
Abstract
The microbial community diversity and succession of Chinese Sichuan sausages during the spontaneous fermentation were demonstrated using high-throughput sequencing technology. The bacterial diversity was abundant and the succession of bacterial community along the direction of Lactobacillus spp. increased and Weissella spp. decreased. While fungal diversity was single and trace fungal population was detected. The core functional microbiota were lactic acid bacteria, including Lactobacillus spp., Weissella spp. and Pediococcus spp. In initial fermentation, Weissella spp. was the dominant bacteria and its relative abundance was 49.84%, but then its relative abundance decreased to 11.96% during fermentation before recovering to 26.74% at the end of fermentation. Meanwhile, Lactobacillus spp. rose from 24.70 to 55.74% and became the dominant genus. Moreover, Pediococcus spp. increased from 0.06 to 18.05% on day 20 but then decreased to 1.89% on day 30. These results revealed that the primary microorganisms contributing to spontaneous fermentation of Chinese Sichuan sausages were bacteria, while eukaryotic microorganisms such as yeast scarcely contributed to fermentation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Chinese Sichuan sausages are Chinese traditional meat products with a long history in China, which are fermented spontaneously for 4–5 weeks at 8–15 °C for the development of distinctive flavors (Chen et al. 2016; Liu et al. 2010; Wang et al. 2013). On the basis of their production process, they are considered as fermented sausages and involve various microorganisms which usually determine their quality and flavors (Chen et al. 2016; Rebecchi et al. 2015). Therefore, it is essential to investigate microbial community characteristics to identify the primary microorganisms and core functional microorganisms for improvement of fermentation management and exploring microbial resource as starter cultures.
In the past several years, the analyses of microbial community in Chinese fermented sausages, mainly focused on bacterial community, especially for lactic acid bacteria, using culture-dependent methods, PCR-temporal temperature gradient gel electrophoresis (PCR-TTGE) and PCR-denaturing gradient gel electrophoresis (PCR-DGGE), but little attention has been paid to fungal diversity (Gao et al. 2014; Wang et al. 2016; Mauriello et al. 2004). Accordingly, a complete understanding of the composition, diversity and succession of microbial community remains elusive, and the core functional microorganisms shift in Chinese Sichuan sausages fermented spontaneously remains unclear.
Nowadays, high-throughput sequencing (HTS) technology conducted by Illumina MiSeq™ analysis has been considered as a powerful tool for exploring complex microbial ecosystems regarding its potentiality to overcome classical microbiology deficiency because it is rapidly improving in quality, speed and cost (Quigley et al. 2012; Dalmasso et al. 2016; Carmen and Mas 2016). Therefore, it is becoming widely used to investigate whole microbial communities in food matrixes, such as Italian salami (Wang et al. 2018; Połka et al. 2015), Paocai (Liang et al. 2018) and Douchi (Yang et al. 2016). However, until now this approach has not been applied to research the microbial community in Chinese Sichuan sausages fermented spontaneously. Thus, in this study, microbial community diversity and succession of Chinese Sichuan sausages during spontaneous fermentation was unraveled by HTS approach. These results will enhance the understanding of the characteristics of microbial community and microbial ecological function of Chinese Sichuan sausages from the point of core functional microorganisms.
Materials and methods
Samples preparation and sampling
Chinese Sichuan sausages formulation includes 750 g/kg lean pork meat, 250 g/kg pork back fat, 25 g/kg NaCl, 2 g/kg sucrose, 5 g/kg chili powder, 50 mg/kg seed powder of Chinese prickly ash, 10 g/kg Spice powder and 100 mg/kg NaNO2. Then the sausages were made and fermented spontaneously according to the methods as described by Wang et al. (2013). Sausages (50 g) were sampled in triplicate from fermentation on day 1, 10, 20 and 30. Then samples were immediately stored at −80 °C for subsequent analysis.
The pH, aw, total lactic acid and total volatile base nitrogen measurement
The pH, aw, total lactic acid and total volatile base nitrogen (TVB-N) were measured in triplicate according to the methods as described by Wang et al. (2018).
DNA extraction
The microbial DNA of samples was extracted using the E.Z.N.A™ Mag-Bind Soil DNA Kit (OMEGA, USA) and DNA quality was monitored using agarose gel electrophoresis according to the methods as described by Wang et al. (2018).
Illumina high-throughput sequencing
Extracted DNA was used as the template and the V4 region of bacterial 16S rRNA and fungal 18S rRNA were amplified by PCR using the universal bacterial primers 515F and 806R with Illumina barcoded adapters (Caporaso et al. 2010; Wang et al. 2018), and the universal fungal primers V43NDF and Euk_V4_R (Wang et al. 2018), respectively. The two step-PCR technology was conducted in which the first PCR step worked 25 cycles using untagged primers, and the second PCR step worked 5 cycles with tagged primers and the first step products used as template. The PCR amplification was carried out according to the methods as described by Wang et al. (2018). Then, the purified PCR products was sequenced with a MiSeq Illumina instrument (Illumina, USA) at Sangon Biotech Co., Ltd (Shanghai, China) (Caporaso et al. 2010; Edgar 2010; Mago et al. 2011).
Data analysis
The operational taxonomic unit (OTU) at an identity threshold of 97% with UPARSE software (Edgar 2010) and the taxonomy based approaches at a set confidence threshold of 80% using Rv 3.0.0 (R Development Core Team 2011) with the Vegan package (Dixon 2003) were performed for the sequence data analysis. Shannon index and Chao1 estimator values were calculated in RDP at 97% sequence similarity (http://pyro.cme.msu.edu/). Alpha diversity was evaluated by Rarefaction curves, Good’s coverage, Simpson and Shannon diversity indices and Chao 1 richness (Grice et al. 2009). Principal component analysis (PCA) was performed to depict the microbial communities composition of samples (Jiang et al. 2013). Venn diagram was used to assess the similarity and difference in microbial communities among all samples (Liu et al. 2017).
Results and discussion
The pH, aw, total lactic acid values and TVB-N concentration
The analyses of pH, aw, total lactic acid values and TVB-N concentration in the Chinese Sichuan sausages during spontaneous fermentation are shown in supplemental Table 1. A slight increase in pH was found in sausages through the whole fermentation course and pH increased from initial 5.49 to 5.73, which may be because of peptides, amino acids, and amines generated by microbial proteolytic activity, resulting in a buffering effect on the organic acids (Ruiz-Moyano et al. 2011). These findings are in agreement with those reported by other authors (Essid and Hassouna 2013; Lorenzo et al. 2014). Interestingly, a sharp increase in total lactic acid values was detected in sausages. The total lactic acid values increased from initial 23.2 to 68.7 mmol/kg on day 10, but then gradually reduced to 48.7 mmol/kg on day 30. These results are in agreement with microbial community structure variety, by which time Lactobacillus spp., which possesses strong acid producing ability and acid resistance (Wang et al. 2013), underwent a sharp increase from 24.70% (day 1) to 63.14% (day 20), then decreased to 55.74% on day 30. The aw value was 0.992 in the initial fermentation, and then gradually decreased to 0.815 along with the fermentation. Such low water activity has some advantages including the inhibition of food-borne pathogenic and spoilage bacteria (Wang et al. 2018). A slight increase reduction in TVB-N concentration was observed from day 1 to day 20, but then increased to 2.73 mg/kg on day 30. The variation trend of TVB-N content as a useful index for spoilage (Hu et al. 2007) was opposite to the change in proportion of core functional microorganisms, including Lactobacillus spp., Weissella spp. and Pediococcus spp. These results indicate that core functional microorganisms becoming dominant microorganisms can conducive to improve the hygienic quality of fermented sausages.
Abundance and diversity of the bacterial and fungal microorganisms
To understand microbial communities composition of samples, OTU reads were performed to depict the bacterial and fungal microorganisms in Chinese Sichuan sausage during spontaneous fermentation. Based on the sequencing results, a total of 259,423 high quality bacterial tags and a total of 283,690 high quality 18S rDNA reads were generated from 4 examined sample sets across the entire sausage fermentation process, respectively. Accordingly, the bacterial sequence reads ranged from 109,844 to 46,110, and fungal sequence reads ranged from 78,592 to 65,637. These sequence reads were clustered into 7609 OTUs at a 97% similarity level, in which 1363 OTUs belonged to bacteria and 6246 OTUs was attributed to eukaryotic microorganism. Moreover, the average number of OTUs was 340 for bacteria and 1561 for eukaryotic microorganisms, respectively. Furthermore, the analyses of the alpha-diversity indices, including the Chao1, Shannon and Simpson diversity indices, are presented in Table 1. The Good’s coverage values were 100% for bacterial OTUs and 99% for fungal OTUs in all samples, suggesting that major bacterial and fungal OTUs had been captured (Grice et al. 2009). The bacterial Chao1 richness estimator decreased from 1 to 20 days fermentation with a range from 560.24 to 304.29, whereas Chao1 richness estimator increased to 580.08 at 30 days fermentation. The fungal Chao1 richness estimator changed little from 1 to 20 days fermentation, except for 20 days fermentation which the Chao1 richness estimator increased to 3730.5. According to the estimated Chao1, the bacterial diversity was clearly tended to decline during the early and mid-term stages of fermentation, and gradually recover at the end of fermentation. Eukaryotic diversity was fluctuating during the early and mid-term fermentation, then a dramatic increase of eukaryotic diversity was observed at the end of fermentation. These results showed that bacteria were prolific at initial stage of fermentation, but suffered along with the fermentation, presumably due to interspecific competition and the decrease of aw during the ripening process, which may seriously inhibit the growth of bacteria, while fungal diversity remained stable during fermentation. In addition, the both bacterial and fungal Simpson indices were the lowest in sample CWC4 which was sampled at the end of fermentation. These results indicated that microbial species increased in the sausages at the end fermentation due to attenuation of core functional microbiota.
Microbial community composition
Bacterial community
Rarefaction analysis based on OTUs at 97% similarity showed approximation to an asymptote as shown in Fig. 1a, which indicated that the sequences commendably represented the bacterial diversity in the 4 samples form different fermentation stages.

Richness rarefaction curves (a) and Venn diagrams (b) of the four samples based on OTUs of bacterial sequences (3% distance). CWC1: sample fermented for 1 day, CWC2: sample fermented for 10 days, CWC3: sample fermented for 20 days, CWC4: sample fermented for 30 days
The Venn diagram was performed to evaluate the distribution of OTUs among the different samples as shown in Fig. 1b. In total, 468, 290, 209 and 396 OTUs were generated from CWC1, CWC2, CWC3 and CWC4 sample, respectively. A total of 46 OTUs were shared by the four samples, which indicated that a low level similarity of bacteria diversity was presented in the four samples. These results suggest that the succession of bacterial communities changed greatly during the fermentation. The shared bacterial OTUs mainly belonged to Firmicutes and Proteobacteria at phylum level, and Lactobacillus spp., Weissella spp., Pediococcus spp. and Staphylococcus spp. at the genus level.
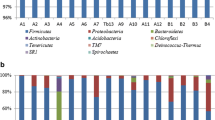
A total of 17 phyla, 35 classes, 69 orders, 111 families and 121 genera were received from bacterial sequences using the RDP classifier. The bacterial population of the samples was investigated at both the phylum (Fig. 2a) and genus levels (Fig. 2b), and a more precise percentage of the bacterial composition at the genus level among four samples is presented in supplemental Table 2. Five bacterial phyla, including Firmicutes, Proteobacteria, Actinobacteria, Bacteroidetes and Chloroflexi, were identified in the initial fermentation, in which Firmicutes, Proteobacteria, Actinobacteria and Bacteroidetes were detected through the whole fermentation process. Furthermore, Firmicutes and Proteobacteria were the predominant phyla, which ranged from 85.65 to 93.96% and from 5.59 to 13.95%, respectively.

Relative abundance of bacteria community proportions at phylum (a) and genus (b) level. Phyla and genera occurred at < 1% abundance in all the samples are defined as “Others”. Taxonomic classification of 97% sequence identity clusters demonstrated at genera classification levels. CWC1: sample fermented for 1 days, CWC2: sample fermented for 10 days, CWC3: sample fermented for 20 days, CWC4: sample fermented for 30 days
In terms of genus, 31, 30, 30 and 30 genera were determined in CWC1, CWC2, CWC3 and CWC4 sample, respectively. In the initial fermentation (sample CWC1), Weissella spp. was the most abundant bacterial genus, comprising about 49.4% of bacteria, followed by Lactobacillus spp., accounting for 24.70% of bacteria. A drastic reduction in bacterial relative abundance of Weissella spp. were detected from day 1 to day 20 during fermentation, and the proportion of Weissella spp. declined from 49.41 to 11.96%. But then Weissella spp. recovered rapidly to reach 26.74% at the end of fermentation (day 30). The decrease of Weissella spp. might be due to interspecific completion with Lactobacillus spp., by which time Lactobacillus spp. underwent a sharp increase from 24.70 to 63.14% and become the most dominant genus, substituting for Weissella spp.. Interestingly, in the initial fermentation, Pediococcus spp. was barely detectable in sample CWC1, but during the subsequent fermentation, this genus increased sharply from 0.06% (day 1) to 18.05% (day 20), then declined dramatically again to 1.89% on day 30. Moreover, other major genera present in the initial fermentation included Brochothrix spp., Psychrobacter spp., Staphylococcus spp., Enterococcus spp., Vibrio spp. and Yersinia spp., each constituting more than 1% of total bacteria. Then, Psychrobacter spp., Enterococcus spp. and Vibrio spp. declined dramatically during the subsequent fermentation and these genera were not detected on fermentation day 30. Brochothrix spp. which belongs to LAB declined dramatically from 7.77% (day 1) to 0.09% (day 20), then recovered rapidly to 4.30% on day 30. Staphylococcus spp. exhibited a similar trend to Brochothrix spp., with the bacterial relative abundance decreased dramatically from 2.74% (day 1) to 0.56% (day 20), then recovered rapidly to 2.52% on day 30.
Fungal community
Fungal diversity of the samples investigated at both the phylum and genus levels is shown in Fig. 3. Most of the reads, occupying for 90% of the total fungal sequences, belonged to unclassified reads. Compared with bacteria, the fungal diversity in sausages was lower during fermentation. At fungal phylum level, only Bangiales and Ascomycota was presented higher than 0.1% throughout the entire fermentation process. At the genus level, Porphyra spp. dominated throughout the entire fermentation process, increasing gradually from 3.75 to 10.75%. Debaryomyces spp. and Saccharomyces spp. were detected with a small amount of quantity, lower than 0.1%.

Relative abundance of fungal community proportions at phylum (a) and genus (b) level. Phyla and genera occurred at < 1% abundance in all the samples are defined as “Others”. Taxonomic classification of 97% sequence identity clusters demonstrated at genera classification levels. CWC1: sample fermented for 1 days, CWC2: sample fermented for 10 days, CWC3: sample fermented for 20 days, CWC4: sample fermented for 30 days
Microbial succession analysis
The similarities in microbial communities in sausages during fermentation process were evaluated by PCA analysis as shown in Fig. 4, showing an obvious separation of the bacterial communities among the four different samples. The first and second axes showed values of cumulative percentage variance of species equal to 88% and 11%, respectively (Fig. 4a). In total, 99% variances of species were explained by the two axes. According to PCA analysis, bacterial communities fluctuated wildly during fermentation from day 1 to 30. However, the fungal communities were relatively stable during fermentation (Fig. 4b).

Multiple samples of PCA analysis according to the bacterial (a) and fungal (b) diversity. CWC1: sample fermented for 1 days, CWC2: sample fermented for 10 days, CWC3: sample fermented for 20 days, CWC4: sample fermented for 30 days
The heat map analysis was performed to compare the differences and similarity in the microbial community structures of sausages during fermentation as shown in Fig. 5 (Rundell et al. 2014). Bacterial community structure of Chinese Sichuan sausages fluctuated greatly during fermentation. In the initial fermentation, Weissella spp. was the most dominant genus, but then decreased from 49.84 to 11.96% during fermentation before recovering to 26.74% at the end of fermentation which remained the predominant genera. The members of Weissella spp. have been regarded as the functional lactic acid bacteria because of their useful metabolites, particularly for acids and bacteriocins, which not only could be conducive to the formation of flavour compounds, but also inhibit the growth of pathogens. During fermentation, Lactobacillus spp. increased from 24.70% (day 1) to 55.74% (day 30) and became the most dominant genus, replacing the dominant position of Weissella spp.. The members of Lactobacillus spp. have been considered to be responsible for development of the distinctive sour taste and rich flavour in sausages due to their capacities for the breakdown of proteins to peptides and amino acids (Georgieva et al. 2009; Luckow and Delahunty 2004). Besides, The proportion of Pediococcus spp. which have been believed to be response for the proteolysis and release of free amino acids increased sharply from 0.06% (day 1) to 18.05% (day 20) but then decreased to 1.89% on day 30 become a minority genus. These findings indicate that Lactobacillus spp., Weissella spp., and Pediococcus spp. are core functional microorganisms in Chinese Sichuan sausages during spontaneous fermentation, which play an impotent role in the formation of the maturity and flavor of the sausages. While low abundance of Staphylococcus spp. and Micrococcus spp. which have been developed as commercial starter cultures for the manufacture of fermented sausages (Leroy et al. 2006; Corbiére Morot-Bizot et al. 2007) were found in Chinese Sichuan sausages during spontaneous fermentation. These findings are in disagreement with those reported by other authors (Zhang et al. 2009) who reported that predominant microorganisms in Chinese traditional dry-cured sausages were Staphylococcus spp., Micrococcus spp. and Lactobacillus spp. with culture-dependant method and Random amplified polymorphic DNA (RADP) molecular marker technology. Moreover, the bacterial distribution of sample CWC1 (at start stage of fermentation) was most abundant, while microbial diversity gradually became lower along with the fermentation (sample CWC2 and CWC3).

Bacterial (a) and fungal (b) community heatmap analysis of Chinese Sichuan sausages during the spontaneous fermentation. CWC1: sample fermented for 1 days, CWC2: sample fermented for 10 days, CWC3: sample fermented for 20 days, CWC4: sample fermented for 30 days
It is more suitable for undesirable microbial reproduction in spontaneous fermentation than inoculated fermentation. In this study, the Enterococcus spp. as one of important pathogens in Gram-positive bacteria with relative abundance of 1.84% was checked in CWC1. The most common clinical infections caused by Enterococcus spp. is urinary tract infection, followed by bacteremia, bacterial endocarditis, diverticulitis and meningitis (Ubeda et al. 2010). Moreover, Gram-negative bacteria with relative abundance of 18.07%, including Brochothrix spp., Pseudomonas spp. and Acinetobacter spp. were found in CWC1. Many members of Gram-negative bacteria are involved as pathogenic and spoilage bacteria. Thus, they are frequently considered as undesirable bacteria in fermented meaty products and a hazardous source of transmissible antibiotic resistance (Greppi et al. 2015). Interestingly, when Lactobacillus spp., Weissella spp., and Pediococcus spp. gradually became the predominant strains, the growth of these undesirable microorganisms was inhibited. Compared with bacteria, trace fungal population except from porphyra spp. was detected in among all the samples. Just low abundance of Debaryomyces spp. (0.39%) was found in CWC1(at start stage of fermentation). Subsequently, the abundance of Debaryomyces spp. gradually decreased along with fermentation. Debaryomyces spp. were found in fermentation sausages by culture-dependant method and Debaryomyces spp., such as Debaryomyces hansenii has been considered that plays an important role in fermentation sausages, which has been widely inoculated in fermented sausages as a starter (Corral et al. 2014; Cano-García et al. 2013; Núñez et al. 2014). However, in the present study, it was showed that Debaryomyces spp. was very rare in Chinese Sichuan sausages fermented spontaneously and did not become a core microorganism. Microbial community diversity and succession of Chinese Sichuan sausages during fermentation indicated that bacteria are the dominant microbial communities while eukaryotic microorganisms are seldom, suggesting that bacteria, especially for lactic acid bacteria including Lactobacillus spp., Weissella spp. and Pediococcus spp., play a leading role in the sausage fermentation process. These results revealed that Lactobacillus spp., Weissella spp. and Pediococcus spp. can be developed as starter cultures.
References
Cano-García L, Rivera-Jiménez S, Belloch C, Flores M (2013) Generation of aroma compounds in a fermented sausage meat model system by Debaryomyces hansenii strains. Food Chem 18(151):364–373
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman F (2010) QIIME allows integration and analysis of high-throughput community sequencing data. Nat Methods 7:335–336
Carmen PM, Mas A (2016) Analysis of microbial diversity and dynamics during wine fermentation of Grenache grape variety by high-throughput barcoding sequencing. LWT Food Sci Technol 72:317–321
Chen X, Li JP, Zhou T, Li JC, Yang JN, Chen WH, Xiong YL (2016) Two efficient nitrite-reducing Lactobacillus strains isolated from traditional fermented pork (Nanx Wudl) as competitive starter cultures for Chinese fermented dry sausage. Meat Sci 121:302–309
Corbiére Morot-Bizot S, Leroy S, Talon R (2007) Monitoring of staphylococcal starters in two French processing plants manufacturing dry fermented sausages. J Appl Microbiol 102:238–244
Corral S, Salvador A, Carmela B, Mónica F (2014) Effect of fat and salt reduction on the sensory quality of slow fermented sausages inoculated with Debaryomyces hansenii yeast. Food Control 45:1–7
Dalmasso A, Maria DSR, Civera T, Pattono D, Cardazzo B, Bottero MT (2016) Characterization of microbiota in Plaisentif cheese by high-throughput sequencing. LWT Food Sci Technol 69:490–496
Dixon P (2003) Vegan, a package of R functions for community ecology. J Veg Sci 14:927–930
Edgar RC (2010) Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461
Essid I, Hassouna M (2013) Effect of inoculation of selected Staphylococcus xylosus and Lactobacillus plantarum strains on biochemical, microbiological and textural characteristics of a Tunisian dry fermented sausage. Food Control 32:707–714
Gao YR, Da Li P, Liu XY (2014) Bacteriocin-producing Lactobacillus sakei C2 as starter culture in fermented sausages. Food Control 35(1):1–6
Georgieva R, Iliev I, Haertle T, Chobert JM, Ivanova I, Danova S (2009) Technological properties of candidate probiotic Lactobacillus plantarum strains. Int Dairy J 19:696–702
Greppi A, Ferrocino I, La SA, Rantsiou K, Ercolini D, Cocolin L (2015) Monitoring of the microbiota of fermented sausages by culture independent rRNA based approaches. Int J Food Microbiol 212:67–75
Grice EA, Kong HH, Conlan S, Deming CB, Davis J, Young AC (2009) Topographical and temporal diversity of the human skin microbiome. Science 324:1190–1192
Hu Y, Xia W, Liu X (2007) Changes in biogenic amines in fermented silver carp sausages inoculated with mixed starter cultures. Food Chem 104:188–195
Jiang XT, Peng X, Deng GH, Sheng HF, Wang Y, Zhou HW, Tem NF (2013) Illumina sequencing of 16S rRNA tag revealed spatial variations of bacterial communities in a mangrove wetland. Microb Ecol 66:96–104
Leroy F, Verluyten J, De Vuyst L (2006) Functional meat starter cultures for improved sausage fermentation. Int J Food Microbiol 106:270–285
Liang HP, Chen HY, Zhang WX, Yu CX, Jia CF, Lin XP (2018) Investigation on microbial diversity of industrial Zhacai paocai during fermentation using high-throughput sequencing and their functional characterization. LWT Food Sci Technol 91:460–466
Liu DC, Wu SW, Tan FJ (2010) Effects of addition of anka rice on the qualities of low-nitrite Chinese sausages. Food Chem 118(2):245–250
Liu WR, Yang DH, Chen WJ, Gu X (2017) High-throughput sequencing-based microbial characterization of size fractionated biomass in an anoxic anammox reactor for low-strength wastewater at low temperatures. Biores Technol 231:45–52
Lorenzo JM, Gomez M, Fonseca S (2014) Effect of commercial starter cultures on physicochemical characteristics, microbial counts and free fatty acid composition of dry-cured foal sausage. Food Control 46:382–389
Luckow T, Delahunty C (2004) Which juice is ‘healthier’? A consumer study of probiotic non-dairy juice drinks. Food Qual Prefer 15:751–759
Mago R, Simkova H, Brown-Guedira G, Breen J, Jin Y, Singh R, Appels R, Lagudah ES, Ellis J, Dolezel J, Spielmeye W (2011) An accurate DNA marker assay for stem rust resistance gene Sr2 in wheat. Theor Appl Genet 122(4):735–744
Mauriello G, Casaburi A, Blaiotta G, Villani F (2004) Isolation and technological properties of coagulase negative staphylococci from fermented sausages of Southern Italy. Meat Sci 67(1):149–158
Núñez F, Lara MS, Peromingo B, Delgado J, Sánchez-Montero L, Andrade MJ (2014) Selection and evaluation of Debaryomyces hansenii isolates as potential bioprotective agents against toxigenic penicillia in dry-fermented sausages. Food Microbiol 46:114–120
Połka J, Rebecchi A, Pisacane V, Morelli L, Puglisi E (2015) Bacterial diversity in typical Italian salami at different ripening stages as revealed by high-throughput sequencing of 16S rRNA amplicons. Food Microbiol 46:342–356
Quigley L, Sullivan O, Beresford TP, Ross RP, Fitzgerald GF, Cotter PD (2012) High-throughput sequencing for detection of subpopulations of bacteria not previously associated with artisanal cheeses. Appl Environ Microbiol 78:5717–5723
Rebecchi A, Pisacane V, Miragoli F, Polka J, Falasconi I, Morelli L, Puglisi E (2015) High-throughput assessment of bacterial ecology in hog, cow and ovine casings used in sausages production. Int J Food Microbiol 212:49–59
Ruiz-Moyano S, Martın A, Benito MJ, Hernandez A, Casquete R, Cordoba MG (2011) Application of Lactobacillus fermentum HL57 and Pediococcus acidilactici SP979 as potential probiotics in the manufacture of traditional Iberian dry-fermented sausages. Food Microbiol 28:839–847
Rundell EA, Banta LM, Ward DV, Watts CD, Birren B, Esteban DJ (2014) 16S rRNA gene survey of microbial communities in winogradsky columns. PLoS ONE 9(8):104–134
R Development Core Team (2011) R: a language and environment for statistical computing, reference index version 2.14.1. R Foundation for Statistical Computing
Ubeda C, Ying T, Jenq RR, Equinda MJ, Son T (2010) Vancomycin-resistant Enterococcus domination of intestinal microbiota is enabled by antibiotic treatment in mice and precedes bloodstream invasion in humans. J Clin Investig 120(12):4332–4341
Wang XH, Ren HY, Liu DY, Zhu WY, Wang W (2013) Effects of inoculating Lactobacillus sakei starter cultures on the microbiological quality and nitrite depletion of Chinese fermented sausages. Food Control 32(2):591–596
Wang XH, Ren HY, Wang W, Xie ZJ (2016) Effects of a starter culture on histamine reduction, nitrite depletion and oxidative stability of fermented sausages. J Food Saf 36(2):195–202
Wang XH, Zhan YL, Ren HY, Zhan Y (2018) Comparison of bacterial diversity profiles and microbial safety assessment of salami, Chinese dry-cured sausage and Chinese smoked-cured sausage by high-throughput sequencing. LWT Food Sin Technol 90:108–115
Yang L, Yang HL, Tu ZC, Wang XL (2016) High-throughput sequencing of microbial community diversity and dynamics during douchi fermentation. PLoS ONE 11(12):e0168
Zhang XM, Yang Y, Liu SL, Qin W, Li C, Pu B (2009) Physico -chemical and microbial properties of Sichuan sausage during the ripening process. Food Ferment Ind 35(11):184–185
Acknowledgements
The research was supported by the China Postdoctoral Science Foundation (2018M631103) and National Natural Science Foundation of China (31772093).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Wang, X., Wang, S. & Zhao, H. Unraveling microbial community diversity and succession of Chinese Sichuan sausages during spontaneous fermentation by high-throughput sequencing. J Food Sci Technol 56, 3254–3263 (2019). https://doi.org/10.1007/s13197-019-03781-y
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13197-019-03781-y