevent false positive detection and denoising sequences using software libraries like DADA2 (Callahan et al. 2016) or Deblur (Amir et al. 2017), (4) filtering sequence table to exclude any samples that have significantly fewer sequences than the majority, (5) clustering sequences to generate operational taxonomic units (OTUs) for taxonomic classification using machine learning algorithms, (6) building phylogenetic trees to generate phylogenetic diversity measures, (7) computing alpha- and beta-diversity measures, (8) alpha rarefaction analysis to determine if samples have been sequenced to a sufficient depth.
Other software packages developed for microbial 16S rRNA analysis include MOTHUR, which has been used for 10 years (Schloss 2020), and USEARCH-UPARSE (Edgar 2013), showing a similar performance in a comparative study (Prodan et al. 2020). It should be noted that the most meaningful difference between these software suites is the choice of algorithms used to cluster sequences (Schloss 2020). With regard to computational pipelines specifically developed for long-read sequencing data, there is a scarcity of bioinformatic tools and protocols. The most extensively used tool to process ONT sequencing data is the cloud service EPI2ME, providing a series of workflows for end-to-end analysis. A typical workflow to process long-read sequencing data involves the following steps (Santos et al. 2020): (1) basecalling, which is the translation of changes in electric currents produced by the passing of DNA strands through a nanopore into a DNA sequence, (2) quality filtering of reads, (3) sequence orientation of mixed forward and reverse sequences that are not complementary to each other resulting from the basecalling process, (4) taxonomic classification using Basic Local Alignment Search Tool (BLAST) (Madden et al. 2018) to the National Center for Biotechnology Information (NCBI) database, (5) abundance table creation, (6) rarefaction, alpha, and beta diversity analysis.
On the other hand, metagenomics allows untargeted sequence of genomes from all microorganisms present in one sample, including bacteria, archaea, virus and unicellular eukaryotic microorganisms. Metagenomics is often used to study the genetic diversity of microbial communities and to determine taxonomic profiles of microorganisms at strain-level, as well as gene families and metabolic pathways (Bharti and Grimm 2021; Dulanto-Chiang and Dekker 2020; Pérez-Cobas et al. 2020). This kind of information could not be obtained by simpler metataxonomic approaches. The pipelines that have been developed for metagenomics analyses are an extension of those that have been developed for whole bacterial genome analysis (Quijada et al. 2019), including genome assembly and annotation steps, as well as the identification of plasmids and mobile elements (Carattoli et al. 2014), and antibiotic resistance genes using specific databases (Bortolaia et al. 2020).
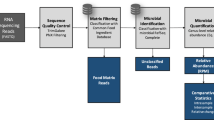
Figure 1 illustrates a typical pipeline to process shotgun metagenomic sequences. The first step in metagenomic data analysis involves quality control of sequences to remove low quality and contaminant reads from the host or food matrix. For this purpose, software packages like Kneaddata and Trimmomatic (Bolger et al. 2014), to trim reads and to perform in silico separation of bacterial reads from contaminant reads, have been developed. These sequences are usually mapped by using Bowtie2 (Langmead and Salzberg 2012) against reference or custom databases containing host contaminant reads. Once sequencing reads have been filtered, assembly-free or assembly-based pipelines can be computed to continue the analysis (Fig. 1).

Typical data analysis workflow of shotgun metagenomic data illustrating both assembly-free and assembly-based methods
Assembly-free methods require less computational power and take less time to complete allowing for a consistent identification of low-abundance species whose genomes could not be assembled. Taxonomic and functional annotation is carried out by mapping reads against reference databases that contain thousands of reference genomes or characteristic marker genes of each clade (Fig. 1). Therefore, assembly-free analysis strongly depends on the information contained in currently available databases, so incomplete databases may yield high false negative rates for those microorganisms that were not previously deposited. Some examples of commonly used software for assembly-free taxonomic and functional analysis include MetaPhlAn and HUMAnN, respectively (Franzosa et al. 2018; Truong et al. 2017). Taxonomic identification relies on clade-specific marker genes from approximately 100,000 reference genomes including 99,500 bacterial and archaeal and 500 eukaryotic genomes while reference protein databases are used to profile the abundance of microbial gene families and metabolic pathways. In addition, assembly-free methods specifically designed for tracking individual strains across samples have been developed. In this sense, StrainPhlAn software performs metagenomic strain-level population genomics by profiling microbes from known species and providing comparative and phylogenetic analyses of strains retrieved from metagenomic samples (Segata et al. 2012; Truong et al. 2017).
With regard to assembly-based methods, metagenome assembly is the process of reconstructing in silico the original genome sequence of a microorganism from small sequenced reads (Fig. 1). During metagenome assembly, sequenced fragments are joined to generate longer and more contiguous sequences called “contigs” by two different methods: (1) without using a previously sequenced reference genome, (2) using a previously sequenced, closely related organism to guide the assembly (Pérez-Cobas et al. 2020). Some examples of common software used for metagenome assembly are MEGAHIT (Li et al. 2016) and metaSPAdes (Nurk et al. 2017). Contigs belonging to the same biological taxon are gathered to generate bins to perform taxonomic classification and functional characterization (Bharti and Grimm 2021; Pérez-Cobas et al. 2020). Common binning algorithms include MaxBin2 (Wu et al. 2016), MetaBAT2 (Kang et al. 2019) and CONCOCT (Alneberg et al. 2013).
Metataxonomics in the food production chain: applications to improve food quality and safety
Metataxonomic approaches are used to know the microbial diversity and abundance of many different fermented foods or their raw materials. They are also considered useful to follow the dynamics of fermentations and ripening of the fermented products (De Filippis et al. 2017). In this sense, NGS data is adequate for developing multivariate statistical and ecological models, useful, for instance, for associating food production processes, or specific final characteristics in the products, to the associated microbial communities, although it is not an appropriate tool for purely taxonomic-descriptive purposes. The most abundant literature is related to the food bacteriome characterization, based on sequencing the 16S rRNA to decipher the members of the bacterial community. Yeasts and molds, that also play a relevant role in food production, were less extensively studied, maybe due to the reduced number of genetic databases for comparison of the amplified genetic regions. This review does not intend to perform an exhaustive collection of food related bacteriomes or mycobiomes, but some representative examples are collected in Table 2. In general, most primers used to explore bacterial diversity target the hypervariable V3–V4 region of the 16S rRNA gene, whereas, the 18S and 28S rRNA and more often ITS DNA regions are used in the case of yeasts and molds. The taxonomic assignment is made by comparing sequences with those annotated in different databases, typically that of the NCBI, or the SILVA ribosomal RNA gene database project, among others. Attempts to have specific databases related to food microbiomes have also been made. This is the case of the FoodMicrobionet database which is a structured collection of food bacteriomes constructed based on the FoodEx2 hierarchical food classification of the EFSA (Parente et al. 2019).
Metataxonomic approaches can be applied to describe food microbiotas along the production chain. In the particular case of the dairy sector, Parente and coworkers have recently reviewed the microbiota of cow’s milk “from the teat to the carton”. They analyzed the evolution of the NGS methodologies applied to study the microbial communities of raw milk, but they put the focus, as well, on the relevance of a good sampling experimental design to gain insight into this complex microbial community. In fact, the choice of the sampling procedure might introduce huge bias in the results obtained in a single milk sample; something simple, such as the way of teat cleaning, might modify the initial number of the bacterial load and, for instance, the success rate of amplification (Parente et al. 2020). Dealing with the issue of low microbial-load samples, McHugh et al. (2021) have compared Illumina and portable Nanopore sequencing platforms in order to propose the implementation of microbiome sequencing for the detection of pathogens in dairy production facilities. The portable platform was comparable, in terms of accurate species assignment, to the lab equipment but it required a higher amount of DNA for sequencing (McHugh et al. 2021). Thus, factors such as the DNA purification protocol, library construction procedures, or the choice of platform for sequencing, among others, together with the pipeline used for the downstream data analysis, have a strong influence on the description of the milk microbiota components (Ruiz et al. 2021). Added to this, other external variables related to the animal (breed, feeding, health, lactation state, etc.), farming environment (management practices, geographical location, season, etc.), milk production (milking type, bulk tanks, etc.), or the transport to the transformation facilities, have a strong influence on the microorganisms that might be present in the raw milk, making it difficult to have a picture of a “standard” or a “core” microbial composition (Doyle et al. 2017; McHugh et al. 2020; Parente et al. 2020; Oliveira et al. 2021). Similarly, the environment of the processing plants also plays a pivotal role in the microbiota finally present in the dairy product. By means of metataxonomic analyses it has been shown that the microbiota of skimmed milk powder, one of the “simplest” products that can be obtained from milk, varied according to the microbial composition of the raw milk and the microorganisms that were selected during manufacturing (McHugh et al. 2020). Cheeses are the most “complex” dairy products that can be made which involve different processing steps that are likely to be similar for all varieties. However, what defines a specific cheese is the microbial community responsible for the fermentation of the curd and for the cheese ripening. Metataxonomics has been useful to study undefined starters driving the initial fermentation step as well as those implicated in spontaneous fermentation processes, still employed in some traditional and artisanal food production processes such as those employed for the production of kimchi, fermented sausages or other raw-milk derived products, among other examples (Maoloni et al. 2020b; Wang et al. 2019; Motato et al. 2017). Remarkably, the knowledge of the communities implicated in spontaneous fermentations may aid towards the design of robust and reproducible starter cocktails, capable of providing desired organoleptic or functional properties to the final product. The 16S rRNA gene was widely used for this purpose, but additional target genes have been used to reach a higher species level resolution of specific bacterial populations (Zotta et al. 2021). This is the case of the purR (purine biosynthesis repressor) for the characterization of Lactococcus lactis subspecies diversity (Saltaji et al. 2020). In the final cheese, certain bacterial populations have been studied by sequencing other genetic regions, such as is the case of the ITS-bifidobacterial amplicons, which has been demonstrated to improve the resolution of bifidobacterial species assignation as compared to other 16Sr RNA regions (Milani et al. 2019). Further, interactions among different microbial communities can be monitored by means of these approaches. In an in vitro model, Wolfe and co-authors have evaluated the evolution of surface bacteria and fungi during the rind aging of different cheese varieties, which has gained insight into the ecology of these particular communities including dissection of particular fungal-bacterial interactions which can influence the attributes of the final product (Wolfe et al. 2014). Thus, upon development of appropriate standardized procedures for food and food-related samples, the application of NGS may play a relevant role in the food industry in the near future.
Shotgun metagenomics in the food production chain: applications to improve food quality and safety
Metagenomics and metataxonomics display a different, although complementary, perspective. While metataxonomics does not provide information about the functional and metabolic features of the microorganisms and it is limited to depicting a profile of the members of the community, metagenomics exploits the information present in the whole genetic content of the community (the metagenome), usually by directly sequencing the total DNA pool of the microbial population, avoiding the bias introduced by the amplification of specific DNA fragments. The sequencing of all microbial DNA present in a sample has been defined as “shotgun metagenomics”, which currently is the gold standard to analyse complex microbial communities (Quince et al. 2017). One step further approach is represented by metatranscriptomics, which sequences all the mRNA present in a sample and thus may add information on the functions and populations that are metabolically active at a given sampling point. However, the cost of metatranscriptomics analyses and quick RNA decay have strongly limited its application, which to date has been very scarce in food-related samples. For this reason, this section will focus on shotgun metagenomics analyses of DNA samples.
The sequences obtained from shotgun metagenomics analyses can be assembled and annotated, providing qualitative and quantitative functional and taxonomic data. Even though there are still some unsolved challenges in metagenomics analysis, such as those derived from the incomplete or absent lysis of some recalcitrant members of the community, the lack of adequate software pipelines to analyse unknown microorganisms or microbial genes, or the fact that these procedures do not distinguish between genetic material from functionally active or inactive cells, metagenomics represents the most complete and reliable approach to the description of microbial populations. Shotgun sequencing generates raw data that can be exploited in many different ways, including taxonomic profiling and metabolic potential of the microbiota, recovery of genome sequences (metagenome-assembled genomes—MAGs), tracking of strains in complex environments or analysis of gene subclusters with specific functions within the whole metagenome, such as antibiotic resistance genes.
Shotgun metagenomics has been extensively used to depict the microbiomes of different environments, including those associated with foods and the food industry. The food metagenome has been studied through shotgun sequencing in both non-fermented foods, such as milk and honey (Bovo et al. 2020; Mchugh et al. 2020), and fermented foods, the latter being the ones that have received the most attention because their microbial load is normally high. Among fermented foods, shotgun sequencing methods have been applied in the cheese industry to assess the functional features of the microbiota of cow’s milk artisanal cheeses from Northwestern Argentina, which has contributed to the isolation of bacteriocin-producing bacteria against Listeria monocytogenes (Suárez et al. 2020). Shotgun methods have also contributed to clarifying the role of individual species during the ripening of surface-ripened cheeses, and their impact on flavor development (Bertuzzi et al. 2018), as well as unravelling the carotenoid-producing microorganism responsible for the pink discoloration defect of Continental-type cheeses (Quigley et al. 2016), among other applications. The most comprehensive metagenomic analysis of different cheese types has recently been published, showing the usefulness of shotgun sequencing to link different bacterial functionalities, such as the synthesis of volatile compounds during ripening or bacteriocin-production, with genes or bacteria present in the cheese microbiota, providing a tool to improve cheese production processes (Walsh et al. 2020). Interestingly, Mchugh et al. (2020) showed the usefulness of untargeted metagenomic sequencing approaches in food safety and quality by tracing microbial species in the dairy industry, during the whole process of skimmed milk-production, showing that the dairy microbiota strongly depends on the initial characteristics of the raw milk.
In addition, metagenomes of fermented-meat and meat-processing industries have also been investigated. The potential functions associated with meat fermentation processes have been studied in sausages, highlighting the key role of the starter cultures in the organoleptic properties of fermented products (Ferrocino et al. 2018). Also, the pathogen populations have been monitored in environmental samples at different points of the beef production chain, from feedlot to the end of the fabrication system, indicating that metagenomic data can be used to track a wide variety of pathogens in the cattle related food chain (Yang et al. 2016). Similar methods have also been applied to industrial facilities producing fermented vegetables, namely traditionally fermented sauerkraut, identifying the raw vegetables and environmental surfaces as the potential sources of the microorganisms carrying out the spontaneous fermentations (Einson et al. 2018).
Overall, as demonstrated in previous examples, the knowledge generated through shotgun metagenomics on food and food processing environments can help towards the selection of starter and adjunct bacterial cultures capable of conferring desired quality attributes to the final product, either in terms of improved nutritional, functional, or organoleptic properties. But, it can also help to improve its safety through selecting microorganisms capable of extending their shelf-life and to guarantee the absence of spoilage or pathogenic bacteria in a range of food products.
Finally, it is important to note that although shotgun analysis offers unprecedented opportunities to analyse food metagenomes from a broad ecological perspective, there are still great challenges to be solved in this field, including the difficulties of analysing foods and food-related environments with a low microbial load with the currently available methodologies, as well as the lack of specific bioinformatics pipelines adapted to the study of food microbiomes. Therefore, there is a need to fine tune current shotgun approaches to fully explore the potential of these applications and implement these new methodologies in the food industry, which will undoubtedly contribute to the increase of quality and safety of food.
Targeted microbiome analysis of datasets: the example of the resistome and its relevance in the food production chain
Targeted microbiome analyses are those which include a selection step in the analytical pipeline to enrich or filter microbial sequences of interest; hence, they are designed to provide tailored information about relevant biological questions beyond taxonomic assignment. Some of these targeted approaches are based on a selection process taking place before library preparation and sequencing, with the aim of enhancing the sensitivity of detection of genetic determinants avoiding the “needle in the haystack” limitation (Mitchell and Simner 2019). Thus, approaches involving PCR amplification of specific sets of genes before library preparation have been followed to, for instance, survey bacterial histidine and tyrosine decarboxylases in raw milk cheeses (O’Sullivan et al. 2015), or integrons and integron-like gene cassettes in marine environments (Elsaied et al. 2007). In other cases, platforms based on probes for the hybridization and capture of selected genes can be used for sequence enrichment after library preparation and sequencing, such as for antibiotic resistance genes or relaxase genes in the report by Lanza et al. (2018).
However, with the continuous temporal decrease in sequencing costs and increase in computers’ performance, and the associated shift from amplicon sequencing to shotgun whole metagenome sequencing approaches, targeted microbiome analyses nowadays mainly rely on the direct query of databases (consolidated and curated, or tailor-made in-house databases) using as input raw reads or assembled reads obtained from the shotgun sequencing of the target sample. This allows information to be obtained on the complete pool of genes in that sample related to any given function of interest, like antimicrobial resistance or bacterial virulence (Walsh et al. 2017).
The resistome, i.e., the collection of all the antimicrobial resistance genes, is the dataset most widely studied through targeted microbiome approaches. While the transmission of antimicrobial resistances (AMR) through food systems is currently under investigation (Bengtsson-Palme, 2017; Oniciuc et al. 2019), it is generally considered that controlling the spread of resistance genes or resistant bacteria in primary food production and food processing must be a priority to reduce the burden associated with infections caused by resistant bacteria in humans. Indeed, current regulatory policies are focused on reducing the use of antimicrobials in crop fields and farms, but the food chain still represents an important reservoir of antibiotic-resistant organisms (Schmithausen et al. 2018). The analysis of the resistome generally involves the detection of known resistance determinants among raw reads, assembled contigs or metagenome assembled genomes by using the BLAST algorithm or bowtie2 alignment (Langmead and Salzberg 2012) to find all possible matches between the database and the query sequences (Alvarez-Molina et al. 2020). Custom or publicly available databases, such as CARD (Alcock et al. 2020), ARG-Annot (Gupta et al. 2014), or ResFinder (Bortolaia et al. 2020), can be used for this purpose. The utility of shotgun metagenomic approaches to understand the factors shaping the resistome abundance and diversity has been highlighted in several recent studies addressing different microbial ecosystems within the food supply chain (Mencía-Ares et al. 2020; Munk et al. 2018; Pitta et al. 2016; Xue et al. 2021b). This has dramatically expanded the information obtained from surveys which previously were exclusively focused on the isolation and characterization of antimicrobial-resistant microorganisms from a limited number of pathogenic or indicator bacterial species and has shown that targeted resistome analyses have the potential to replace or complement culture-dependent approaches in antimicrobial resistance monitoring initiatives (EFSA 2019).
The main limitations of resistome analyses derive from the lack of harmonized methods or the fact that the results obtained strongly depend on the choice of wet lab methods, like DNA isolation methods, and databases and bioinformatics pipelines. In addition, the characterization of resistomes, which is commonly performed using short-read sequencing technology, does not generally allow the attribution of the identified resistance genes to specific taxa or strains and their identification as transferable or non-transferable resistance determinants, which can hamper the assessment of the actual risk posed by such resistance determinants (Oniciuc et al. 2018). However, in recent years it is becoming apparent that the use of long read sequencing approaches will increase the resolution of complex genomic regions in metagenomes, allowing the location of resistance genes within mobile genetic elements to be unraveled (Che et al. 2019), and enabling host cell taxonomic classification of both resistance genes and mobile genetic elements (Beaulaurier et al. 2018). These advances, coupled to the recent improvements of assembly and binning algorithms which are facilitating the mining of thousands of individual genomes from metagenomes (Pasolli et al. 2019), will ensure that the most detailed information will be obtained in future resistome surveys. Overall, these and other future technological and analytical developments will surely further the development and enhance the implementation of targeted microbiome and resistome analyses in the food industry. This will allow the tracking of resistance genes and mobile genetic elements and will provide unique insights into hotspots, the mechanisms of selection for, and the spread of, AMR in food-related settings, ultimately leading to the development of knowledge-based interventions aimed at reducing dispersal of multidrug resistant microorganisms in the food industry (Oniciuc et al. 2018; de Filippis et al. 2021).
Conclusions and future perspectives
The rise in sequencing technologies has allowed the mapping of microbial communities associated with the production of specific foods, these being valuable tools to know “who” are in this niche and also “what” might be their role in the characteristics of the final product (De Filippis et al. 2021). However, there is a scarcity of food microbiome data and publicly available sequences compared to the large amount of information that has been generated in recent years concerning the human microbiome. In this regard, food microbiology should benefit from the methodologies already developed for the study of the human microbiome, and be able to adapt analytical and computational tools in order to enhance the knowledge of food microbial communities. There is an urgent need to create dedicated food microbiome databases, as well as to develop bioinformatic pipelines to interrogate food metagenomes, in order to explore the genetic and functional information of food micro-ecosystems. The current challenge is how to implement these metagenomic technologies in the industrial chain, “from farm to fork”, in the context of sustainable food and feed production, and also taking into account regulatory issues.
Availability of data and materials
N/A.
Code availability
N/A.
References
Alcock BP, Raphenya AR, Lau TTY et al (2020) CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res 48(D1):D517–D525. https://doi.org/10.1093/nar/gkz935
Alneberg J, Bjarnason BS, de Bruijn I, Schirmer M, Quick J, Ijaz UZ, Loman NJ, Andersson AF, Quince C (2013) CONCOCT: clustering contigs on coverage and composition. arXiv preprint
Álvarez-Molina A, de Toro M, Alexa EA, Alvarez-Ordóñez A (2020) Applying genomics to track antimicrobial resistance in the food chain. In: Comprehensive Foodomics. A. Cifuentes (Ed.), Elsevier. 1–24 ISBN:9780128163955
Amir A, McDonald D, Navas-Molina JA et al (2017) Deblur rapidly resolves single-nucleotide community sequence patterns. mSystems 2:e00191-16. https://doi.org/10.1128/mSystems.00191-16
Anagnostopoulos DA, Kamilari E, Tsaltas D (2019) Contribution of the microbiome as a tool for estimating wine’s fermentation output and authentication. In book: Advances in Grape and Wine Biotechnology, IntechOpen. https://doi.org/10.5772/intechopen.85692
Beaulaurier J, Zhu S, Deikus G et al (2018) Metagenomic binning and association of plasmids with bacterial host genomes using DNA methylation. Nat Biotechnol 36:61–69. https://doi.org/10.1038/nbt.4037
Belleggia L, Ferrocino I, Realec A et al (2020) Portuguese cacholeira blood sausage: a first taste of its microbiota and volatile organic compounds. Food Res Int 136:109567. https://doi.org/10.1016/j.foodres.2020.109567
Bengtsson-Palme J (2017) Antibiotic resistance in the food supply chain: where can sequencing and metagenomics aid risk assessment? Curr Opin Food Sci 14:66–71. https://doi.org/10.1016/j.cofs.2017.07.010
Benítez-Cabello A, Romero-Gil V, Medina E, Sánchez B, Calero-Delgado B, Bautista-Gallego J, Jiménez-Díaz R, Arroyo-López FN (2019) Metataxonomic analysis of the bacterial diversity in table olive dressing components. Food Control 105:190–197. https://doi.org/10.1016/j.foodcont.2019.05.036
Benítez-Cabello A, Romero-Gil V, Medina-Pradas E, Garrido-Fernández A, Arroyo-López FN (2020) Exploring bacteria diversity in commercialized table olive biofilms by metataxonomic and compositional data analysis. Sci Rep 10:11381. https://doi.org/10.1038/s41598-020-68305-7
Bertuzzi AS, Walsh AM, Sheehan JJ, Cotter PD, Crispie F, McSweeney PLH, Kilcawley KN, Rea MC (2018) Omics-based insights into flavor development and microbial succession within surface-ripened cheese. mSystems 3:e00211-17. https://doi.org/10.1128/mSystems.00211-17
Bharti R, Grimm DG (2021) Current challenges and best-practice protocols for microbiome analysis. Brief Bioinform 22:178–193. https://doi.org/10.1093/bib/bbz155
Bhutia MO, Thapa N, Shangpliang HNJ, Tamang JP (2021) Metataxonomic profiling of bacterial communities and their predictive functional profiles in traditionally preserved meat products of Sikkim state in India. Food Res Int 140:110002. https://doi.org/10.1016/j.foodres.2020.110002
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. https://doi.org/10.1093/bioinformatics/btu170
Bolyen E, Rideout JR, Dillon MR et al (2019) Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol 37:852–857. https://doi.org/10.1038/s41587-019-0209-9
Bortolaia V, Kaas RS, Ruppe E et al (2020) ResFinder 4.0 for predictions of phenotypes from genotypes. J Antimicrob Chemother 75:3491–3500. https://doi.org/10.1093/jac/dkaa345
Bovo S, Utzeri VJ, Ribani A, Cabbri R, Fontanesi L (2020) Shotgun sequencing of honey DNA can describe honey bee derived environmental signatures and the honey bee hologenome complexity. Sci Rep 10:9279. https://doi.org/10.1038/s41598-020-66127-1
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP (2016) DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods 13:581–583. https://doi.org/10.1038/nmeth.3869
Carattoli A, Zankari E, García-Fernández A, Larsen MV, Lund O, Villa L, Aarestrup FM, Hasman H (2014) In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob Agents Chemother 58:3895–3903. https://doi.org/10.1128/AAC.02412-14
Carr VR, Shkoporov A, Hill C, Mullany P, Moyes DL (2021) Probing the mobilome: discoveries in the dynamic microbiome. Trends Microbiol 29:158–170. https://doi.org/10.1016/j.tim.2020.05.003
Castro I, Alba C, Aparicio M, Arroyo R, Jiménez L, Fernández L, Arias R, Rodríguez JM (2019) Metataxonomic and immunological analysis of milk from ewes with or without a history of mastitis. J Dairy Sci 102:9298–9311. https://doi.org/10.3168/jds.2019-16403
Che Y, Xia Y, Liu L, Li AD, Yang Y, Zhang T (2019) Mobile antibiotic resistome in wastewater treatment plants revealed by Nanopore metagenomic sequencing. Microbiome 7:44. https://doi.org/10.1186/s40168-019-0663-0
Chibuzor-Onyema IE, Ezeokoli OT, Sulyok M, Notununu I, Petchkongkaew A, Elliott CT, Adeleke RA, Krska R, Ezekiel CN (2021) Metataxonomic analysis of bacterial communities and mycotoxin reduction during processing of three millet varieties into ogi, a fermented cereal beverage. Food Res Int 143:110241. https://doi.org/10.1016/j.foodres.2021.110241
Cocolin L, Alessandria V, Dolci P, Gorra R, Rantsiou K (2013) Culture independent methods to assess the diversity and dynamics of microbiota during food fermentation. Int J Food Microbiol 167:29–43. https://doi.org/10.1016/j.ijfoodmicro.2013.05.008
De Filippis F, Parente E, Ercolini D (2017) Metagenomics insights into food fermentations. Microb Biotechnol 10:91–102. https://doi.org/10.1111/1751-7915.12421
De Filippis F, Valentino V, Alvarez-Ordoñez A, Cotter PD, Ercolini D (2021) Environmental microbiome mapping as a strategy to improve quality and safety in the food industry. Cur Opin Food Sci 38:168–176. https://doi.org/10.1016/j.cofs.2020.11.012
Dertli E, Çon AH (2017) Microbial diversity of traditional kefir grains and their role on kefir aroma. LWT Food Sci Tech 85:151–157. https://doi.org/10.1016/j.lwt.2017.07.017
Desdouits M, de Graaf M, Strubbia S, Oude Munnink BB, Kroneman A, Le Guyader FS, Koopmans MPG (2020) Novel opportunities for NGS-based one health surveillance of foodborne viruses. One Health 14. https://doi.org/10.1186/s42522-020-00015-6
Doyle CJ, Gleeson D, O’Toole PW, Cotter PD (2017) High-throughput metataxonomic characterization of the raw milk microbiota identifies changes reflecting lactation stage and storage conditions. Int J Food Microbiol 255:1–6. https://doi.org/10.1016/j.ijfoodmicro.2017.05.019
Dulanto-Chiang A, Dekker JP (2020) From the pipeline to the bedside: advances and challenges in clinical metagenomics. J Infect Dis 221:S331–S340. https://doi.org/10.1093/infdis/jiz151
Edgar RC (2013) UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods 10:996–998. https://doi.org/10.1038/nmeth.2604
EFSA (2019) Whole genome sequencing and metagenomics for outbreak investigation, source attribution and risk assessment of food-borne microorganisms. EFSA J 17:e05898
Einson JE, Rani A, You X et al (2018) Vegetable fermentation facility hosts distinct microbiomes reflecting the production environment. Appl Environ Microbiol 84:e01680-e1718. https://doi.org/10.1128/AEM.01680-18
Elsaied H, Stokes HW, Nakamura T, Kitamura K, Fuse H, Maruyama A (2007) Novel and diverse integrin integrase genes and integrin-like gene cassettes are prevalent in deep-sea hydrothermal vents. Environ Microbiol 9:2298–2312. https://doi.org/10.1111/j.1462-2920.2007.01344.x
Ferrocino I, Bellio A, Giordano M, Macori G, Romano A, Rantsiou K, Decastelli L, Cocolin L (2018) Shotgun metagenomics and volatilome profile of the microbiota of fermented sausages. Appl Environ Microbiol 84:e02120-e2217. https://doi.org/10.1128/AEM.02120-17
Franzosa EA, McIver LJ, Rahnavard G et al (2018) Species-level functional profiling of metagenomes and metatranscriptomes. Nat Methods 15:962–968. https://doi.org/10.1038/s41592-018-0176-y
Gupta SK, Padmanabhan BR, Diene SM, Lopez-Rojas R, Kempf M, Landraud L, Rolain J-M (2014) ARG-annot, a new bioinformatic tool to discover antibiotic resistance genes in bacterial genomes. Antimicrob Agents Chemother 58:212–220. https://doi.org/10.1128/AAC.01310-13
Hall M, Beiko RG (2018) 16S rRNA gene analysis with QIIME2. In Microbiome analysis. Humana Press, New York, pp 113–129
Haynes E, Jiménez E, Pardo MA, Helyar SJ (2019) The future of NGS (Next Generation Sequencing) analysis in testing food authenticity. Food Control 101:134–143. https://doi.org/10.1016/j.foodcont.2019.02.010
Heather JM, Chain B (2016) The sequence of sequencers. The History of Sequencing DNA Genomics 107:1–8. https://doi.org/10.1016/j.ygeno.2015.11.003
Johnson JS, Spakowicz DJ, Hong BY et al (2019) Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat Commun 10:5029. https://doi.org/10.1038/s41467-019-13036-1
Kamilari E, Tomazou M, Antoniades A, Tsaltas D (2019) High throughput sequencing technologies as a new toolbox for deep analysis, characterization and potentially autentication of protection designation of origin cheeses? Int J Food Sci 2019:5837301. https://doi.org/10.1155/2019/5837301
Kamimura BA, De Filippi F, Sant’Ana AA, Ercolini D, (2019) Large-scale mapping of microbial diversity in artisanal Brazilian cheeses. Food Microbiol 80:40–49. https://doi.org/10.1016/j.fm.2018.12.014
Kang DD, Li F, Kirton E, Thomas A, Egan R, An H, Wang Z (2019) MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 7:e7359. https://doi.org/10.7717/peerj.7359
Karst SM, Dueholm MS, McIlroy SJ, Kirkegaard RH, Nielsen PH, Albertsen M (2018) Retrieval of a million high-quality, full-length microbial 16S and 18S rRNA gene sequences without primer bias. Nat Biotech 36:190–195. https://doi.org/10.1038/nbt.4045
Kazou M, Tzamourani A, Panagou EZ, Tsakalidou E (2020) Unraveling the microbiota of natural black cv. Kalamata fermented olives through 16S and ITS metataxonomic analysis. Microorganisms 8:672. https://doi.org/10.3390/microorganisms8050672
Kono N, Arakawa K (2019) Nanopore sequencing: review of potential applications in functional genomics. Dev Growth Differ 61(5):316–326. https://doi.org/10.1111/dgd.12608
Koutsoumanis K, Allende A, Alvarez-Ordonez A et al (2019) Whole genome sequencing and metagenomics for outbreak investigation, source attribution and risk assessment of food-borne microorganisms. EFSA J 17:5898. https://doi.org/10.2903/j.efsa.2019.5898
Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. https://doi.org/10.1038/nmeth.1923
Lanza VF, Baquero F, Martínez JL et al (2018) In-depth resistome analysis by targeted metagenomics. Microbiome 6:11. https://doi.org/10.1186/s40168-017-0387-y
Li D, Luo R, Liu CM, Leung CM, Ting HF, Sadakane K, Yamashita H, Lam TW (2016) MEGAHIT v1. 0: a fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 102:3–11. https://doi.org/10.1016/j.ymeth.2016.02.020
Madden TL, Busby B, Ye J (2018) Reply to the paper: Misunderstood parameters of NCBI BLAST impacts the correctness of bioinformatics workflows. Bioinformatics 35:2699–2700. https://doi.org/10.1093/bioinformatics/bty1026
Maoloni A, Blaiotta G, Ferrocino I et al (2020a) Microbiological characterization of Gioddu, an Italian fermented milk. Int J Food Microbiol 323:108610. https://doi.org/10.1016/j.ijfoodmicro.2020.108610
Maoloni A, Ferrocino I, Milanovic V et al (2020b) The microbial diversity of mon-Korean Kimchi as revealed by viable counting and metataxonomic sequencing. Foods 9:1568. https://doi.org/10.3390/foods9111568
McHugh AJ, Feehily C, Fenelon MA, Gleeson D, Hill C, Cotter PD (2020) Tracking the dairy microbiota from farm bulk tank to skimmed milk powder. mSystems 5:e00226-20. https://doi.org/10.1128/mSystems.00226-20
McHugh AJ, Yap M, Crispie F, Feehili C, Hill C, Cotter PD (2021) Microbiome-based environmental monitoring of a dairy processing facility highlights the challenges associated with low microbial-load samples. NPJ Sci Food 5:4. https://doi.org/10.1038/s41538-021-00087-2
Mencía-Ares O, Cabrera-Rubio R, Cobo-Díaz JF et al (2020) Antimicrobial use and production system shape the faecal, environmental and slurry resistomes of pig farms. Microbiome 8:164. https://doi.org/10.1186/s40168-020-00941-7
Mezzasalma V, Sandionigi A, Bruni I, Bruno A, Lovicu G, Casiraghi M, Labra M (2017) Grape microbiome as a reliable and persistent signature of field origin and environmental conditions in Cannonau wine production. PLoS ONE 12:e0184615. https://doi.org/10.1371/journal.pone.0184615
Milani C, Alessandri G, Mancabelli L et al (2019) Bifidobacterial distribution across Italian cheeses produced from raw milk. Microorganisms 7:599. https://doi.org/10.3390/microorganisms7120599
Mitchell SL, Simner PJ (2019) Next-generation sequencing in clinical microbiology: Are we there yet? Clin Lab Med 39:405–418. https://doi.org/10.1016/j.cll.2019.05.003
Mota-Gutierrez J, Botta C, Ferrocino I, Giordano M, Bertolino M, Dolci P, Cannoni M, Cocolin L (2018) Dynamics and biodiversity of bacterial and yeast communities during fermentation of cocoa beans. Appl Environ Microbiol 84:e01164-e1218. https://doi.org/10.1128/AEM.01164-18
Motato KE, Milani C, Ventura M, Valencia FE, Ruas-Madiedo P, Delgado S (2017) Bacterial diversity of the Colombian fermented milk “Suero Costeño” assessed by culturing and high-throughput sequencing and DGGE analysis of 16S rRNA gene amplicons. Food Microbiol 68:129–136. https://doi.org/10.1016/j.fm.2017.07.011
Munk P, Knudsen BE, Lukjancenko O et al (2018) Abundance and diversity of the faecal resistome in slaughter pigs and broilers in nine European countries. Nat Microbiol 3:898–908. https://doi.org/10.1038/s41564-018-0192-9
Munroe DJ, Harris TJR (2010) Third generation sequencing fireworks at Marco Island. Nat Biotechnol 28:426–428. https://doi.org/10.1038/nbt0510-426
Nurk S, Meleshko D, Korobeynikov A, Pevzner PA (2017) metaSPAdes: a new versatile metagenomic assembler. Genome Res 2:824–834. https://doi.org/10.1101/gr.213959.116
O’Sullivan DJ, Fallico V, O’Sullivan O, McSweeney PLH, Sheehan JJ, Cotter PD, Giblin L (2015) High-throughput DNA sequencing to survey bacterial hisitidine and tyrosine decarboxylases in raw milk cheeses. BMC Microbiol. 15:0596. https://doi.org/10.1186/s12866-015-0596-0
Oliveira ACD, Nogueira Souza F, de Sant’Anna FM et al (2021) Temporal and geographical comparison of bulk tank milk and water microbiota composition in Brazilian dairy farms. Food Microbiol. 98:103793. https://doi.org/10.1016/j.fm.2021.103793
Oniciuc EA, Likotrafiti E, Alvarez-Molina A, Prieto M, Santos JA, Álvarez-Ordóñez A (2018) The present and future of whole genome sequencing (WGS) and whole metagenome sequencing (WMS) for surveillance of antimicrobial resistant microorganisms and antimicrobial resistance genes across the food chain. Genes 9:268. https://doi.org/10.3390/genes9050268
Oniciuc EA, Likotrafiti E, Alvarez-Molina A, Prieto M, López M, Alvarez-Ordoñez A (2019) Food processing as a risk factor for antimicrobial resistance spread along the food chain. Curr Opin Food Sci 30:21–26. https://doi.org/10.1016/j.cofs.2018.09.002
Parente E, De Filippis F, Danilo E, Ricciardi A, Zotta T (2019) Advancing integration of data on food microbiome studies: FoodMicrobionet 3.1, a major upgrade of the FoodMicrobionet database. Int J Food Microbiol. 305:108249. https://doi.org/10.1016/j.ijfoodmicro.2019.108249
Parente E, Ricciardi A, Zotta T (2020) The microbiota of dairy milk: A review. Int Dairy J 107:104714. https://doi.org/10.1016/j.idairyj.2020.104714
Pasolli E, Asnicar F, Manara S et al (2019) Extensive unexplored human microbiome diversity revealed by over 150,000 genomes from metagenomes spanning age, geography, and lifestyle. Cell 176:649–662. https://doi.org/10.1016/j.cell.2019.01.001
Pearman WS, Freed NE, Silander OK (2020) Testing the advantages and disadvantages of short-and long-read eukaryotic metagenomics using simulated reads. BMC Bioinformatics 21:1–15. https://doi.org/10.1128/mSystems.00226-20
Penland M, Falentin H, Parayre S, Pawtowski A, Maillard MB, Thierry A, Mounier J, Coton M, Deutsch SM (2021) Linking Pélardon artisanal goat cheese microbial communities to aroma compounds during cheese-making and ripening. Int J Food Microbiol 345:109130. https://doi.org/10.1016/j.ijfoodmicro.2021.109130
Pérez-Cobas AE, Gomez-Valero L, Buchrieser C (2020) Metagenomic approaches in microbial ecology: an update on whole-genome and marker gene sequencing analyses. Microb Genom 6:mgen.0.000409. https://doi.org/10.1099/mgen.0.000409
Pitta DW, Dou Z, Kumar S, Indugu N, Toth JD, Vecchiarelli B, Bhukya B (2016) Metagenomic evidence of the prevalence and distribution patterns of antimicrobial resistance genes in dairy agroecosystems. Foodborne Pathog Dis 13:296–302. https://doi.org/10.1089/fpd.2015.2092
Pogacic T, Kelava N, Zamberlin S, Dolenčić-Špehar I, Samaržija D (2010) Methods for culture-independent identification of lactic acid bacteria in dairy products. Food Tech Biotech 48:3–10
Prodan A, Tremaroli V, Brolin H, Zwinderman AH, Nieuwdorp M, Levin E (2020) Comparing bioinformatic pipelines for microbial 16S rRNA amplicon sequencing. PLoS ONE 15:e0227434. https://doi.org/10.1371/journal.pone.0227434
Quigley L, O’Sullivan DJ, Daly D et al (2016) Thermus and the pink discoloration defect in cheese. mSystems 1:e00023-16. https://doi.org/10.1128/mSystems.00023-16
Quijada NM, Rodríguez-Lázaro D, Eiros JM, Hernández M (2019) TORMES: an automated pipeline for whole bacterial genome analysis. Bioinformatics 35:4207–4212. https://doi.org/10.1093/bioinformatics/btz220
Quince C, Walker AW, Simpson JT, Loman NJ, Segata N (2017) Shotgun metagenomics, from sampling to analysis. Nat Biotechnol 35:833–844. https://doi.org/10.1038/nbt.3935
Raimondi S, Amaretti A, Rossi M, Fall PA, Tabanelli G, Gardini F, Montanari C (2017) Evolution of microbial community and chemical properties of a sourdough during the production of Colomba, an Italian sweet leavened baked product. LWT Food Sci Technol 86:31–39. https://doi.org/10.1016/j.lwt.2017.07.042
Ritschard JS, Amato L, Kumar Y, Müller B, Meile L, Schuppler M (2018) The role of the surface smear microbiome in the development of defective smear on surface-ripened red-smear cheese. AIMS Microbiology 4:622–641. https://doi.org/10.3934/microbiol.2018.4.622
Rui Y, Wan P, Chen G, Xie M, Sun Y, Zeng X, Liu Z (2019) Analysis of bacterial and fungal communities by Illumina MiSeq platforms and characterization of Aspergillus cristatus in Fuzhuan brick tea. LWT Food Sci Tech 110:168–174. https://doi.org/10.1016/j.lwt.2019.04.092
Ruiz L, Alba C, Garcia-Carral C et al (2021) Comparison of two approaches for the metataxonomic analysis of the human milk microbiome. Front Cell Infect Microbiol 11:622550. https://doi.org/10.3389/fcimb.2021.622550
Saltaji S, Rué O, Sopena V, Sablé S, Tambadou F, Didelot S, Chevrot R (2020) Lactococcus lactis diversity revealed by targeted amplicon sequencing of purR gene, metabolic comparisons and antimicrobial properties in an undefined mixed starter culture used for soft-cheese manufacture. Foods 9:622. https://doi.org/10.3390/foods9050622
Santos A, van Aerle R, Barrientos L, Martinez-Urtaza J (2020) Computational methods for 16S metabarcoding studies using Nanopore sequencing data. Comput Struct Biotech J 18:296–305. https://doi.org/10.1016/j.csbj.2020.01.005
Schloss PD (2020) Reintroducing mothur: 10 years later. Appl Environ Microbiol 86:e02343-e2419. https://doi.org/10.1128/AEM.02343-19
Schmithausen RM, Schulze-Geisthoevel SV, Heinemann C, Bierbaum G, Exner M, Petersen B, Steinfoff-Wagner J (2018) Reservoirs and transmission pathways of resistant indicator bacteria in the biotope pig stable and along the food chain: a review from a one health perspective. Sustainability 10(11):3967. https://doi.org/10.3390/su10113967
Segata N, Waldron L, Ballarini A, Narasimhan V, Jousson O, Huttenhower C (2012) Metagenomic microbial community profiling using unique clade-specific marker genes. Nat Methods 9:811–814. https://doi.org/10.1038/nmeth.2066
Song HS, Whon TW, Kim J, Lee HE, Kim JY, Kim YB, Choi HJ, Rhee JK, Roh SW (2020) Microbial niches in raw ingredients determine microbial community assembly during kimchi fermentation. Food Chem 318:126481. https://doi.org/10.1016/j.foodchem.2020.126481
Stasiewicz MJ, Oliver HF, Wiedmann M, den Bakker HC (2015) Whole-genome sequencing allows for improved identification of persistent Listeria monocytogenes in food-associated environments. Appl Environ Microbiol 8:6014–6037. https://doi.org/10.1128/AEM.01049-15
Stefanini I, Carlin S, Tocci N et al (2017) Core microbiota and metabolome of Vitis vinifera L. cv. Corvina grapes and musts. Front Microbiol 8:457. https://doi.org/10.3389/fmicb.2017.00457
Suárez N, Weckx S, Minahk C, Hebert EM, Saavedra L (2020) Metagenomics-based approach for studying and selecting bioprotective strains from the bacterial community of artisanal cheeses. Int J Food Microbiol 335:108894. https://doi.org/10.1016/j.ijfoodmicro.2020.108894
Truong DT, Tett A, Pasolli E, Huttenhower C, Segata N (2017) Microbial strain-level population structure and genetic diversity from metagenomes. Genome Res 27:626–638. https://doi.org/10.1101/gr.216242.116
Walsh AM, Crispie F, Kilcawley K, O’Sullivan O, O’Sullivan MG, Claesson MJ, Cotter PD (2016) Microbial succession and flavor production in the fermented dairy beverage kefir. mSystems 1(5):e00052-16 (mSystems.00052-16)
Walsh AM, Crispie F, Claesson MJ, Cotter PD (2017) Translating omics to food microbiology. Annu Rev Food Sci Technol 8:113–134. https://doi.org/10.1146/annurev-food-030216-025729
Walsh AM, Macori G, Kilcawley KN et al (2020) Meta-analysis of cheese microbiomes highlights contributions to multiple aspects of quality. Nat Food 1:500–510. https://doi.org/10.1038/s43016-020-0129-3
Wang X, Wang S, Zhao H (2019) Unraveling microbial community diversity and succession of Chinese Sichuan sausages during spontaneous fermentation by high-throughput sequencing. J Food Sci Technol 56:3254–3263. https://doi.org/10.1007/s13197-019-03781-y
Wolfe BE, Button JE, Santarelli M, Dutton RJ (2014) Cheese rind communities provide tractable systems for in situ and in vitro studies of microbial diversity. Cell 158:422–433. https://doi.org/10.1016/j.cell.2014.05.041
Wu YW, Simmons BA, Singer SW (2016) MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 32:605–607. https://doi.org/10.1093/bioinformatics/btv638
Xue Z, Brooks JT, Quart Z, Stevens ET, Kable ME, Heidenreich J, McLeod J, Marco ML (2021) Microbiota assessments for the identification and confirmation of slit defect-causing bacteria in milk and Cheddar cheese. mSystems 6:e01114-20. https://doi.org/10.1128/mSystems.01114-20
Xue MY, Xie YY, Zhong YF, Liu JX, Guan LL, Sun HZ (2021b) Ruminal resistome of dairy cattle is individualized and the resistotypes are associated with milking traits. Anim Microbiome 3:18. https://doi.org/10.1186/s42523-021-00081-9
Yang X, Noyes NR, Doster E et al (2016) Use of metagenomic shotgun sequencing technology to detect foodborne pathogens within the microbiome of the beef production chain. Appl Environ Microbiol 82:2433–2443. https://doi.org/10.1128/AEM.00078-16
Zotta T, Ricciardi A, Condelli N, Parente E (2021) Metataxonomic and metagenomic approaches for the study of undefined strain starters for cheese manufacture. Crit Rev Food Sci Nutr 18:1–15. https://doi.org/10.1080/10408398.2020.1870927
Funding
The work in our research groups was funded by the European Union’s Horizon 2020 research and innovation programme under grant agreement No. 818368 (MASTER), and the grants RTI2018-095021-J-I00 (funded by (MCIU/AEI/FEDER, UE), AGL2016-78085-P and AGL2016-78311-R (funded by (MINECO/AEI/FEDER, UE). Carlos Sabater acknowledges his Postdoctoral research contract funded by the Instituto de Investigación Sanitaria del Principado de Asturias (ISPA) and Postdoctoral research contract Juan de la Cierva-Formación from Spanish Ministry of Science and Innovation (FJC2019-042125-I).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Sabater, C., Cobo-Díaz, J.F., Álvarez-Ordóñez, A. et al. Novel methods of microbiome analysis in the food industry. Int Microbiol 24, 593–605 (2021). https://doi.org/10.1007/s10123-021-00215-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10123-021-00215-8