
Abstract
New methodologies have been proposed to be incorporated in predictive microbiology in foods and quantitative microbial risk assessment (QMRA) to achieve more reliable models and facilitate predictive model applications. The meta-analysis is one of the proposed strategies focused on a systematic analysis of a large collection of data with the intention of generating standardized and summarized information to produce a global estimate. This data analysis approach can be applied to better understand the relationship between environmental factors and kinetic parameters or to input QMRA studies to assess the effect of a particular intervention or treatment concerning food safety. The emergence of systems biology is also affecting predictive microbiology, offering new and more mechanistic approaches to yield more reliable and robust predictive models. The so-called genomic-scale models are built on a molecular and genomic basis supported by experimental data obtained from the genomic, proteomic, and metabolomic research areas. Although the existing gene-scale models are promising regarding prediction capacity, they are still few and limited to specific model microorganisms and situations. Further research is needed, in the coming decades, to complete omics information and thus to produce more suitable models to be applied to real-world situations in food safety and quality.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Meta-analysis
- Stepwise process
- Data bases
- Systems biology
- Genomic-scale models
- Flux balance analysis (FBA)
- Network analysis
- Objective function
7.1 Introduction
7.1.1 Meta-Analysis Approach and Benchmarking Data
As previously explained, Quantitative Microbial Risk Assessment (QMRA) is an iterative process that gives insight into setting microbiological criteria and identifying the most relevant factors along the food chain. However, it is recognized that the great amount of data required is the most important drawback to be implemented. Also, as a multidisciplinary area, data processing is becoming more difficult as information is reported in a heterogeneous form. The need to account for variability and uncertainty sources together with the characterization of the main statistical distributions to describe the data leads to the creation of alternative tools to integrate these findings and provide a global estimate. A meta-analysis is a systematic analysis of a large collection of data from individual studies aiming to integrate the information generated in a QMRA study and to produce a global estimate of the effect of a particular intervention or treatment (van Besten and Zwietering 2012). This technique has been more extensively used in food microbiology and can give an improved understanding of main and side effects on microbiological kinetics (Ross et al. 2008).
To start the application of a meta-analysis approach, a sufficient number of data should be generated. Gonzales-Barron and Butler (2011) suggested a stepwise procedure to meta-analysis consisting of (1) systematic review; (2) data extraction to collate quantitative and qualitative information from the primary studies; (3) selection of the appropriate effect size parameter to describe, summarize, and compare the data of the primary studies, and when needed, subsequent translation of the reported findings of the individual studies into the parameter; (4) estimation of the overall effect size by combining the primary studies; (5) assessment of heterogeneity among the studies; and, finally, (6) the presentation of the meta-analysis results.
Selection of data coming from primary studies can begin with experimental data from research institutions or extra data available in scientific data bases. However, individual results must be incorporated into the meta-analysis when they are properly defined, structured, and transparently reported.
In the systematic review process the information to be included in the meta-analysis has to be sufficiently accurate to answer the embedded question of a given case study. For instance, in a lettuce disinfection process, one can measure several heads of lettuce to see if there is contamination by Escherichia. coli. The data introduced in the meta-analysis approach should justify if the intervention (disinfection) makes a causal inference on the outcome (presence/absence of E. coli) and, if so, how large the effect is.
The data extraction from the primary studies should provide the information necessary for summarizing and synthesizing the results and include both numeric and nonnumeric data.
Effect size refers to the degree to which the phenomenon is present in the population (reduction of E. coli numbers by disinfection). For the primary studies, meta-analysis converts the effect size into a ‘parameter’ that allows direct comparison and summation of the primary studies. There are many types of effect size parameters: (1) binary or dichotomous, for example, indicating the presence or absence of the event of interest in each subject, (2) continuous, and (3) ordinal, where the outcome is measured on an ordered categorical scale.
For the estimation of the overall size effect, primary studies may be weighted to reflect sample size, quality of research design, or other factors influencing their reliability. A relevant factor in precision is the sample size, with larger samples yielding more precise estimates than smaller samples. Another factor affecting precision is the study design, with matched groups yielding more precise estimates (as compared with independent groups) and clustered groups yielding less precise estimates. This consideration can also imply that the obtaining of a lower variance in the primary studies leads to a more accurate global estimate.
On the other hand, it is necessary to make a heterogeneity test among the primary samples to assess the extra-variation in the meta-analysis approach. Generally speaking, individual samples are weighted and statistically compared with aiming at quantifying the variability associated to heterogeneity. In food microbiology, most of the microbial data have been generated in culture media and the effect of environmental factors may not necessarily reflect what might happen in an actual food. Also, results of different studies on factors influencing microbial kinetics are not always similar or may be even contradictory. Variations among microbial strains, individual cell studies, or model estimations contribute positively to increase variability in results. Therefore, quantitative information about the influence of various factors on microbial kinetics is often not adequate under specific conditions, and also often is not available in the published literature.
Finally, results coming from the meta-analysis are presented into several graph types, such as bubble plots, which display point estimates and confidence intervals of each primary study and the overall effects in the global estimate.
The use of data bases in predictive microbiology can provide thousands of records of microbial growth or inactivation kinetics under a wide range of environmental conditions. A systematic and critical analysis of the literature followed by integration of the gathered data results in global estimates of kinetic parameters with their variability, and these can be used to benchmark the latest published data (van Asselt and Zwietering 2006). Meta-analysis has been used in various QMRA studies for relating the microbial concentration of a given hazard to a public health outcome (Pérez-Rodríguez et al. 2007b). However, large variability sources are expected in some cases, mainly because of heterogeneity in primary data. Additionally, overlapping problems are generated when the same information of one variable is obtained from different studies. In spite of these disadvantages, when a large dataset is manipulated, meta-analysis can provide useful links to discern between explanatory variables on the global estimate. The construction of updated data bases on the reviewed question or parameter can also reveal the present knowledge, can highlight default areas where there is a lack of information on factors that might affect the parameter of interest, and can therefore provide direction for future research.
7.2 Mechanistic Predictive Models
Advances in molecular biology, particularly in genome sequencing and high-throughput measurements, enable us to obtain comprehensive data on the cellular system and gain information on the underlying molecules (Kitano 2002). This genomics revolution has in the past years provided researchers with the option to look genome wide for cellular responses at the level of gene expression (Keijser et al. 2007) and protein presence (Wolff et al. 2006; Hahne et al. 2010). The need of integrating all this complex information has contributed to an emerging scientific field, so-called systems biology, aimed at understanding complex biological systems at the systems level (Kitano 2001). The fundamental idea behind the systems biology approach is that biological systems are hierarchically organized with influences going both up and down through the hierarchy (Brul et al. 2008).
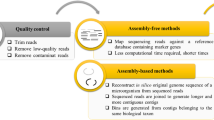
The great avalanche of ‘omics’ data (i.e., genomic and proteomic data) in systems biology necessitates applying mathematical methods to better understand the interactions and relationships among the different elements within the studied system (Fig. 7.1)

Scheme of the workflow applied to systems biology
. Stelling (2004) classified mathematical models applied in systems biology in interaction-based models, constraint-based models, and mechanism-based models. The interaction-based models refer to network topology analysis in which interactions between the different elements in the system, for example, metabolic reactions, protein–protein interactions, and gene regulation, are accounted for by graphical networks. In constraint-based models, physicochemical properties such as reaction stoichiometries and reversibilities impose constraints on network function in addition to network topology. This network reconstruction process ultimately results in the generation of a biochemically, genomically, and genetically (BiGG) structured data base that can be further utilized for both mathematical computation and analysis of high-throughput data sets. The network spans the set of metabolic reactions taking place in a specific biological system, assuming a stationary state (Hertog et al. 2011) in which each reaction is referred to as a flux. The methodologies developed in metabolic engineering such as metabolic control analysis and metabolic flux analysis are applied to analyze steady-state fluxes, although these may also be used to explain oscillatory systems so long as average fluxes are considered (Schuster et al. 2002). More quantitative models can be addressed based on kinetic rates of metabolic reactions included in the biological networks. In this approach, a system of linear differential equations is used to account for reactions rate of the quasi-dynamic or dynamic state fluxes (Hertog et al. 2011). As new genomic data become available, these may aid in the parameterization of metabolic models (Voit 2002). However, one weakness of this approach is that it ignores the variability and noise found in biological networks, which may have important implications in their function (Heath and Kavraki 2009). To overcome this limitation, a stochastic approach has been proposed that basically consists of adding a noise term to the differential equations. Similarly, gene expression regulation (i.e., transcription and translation) and signaling networks have a probabilistic nature that should be accounted for by applying a stochastic approach (Treviño Santa Cruz et al. 2005; McAdams and Arkin 1997).
The latter type of model mentioned by Stelling (2004) refers to mechanism-based models. The author means that with this type of model, models can predict the system dynamics by integrating detailed mechanisms operating in metabolism, signal processing, and gene regulation. The success of this mechanistic approach, that is, integrated modeling, relies largely on the availability of information about the complete mechanism and attendant parameters.
Traditionally, in the field of predictive microbiology applied to foods, the scarce information on the mechanisms involved in the cellular functions has hampered microbiologists from undertaking more mechanistic models, albeit some mechanistic parameters has been introduced in specific cases (Baranyi and Roberts 1995). The emergence of systems biology is creating a new path for microbiologists in predictive microbiology, offering new and more mechanistic approaches to give rise to more reliable and robust models (Brul et al. 2008). In so doing, predictive microbiology will be able to move from the most used empirical modeling, that is, black box models, toward so-called white or gray box models, based on an better understanding of the biological functions in cells, enabling providing more accurate predictions under specific physical and chemical changes and even extending the model outside the range of space bounded by observations. Such data not only allow for a better fine-tuning of growth/no growth boundaries but will also begin to strengthen die-off/survival models (Brul et al. 2008). Several computer models have been developed on the basis of information derived from systems biology studies and wealthy databases. However, many of the mechanistic studies have been done under conditions and in model microorganisms with relatively low practical relevance (Brul et al. 2008). One of the most studied microorganisms is E. coli, as much is known about its metabolism, regulation, and genome, enabling the development of more mechanistic and reliable in silico models for this model microorganism (Reed and Palsson 2003). The experience obtained with E. coli has served to be applied to other microorganisms such as Haemophilus influenzae (Edward and Palsson 1999), Helicobacter pylori, and Saccharomyces cerevisiae (Petranovic and Vemuri 2009).
To date, few systems biology-based models have been explored or developed within the area of predictive microbiology in foods (Brul et al. 2008). However, constraint-based models seem to be the first choice by microbiologists to understand the behavior of microorganisms in food-related environments (Métris et al. 2011; Peck et al. 2011). The most significant kinetic reactions constituting the metabolism of the model bacterium are modeled and simulated to know which specific metabolic processes are related to a determined bacterial response (e.g., outgrowth, adaptation, survival). These models consist of describing the fluxes that make up a metabolic network in which each flux accounts for a metabolic reaction as concentration change per time unit for the substrate and product. The reactions can be described by a system of linear differential equations in which stoichiometric coefficient of equations are assumed to be constant because the model represents
Here, x defines a vector of the intermediate concentrations of metabolites at a specific time, S is the stoichiometric matrix describing all the metabolic reactions, and S ij corresponds to the ith stoichiometric coefficient in the jth reaction. The thermodynamic constraints and enzyme capacity constraints are represented by vector v = [v1, … vj], which includes the reaction rates of each metabolic reaction or flux. Setting Eq. (7.1) to 0 means that conservation laws apply in the production and consumption rates (i.e., rateconsumption = rateproduction). A simplified example of the steady-state flux might be the well-known coenzyme nicotinamide adenine dinucleotide (NAD), involved in many metabolic routes as an electron donator. In this case, the reaction would be
According to the law of conservation, NAD + NADH = a constant, which means that the sum of concentrations of NAD and NADH does not change with time.
The derivation of the reaction rate equations is another important aspect and should be based on an appropriate metabolic network, which should be completely known and closed. The quasi-steady-state and rapid equilibrium approaches can be used to obtain the reaction rate equations. With regard to the kinetic parameters, these might be estimated by using sources such as literature data, electronic data bases, experimental data for dependencies between initial reactions rates and products, inhibitors, substrates, and activators, and finally time-series data for enzyme kinetics and whole pathways (Demin et al. 2005).
Because the system of equations has more fluxes than metabolites, the system is underdetermined (Kauffman et al. 2003), which means that the system has multiple solutions. To reduce the solution space of the system, the model is constrained by imposing different rules, which are often related to thermodynamic feasibility, enzymatic capacity, and mass balance. Model solutions that do not comply with such criteria are excluded from the solution space of the model (Reed and Palsson 2003). Once constraints are defined for the model, the corresponding solution space should be determined. To this end, several mathematical approaches can be taken such as linear optimization, elementary modes and extreme pathways, phenotypic phase plane analysis, gene deletions, or finding objective functions. The linear optimization, which is referred to as flux balance analysis (FBA), is based on an objective function, which is utilized to define the solution space by maximizing or minimizing the defined objective function (Feist and Palsson 2010; Varma and Palsson 1994). The most used objective functions include ATP production, production of a specific by-product, and biomass production (i.e., growth rate) (Van Impe et al. 2011; Reed and Palsson 2003). In this respect, using a biomass production objective function can accurately estimate the growth rate of E. coli, as evidenced by the work by Feist et al. (2007).
Métris et al. (2011) performed in silico simulations based on the model of E. coli K12 MG1655 previously developed by Feist et al. (2007) considering 1,387 metabolic reactions and 1,260 genes. This study can be considered as one of the first approaches of predictive microbiology in the foods area to systems biology modeling. This model applied the most often used objective function based on optimizing the biomass production, which is associated with growth-associated maintenance (GAM) energy and non-growth-associated maintenance (NGAM) energy. Their values were derived from experiments in a chemostat without added NaCl, which we refer to as the control conditions (Feist et al. 2007). The model was modified to consider exposure to osmotic stress by including changes of concentrations in an osmoprotectant associated with osmolarity changes. The work did not find definitive results relating the changes of these substances with a decrease of the growth rate. Similarly, the model was tested to ascertain if biomass composition derived from osmotic stress might explain the decrease of growth rates observed in experiments; however, again the results were not conclusive. Finally, the authors suggested that more specific objective functions should be developed to explain the chemicophysical limitations of the growth rate. For that, authors suggested including gene regulation, crowding, and other additional cell resources such as ribosomal content and some tradeoff observed under osmotic stress. This work and its results provide evidence that a new modeling approach is emerging, although still with important gaps and limitations. Nonetheless, it might provide the necessary theoretical basis to develop more mechanistic predictive models in foods (Fig. 7.1).
References
Baranyi J, Roberts TA (1995) Mathematics of predictive food microbiology. Int J Food Microbiol 26:199–218
Brul S, Mensonides FIC, Hellingwerf KJ, Teixeira de Mattos MJ (2008) Microbial systems biology: new frontiers open to predictive microbiology. Int J Food Microbiol 128:16–21. doi:10.1016/j.ijfoodmicro.2008.04.029
Demin OV, Plyusnina TY, Lebedeva GV, Zobova EA, Metelkin EA, Kolupaev AG, Goryanin II,Tobin F (2005) Kinetic modelling of the Escherichia coli metabolism. Top Curr Genet 13. In: Alberghina L, Westerhoff HV (eds) Systems Biology
Edward JS, Palsson BO (1999) J Biol Chem 274:17410–17416
Feist AM, Palsson BØ (2010) The biomass objective function. Curr Opin Microbiol 13:344–349. doi:10.1016/j.mib.2010.03.003
Feist AM, Henry CS, Reed JL, Krummenacker M, Joyce AR, Karp PD, Broadbelt LJ, Hatzimanikatis V, Palsson BO (2007) A genome-scale metabolic reconstruction for Escherichia coli K-12 MG1655 that accounts for 1260 ORFs and thermodynamic information. Mol Syst Biol 3:121. doi:10.1038/msb4100155
Gonzales-Barrón U, Butler F (2011) The use of meta-analytical tools in risk assessment for food safety. Food Microbiol 28:823–827. doi:10.1016/j.fm.2010.04.007
Hahne H, Mäder U, Otto A, Bonn F, Steil L, Bremer E, Hecker N, Becher D (2010) A comprehensive proteomics and transcriptomics analysis of Bacillus subtilis salt stress adaptation. J Bacteriol 192:870–882. doi:10.1128/JB.01106-09
Heath AP, Kavraki LE (2009) Computational challenges in systems biology. Comput Sci Rev 3:1–17. doi:10.1016/j.cosrev.2009.01.002
Hertog MLATM, Rudell DR, Pedreschi R, Schaffer RJ, Geeraerd AH, Nicolai BM, Ferguson I (2011) Where systems biology meets postharvest. Postharvest Biol Tec 62:223–237. doi:10.1016/j.postharvbio.2011.05.007
Kauffman KJ, Prakash P, Edwards JS (2003) Advances in flux balance analysis. Curr Opin Biotechnol 14:491–496. doi:10.1016/j.copbio.2003.08.001
Kitano H (2002) Systems biology: a brief overview. Science, New York 295:1662–1664. doi: http://10.1126/science.1069492
McAdams H, Arkin A (1997) Stochastic mechanisms in gene expression. Proc Natl Acad Sci USA 94:814–819. doi:10.1073/pnas.94.3.814
Métris A, George S, Baranyi J (2011) Modelling osmotic stress by flux balance analysis at the genomic scale. Int J Food Microbiol 152:123–128. doi:10.1016/j.ijfoodmicro.2011.06.016
Peck MW, Stringer SC, Carter AT (2011) Clostridium botulinum in the post-genomic era. Food Microbiol 28:183–191. doi:10.1016/j.fm.2010.03.005
Pérez-Rodríguez F, van Asselt ED, Garcia-Gimeno RM, Zurera G, Zwietering MH (2007b) Extracting additional risk managers information from a risk assessment of Listeria monocytogenes in deli meats. J Food Prot 70:1137–1152
Petranovic D, Vemuri GN (2009) Impact of yeast systems biology on industrial biotechnology. J Biotechnol 144:204–211. doi:10.1016/j.jbiotec.2009.07.005
Reed JL, Palsson BØ (2003) Thirteen years of building constraint-based in silico models of Escherichia coli MINIREVIEW Thirteen Years of Building Constraint-Based In Silico Models of Escherichia coli. Society. doi:10.1128/JB.185.9.2692
Ross T, Zhang D, Mc Questin OJ (2008) Temperature governs the inactivation rate of vegetative bacteria under growth-preventing conditions. Int J Food Microbiol 128:129–135. doi:10.1016/j.ijfoodmicro.2008.07.023
Schuster S, Klamt S, Weckwerth W, Moldenhauer F, Pfieffer T (2002) Use of network analysis of metabolic systems in bioengineering. Bioprocess Biosyst Eng 24:363–372. doi:10.1016/S0167-7799(02)02026-7
Stelling J (2004) Mathematical models in microbial systems biology. Curr Opin Microbiol 7:513–518. doi:10.1016/j.mib.2004.08.004
Treviño Santa Cruz MB, Genoud D, Métraux JP, Genoud T (2005) Update in bioinformatics. Toward a digital database of plant cell signalling networks: advantages, limitations and predictive aspects of the digital model. Phytochemistry 66:267–276. doi:10.1016/j.phytochem.2004.11.020
Van Besten HMW, Zwietering MH (2012) Meta-analysis for quantitative microbiological risk assessments and benchmarking data. Trends Food Sci Technol 25:34–39. doi:10.1016/j.tifs.2011.12.004
Van Impe JF, Vercammen D, Van Derlinden E (2011) Developing next generation predictive models: a systems biology approach. Proc Food Sci 1:965–971. doi:10.1016/j.profoo.2011.09.145
Varma A, Palsson BO (1994) Metabolic flux balancing: basic concepts, scientific and practical use. Biotechnology 12:994–998. doi:10.1038/nbt1094-994
Voit EO (2002) Models-of-data and models-of-processes in the post-genomic era. Math Biosci 180:263–274. doi:10.1016/S0025-5564(02)00115-3
Wolff S, Antelmann H, Albrecht D, Becher D, Bernhardt J, Bron S, Bütner K, van Dijl JM, Eymann C, Otto A, Tam LT, Hecker M (2006) Towards the entire proteome of the model bacterium Bacillus subtilis by gel-based and gel—free approaches. J Chromatogr B 849:129–140
Author information
Authors and Affiliations
Rights and permissions
Copyright information
© 2013 Fernando Pérez-Rodríguez and Antonio Valero
About this chapter
Cite this chapter
Pérez-Rodríguez, F., Valero, A. (2013). Future Trends and Perspectives. In: Predictive Microbiology in Foods. SpringerBriefs in Food, Health, and Nutrition, vol 5. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-5520-2_7
Download citation
DOI: https://doi.org/10.1007/978-1-4614-5520-2_7
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-5519-6
Online ISBN: 978-1-4614-5520-2
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)