
Abstract
Background
Sporadic amyotrophic lateral sclerosis (sALS) is a severe neurodegenerative disease characterized by continuous diminution of motor neurons in the brain and spinal cord. Earlier studies indicated that the DPP6 gene variant has a role in the development of sALS. This meta-analysis was designed to uncover the role of rs10260404 polymorphism of the DPP6 gene and its association with sALS.
Methods
All case–control articles published prior to October 2022 on the association between DPP6 (rs10260404) polymorphism and sALS risk were systematically extracted from different databases which include PubMed, PubMed Central, and Google Scholar. Overall odds ratios (ORs) and “95% confidence intervals (CIs)” were summarized for various genetic models. Subgroup and heterogeneity assessments were performed. Egger’s and “Begg’s tests were applied to evaluate publication bias. Trial sequential analysis (TSA) and false-positive report probability (FPRP) were performed.
Results
Nine case–control studies containing 4202 sALS cases and 4444 healthy controls were included in the meta-analysis. A significant association of the DPP6 (rs10260404) variant with an increased sALS risk in overall pooled subjects under allelic model [C allele vs. T allele, OR = 1.149, 95% CI (1.010–1.307), p-value = 0.035], dominant model [CC + CT vs. TT, OR = 1.165, 95% CI (1.067–1.273), p-value = 0.001], and homozygote comparison [CC vs. TT, OR = 1.421, 95% CI (1.003–2.011), p-value = 0.048] were observed. Moreover, in subgroup analysis by nationality, remarkable associations were detected in Dutch, Irish, American, and Swedish under allelic, dominant, and homozygote models. Additionally, stratification analysis by ethnicity exhibited an association with sALS risk among Caucasians and Americans under different genetic models. Interestingly, none of the models found any significant association with Asians.
Conclusion
The present meta-analysis indicates that DPP6 (rs10260404) polymorphism could be a candidate risk factor for sALS predisposition.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Amyotrophic lateral sclerosis (ALS) is a severe disabling and lethal disorder characterized by progressive death of motor neurons in the spinal cord, brainstem, and cerebral cortex. The loss of motor neurons narrows the central nervous system (CNS) ability to control voluntary muscle movements, which leads to muscle decimating and eventually death due to respiratory failure [1]. The peak time of ALS onset lies between age 50 and 75 [2] and most ALS patients survive about 3–5 years after disease onset [3]. ALS can be descended genetically from ancestors as an autosomal dominant, autosomal recessive, or X-linked manner [4]. Approximately 5–10% of cases are thought as familial ALS (fALS), whereas the remaining cases seem to be sporadic ALS (sALS) with no family history of ALS [5]. The cause of sporadic ALS (sALS) is primarily unknown, although familial and epidemiological statistics reveal that genetic components may promote its pathogenesis [6]. As of now, several modifier loci and related genes have been involved and a number of polymorphic variants have been suggested as risk factors for developing sALS [7]. Despite this, no particular gene has been clearly shown to cause sALS, as endeavors to determine genetic variants associated with sporadic ALS utilizing candidate gene approaches have often generated dissatisfying results [8]. The pathogenesis of sporadic ALS remains an ambiguity [9].
Environmental and genetic ingredients consider as the acknowledged pathogens of sALS. Infections through viruses and bacteria are appraised as the potential environmental factors for the development of sALS [10,11,12]. Other environmental factors such as organophosphate, organochlorine [13, 14], heavy metal exposure [15], intense physical activity [16], smoking, electromagnetic fields, electric shocks, cyanotoxins, and military service [17] may also produce significant impact for the pathogenesis of sALS. However, none of the recognized environmental risk elements has been conclusively certified, and no definite conclusions have been worked out until now [18]. If environmental factors are precisely a conducive risk factor in sALS occurrence, the genetic predisposition would be expected to amplify the possibility of sALS development because of exposure to environmental agents [19]. Therefore, genetic factors have drawn considerable attention in the investigation of sALS pathogenesis since the revelation of dipeptidyl-peptidase 6 (DPP6) mutations in sALS. During the last decade, the advancement of molecular genetic technologies has rapidly expanded our knowledge relating to the genetic pathogenesis of sALS. The occurrence of fALS has been ascribed to mutations in at least 24 independent genes. Specific mutations responsible for fALS generation have been detected also in patients with sALS [20]. Thus, sALS has been thought a complicated gene-related disease. However, recently it has been determined that the rs10260404 polymorphism in the DPP6 gene is strongly merged with the susceptibility to sALS among diverse populations of European origin and in a group of American patients [21]. The rs10260404 is located in the centre of intron 3, at position 154,513,713 of the DPP6 gene on chromosome 7 encoding the dipeptidyl-peptidase 6 protein. The rs10260404 polymorphism in the DPP6 gene was reported to be a T > C variation that plays a crucial function in sALS susceptibility and progression [3]. In particular, the CC genotype and the C allele were excessive representation in patients compared to healthy participants and identified with an increased possibility of sALS in recessive association and allelic tests [5]. This observation was also inconsistent [3], remained inconclusive, and varied across studies. Therefore, a meta-analysis of all appropriate studies for the rs10260404 polymorphism of the DPP6 gene and its association with the sALS risk was done to clarify a more rigorous assessment of this association and to scrutinize the root of heterogeneity and any hypothetical bias within these reports.
Materials and methods
In silico data analysis
Analysis of gene sequence of the DPP6 gene, its promoter, transcript variants because of alternative splicing, and synonyms was performed by Ensembl (www.ensembl.org), NCBI genome database (https://www.ncbi.nlm.nih.gov/genome/), Eukaryotic promoter database (https://epd.epfl.ch/cgi-bin/), and Genecards (www.genecards.org), respectively. UniProt/SwissProt (www.uniprot.org) and Protter (http://wlab.ethz.ch/protter/start/) bio tools were used to retrieve predicted secondary structure of DPP6 protein, conserved domains, and essential functional motifs, whereas compartment software (http://compartments.jensenlab.org/) was utilized to obtain subcellular localization of gene and protein isoforms. String database version 11.5 (https://string-db.org/) was used to determine functional annotation of DPP6 protein along with functional protein–protein association networks with gene ontology of DPP6. Additionally, several other programs such as Ensembl.org, SNPedia, dbSNP, and GWAS catalogue were used to analyze genomic variance. Missense coding mutations as well as their distribution and molecular modifications were also analyzed.
Study search and selection
This study was prepared to follow the guidelines of “Meta-analysis of Observational Studies in Epidemiology (MOOSE)” [22]. The study results were reported in accordance with “Preferred reporting items for systematic reviews and meta-analyses protocols (PRISMA-P)” [23]. An independent duplicated systematic exploration was conducted by dual researchers. The PubMed, PubMed Central, and Google Scholar databases were explored to retrieve the articles related to the genetic polymorphisms/variants of DPP6 (rs10260404) with an increased probability of sALS prior to October 2022. The search procedure was accomplished by pursuing a combination of the following terms and Medical Subjects Headings (MeSHs) including (1) DPP6, (2) GWAS, (3) polymorphisms, (4) genetic variant, (5) mutation or SNPs, (6) rs10260404, (7) case–control study, and (8) sALS. No limitations for publication time, language, territory, sample size, and ethnicity were confirmed to curb the impact of publication bias. Manual exploration for the reference records of all the published papers and assessments was executed to identify and consider other related articles.
Eligibility criteria
Titles and abstracts of all the published articles were searched independently by two authors. Impertinent and incompatible articles were omitted primarily. The assembled publications were selected based on the following explicit criteria: (1) “case–control” reports, (2) the study population was defined accurately, (3) the studies were on human beings, (4) the studies evaluated the relation of DPP6 (rs10260404) variant with sALS risk, (5) the articles were a genome-wide association study (GWAS), (6) complete information of allele frequency, (7) efficient data for “odds ratios” and 95% confidence intervals (ORs, 95% CIs)” estimation, and (9) the year of the study conducted was specified. However, the articles were eliminated (1) when the studies did not harmonize with the “case–control” presentation, (2) with identical reports from previous studies, (3) when the publications were “reviews, editorials, abstracts, meta-analysis, conference meetings, case report, and non-human researches”, and (4) with insufficient genotype data.
Data extraction
A structured data compilation form was executed to characterize retrieved publications. The extracted data were organized from the selected text articles, where each study includes (1) the name of the first author, (2) the year of the published article, (3) the nationality, (4) ethnic origin, (5) the population size of the sALS cases and controls for the investigated variant, (6) the method of the genotyping, (7) the source of the control (“Hospital-based” or “Population-based”), (8) the genotypes and alleles frequencies for the identified SNP among sALS cases, and the healthy volunteers, and (9) p-value of the Hardy–Weinberg equilibrium (HWE) within study controls, and also (10) the quality score.
Quality score assessment
The methodological quality of retrieved articles was evaluated independently by dual investigators utilizing a set of established criteria based on the scale that was extracted and amended from a prior meta-analysis of molecular association studies [24,25,26]. These revised sets encompassed the representativeness of sALS, reliability of controls, ascertainment of sALS and controls, ALS Functional Rating Scale (ALSFRS) examination, matching of the case–control, quality control of the method of genotyping, the assessment of genotyping, specimens applied for confirming genotypes, HWE in controls, total population size, and association analysis. The quality scores varied from 0 (lowest) to 14 (highest), and the studies secured scores < 10 were categorized as “low quality,” while those with scores ≥ 10 were classified as “high quality” (Supplementary data, Table S1).
Trial sequential analysis (TSA)
TSA software (version 0.9.5.10 beta) [27] was applied to determine the statistical accuracy of the meta-analysis by integrating cumulative specimens of all the published studies, and to analyze the unexpected miscalculations and improve the weight of conclusions [28, 29]. Power fixed at 5% and 80% and two-sided tests with “type I error (α)” were employed [27, 30]. A significant level of accuracy is achieved and no additional trials are fundamental, if the “cumulative Z-curve” cut across the TSA supervising boundaries. Conversely, if the “Z- curve” fails to adjoin with the boundary edges, the evaluated sample size has not gained the expected threshold to draw satisfactory outcomes and additional investigations are required.
Statistical analysis
Initially, genotype, allele, and allele frequencies were determined and recorded from selected publications of different ethnic populations. HWE test in cases and controls population was calculated using the online “Association, Odds Ratios, and Relative Risks (AssociatORRR)” software (http://www.genecalcs.weebly.com/associatorrr.html) and validated through the chi-square experiment with p-value < 0.05 was assessed to be a disequilibrium condition [31]. After that, the DPP6 (rs10260404) polymorphism impact on sALS risk was evaluated by “a logistic regression approach.” The steps such as crude odds ratios (ORs) and corresponding 95% confidence intervals (CIs) were calculated to assess the association of DPP6 (rs10260404) with the risk of sALS. Pooled ORs were employed by utilizing a combination of various genetic models encompassing allelic model, recessive model, dominant comparison, homozygote comparison, and heterozygote comparison. Moreover, pooled OR significance was evaluated by the “Z-test”, and p < 0.05 was appraised as statistically effective. Heterogeneity among different publications was estimated using the “chi-square-based Q-statistic” [32] and quantified using the I2 index [33]. p-value ≥ 0.10 for the “Q-test” and/or I2 index < 50% were identified as no appreciable heterogeneity, thus “fixed-effects model” was utilized to determine the pooled odds ratios (ORs) of each article [34]; if not, the “random-effects model” was taken in consideration to calculate within-study sampling inaccuracies and between-study variances [35]. Subgroup analyses were carried out by nationality, ethnicity, and source of controls. Moreover, “Begg’s funnel plot” and “Egger’s linear regression” assessments were executed to evaluate quantitative evidence of publication bias [36]. Finally, false-positive report probability (FPRP) was studied to reveal whether any associations published earlier were false positives. All tests were conducted using “Comprehensive Meta-Analysis (CMA)” version 3.0 software [37].
Results
In silico data analysis
Dipeptidyl-peptidase 6 protein is encoded by the DPP6 gene (ENSG00000130226), which is positioned on chromosome 7 long arm (7q36.2) (Supplementary data, Fig. S1), comprising by 26 exons (Fig. 1D), and spanning 1,146,153 bases long on the plus strand (genomic site: chromosome 7: 153,748,133–154,894,285, according to GRCh38/hg38). The gene has multiple synonyms, including DPPX, DPL1, dipeptidyl aminopeptidase-like protein 6, DPPVI, DPP6p, hDPP6, E9PDL2, E9PF59, MRD33, and VF2. The gene holds 23 transcripts, but only 9 of them can code for protein (Supplementary data, Table S2). 4826 base pairs containing transcript (ID: ENST00000377770.8) of the DPP6 gene has four promoters and position of those promoters are 4282 (P1), 3889 (P2), 3137 (P3), and 408 (P4). Fisher’s linear discriminant (LDF) value of P1 is + 4.926. TATA box position, score, and nucleotide sequence of TATA box are 4250, + 8.954, and TATATAAA, respectively. Thus, LDF value, TATA box position, score, and sequence of TATA box of P2 are + 2.154, 3844, + 4.012, and CATTAAAA. Likewise, LDF score of P3 is + 10.776 that corresponds to more reliable promoter. Similarly, P4 contains LDF score (+ 4.639), TATA box (381), box score (+ 6.000), and nucleotide sequence of TATA box (TATTAAAA). The rs10260404 T > C SNP site with three-frame translate of the DPP6 gene is shown in Fig. 1C. The dipeptidyl-peptidase 6 protein (UniProtKB: P42658) is composed of 865 amino acids and has a molecular weight of 97.588 kilodaltons (kDa). It contains 2 isoforms that are produced by alternative splicing and located mostly in the plasma membrane, membrane, and voltage-gated potassium channel complex with greater confidence (Supplementary data, Table S3 and Fig. S2). The protein functions as a peptidase S9B family domain and dipeptidyl-peptidase IV (DPPIV) N-terminal region at position 195–561 (Fig. 1D). It also contains domains for the prolyl oligopeptidase family (amino acids 642–848) (Fig. 1D). It acts as a locale of membrane-bound protein estimated to be outside the membrane, in the cytoplasm (amino acids 1–195), and the extracellular region (amino acids 118–865). The top gene ontology (GO) annotation for biological processes activates cell surface expression and regulates the activity and gating characteristics of the potassium channel KCND2. The functional annotation of the DPP6 protein and mRNA expression for DPP6 gene in normal human tissues are illustrated in Fig. 2 A and B.

A Hundreds of studies related to the single-nucleotide variants of the DPP6 gene were published in different journals over the last two decades. B Variant inspection indicated amino acid position at 153 of the DPP6 protein (DPP6:D153int) and genomic position at chromosome 7: 154,475,037–154,540,533 of the DPP6 gene are the most cited loci. Approximately 82 published articles are highly involved between DPP6 (rs10260404) polymorphism and sALS risk (source: https://mastermind.genomenon.com/). C DPP6 (rs10260404) variant position at base level (chromosome 7: 154,513,713-T-C) with three-frame translate reveals how amino acids sequence of the DPP6 protein are altered due to the polymorphism. D The tiny green balls of the DPP6 protein indicate missense mutations, yellow balls reflect splice mutations, and light-color black balls represent nonsense, nonstop, frameshift deletion, and frameshift insertion mutations

A Functional annotation of DPP6 protein and its network statistics are- number of nodes: 11, number of edges: 29, average node degree: 5.27, average local clustering coefficient: 0.853, expected number of edges: 11, PPI enrichment p-value: 2.4e-06. B DPP6 gene expression in normal human tissues
Characteristics of qualified studies
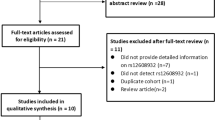
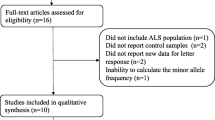
We identified overall 401 appropriate records (PubMed = 86, Google Scholar & Web of Science = 315) based on search strategy and selection specification. Besides, the manual exploration for the quoted references within specified studies recognized twenty-one further records through additional sources. After eliminating 107 simulated records, a total of 315 prospective studies were screened based on their titles and abstracts. After titles and abstracts reviewing, 142 pertinent articles were selected for supplementary full-text investigation. Thereafter, we eliminated 127 records for having no case–control representation, excluded 5 articles for overlapping data and there was no meta-analysis record for exclusion. Finally, a total of 9 publications [1, 3, 5, 6, 21, 38,39,40,41] satisfied the inclusion principle that was applied in the present work for the evaluation of the DPP6 (rs10260404) polymorphism with increasing sALs risk (Fig. 3).

Flowchart of articles search and screen for DPP6 (rs10260404) polymorphism
Scrutiny on the inclusion studies of the DPP6 (rs10260404) polymorphism
A total of nine eligible case–control publications that explained DPP6 (rs10260404) association with sALS susceptibility were scrutinized meticulously to enlist potential data. The data from individual publication was organized as a distinct study. However, two of these selected studies incorporated data from seven different sets [21, 41] and these diverse sets were scrutinized independently. Therefore, the selected 9 publications encompassed 14 comparisons of case–control articles that involve 4202 sALS patients and 4444 controls (Table 1). Among these significant studies, four experiments were conducted on Asians [3, 38,39,40], nine studies were established on Caucasians [1, 5, 6, 21, 41], and one study was examined in American [21]. All evaluated published articles were carried out under certified genotyping procedures involving “Sequenom Massarray, PCR-HRMA, SeqMan, Sequenom iPLEX Assay, BigDye Terminator protocol, TaqMan, Infinium II HumanHap, TaqMan allelic discrimination Assay, Infinium HumanHap, and Allele specific PCR”. The sources of control of ten studies were found as population-based while four articles were hospital based. This meta-analysis investigation exhibited that all articles were accorded with HWE among healthy volunteers, and no studies attained disequilibrium with HWE. Moreover, nine articles obtained a high-quality score whereas, six articles secured a low-quality score.
Meta-analysis of the association between DPP6 (rs10260404) polymorphism and sALS risk
The association of the DPP6 (rs10260404) polymorphism with increasing sALS risk was recapitulated in Table 2. Initially, the relation between DPP6 (rs10260404) variant and sALS risk was examined within the overall population, and then, the results were categorized according to nationality, race/ethnicity, and source of controls. Overall, the pooled ORs for all the subjects suggested that the DPP6 (rs10260404) polymorphism was remarkably related to increasing sALS risk for the allelic model [C allele vs. T allele, OR = 1.149, 95% CI (1.010–1.307), p-value = 0.035] (Fig. 4A); dominant model [CC + CT vs. TT, OR = 1.165, 95% CI (1.067–1.273), p-value = 0.001] (Fig. 4B); and homozygote model [CC vs. TT, OR = 1.421, 95% CI (1.003–2.011), p-value = 0.048] (Fig. 4D). In contrast, there was no substantiation of the DPP6 (rs10260404) association with increasing sALS risk in all pooled subjects under other models, including the recessive model [CC vs. CT + TT, OR = 1.312, 95% CI (0.929–1.852), p-value = 0.124] (Fig. 4C); and heterozygote comparison [CC vs. CT, OR = 1.238, 95% CI (0.879–1.744), p-value = 0.221] (Fig. 4E).

Forest plots of rs10260404 in DPP6 gene and risk of sALS under different genetic models utilizing random effect model
Subgroup analyses performed by nationality
The current meta-analysis exhibited a clear association of the DPP6 (rs10260404) polymorphism and sALS risk among the Dutch population under four genetic models including the allelic model [C allele vs. T allele, OR = 1.290, 95% CI (1.115–1.492), p-value = 0.001]; recessive model [CC vs. CT + TT, OR = 1.438, 95% CI (1.091–1.895), p-value = 0.010]; dominant model [CC + CT vs. TT, OR = 1.376, 95% CI (1.114–1.699), p-value = 0.003]; homozygote comparison [CC vs. TT, OR = 1.664, 95% CI (1.228–2.257), p-value = 0.001]. In addition, the subgroup investigations of the Irish population indicated that the DPP6 (rs10260404) variant was strongly associated with increasing sALS risk in allelic model [C allele vs. T allele, OR = 1.336, 95% CI (1.047–1.704), p-value = 0.020]; dominant model [CC + CT vs. TT, OR = 1.419, 95% CI (1.008–1.998), p-value = 0.045]; homozygote comparison [CC vs. TT, OR = 1.801, 95% CI (1.067–3.040), p-value = 0.028]. Likewise, the outcomes of the subgroup examinations of the American population signified that the DPP6 (rs10260404) polymorphism was highly associated with increasing sALS risk in the allelic model [C allele vs. T allele, OR = 1.411, 95% CI (1.104–1.803), p-value = 0.006]; recessive model [CC vs. CT + TT, OR = 1.671, 95% CI (1.029–2.714), p-value = 0.038]; dominant model [CC + CT vs. TT, OR = 1.518, 95% CI (1.073–2.146), p-value = 0.018]; and homozygote comparison [CC vs. TT, OR = 2.006, 95% CI (1.186–3.392), p-value = 0.009]. Moreover, a significant effect of DPP6 (rs10260404) with an increased sALS risk was detected among the Swedish under allelic model [C allele vs. T allele, OR = 1.293, 95% CI (1.068–1.566), p-value = 0.008]; dominant model [CC + CT vs. TT, OR = 1.371, 95% CI (1.049–1.790), p-value = 0.021]; homozygote comparison [CC vs. TT, OR = 1.672, 95% CI (1.108–2.524), p-value = 0.014] (Table 2).
On the other hand, the analyses data for the DPP6 (rs10260404) polymorphism suggested that no remarkable effect was determined among Chinese, Italian, Belgian, and Polish under different genetic models such as, for Chinese, allelic model [C allele vs. T allele, OR = 0.906, 95% CI (0.664–1.236), p-value = 0.533]; recessive model [CC vs. CT + TT, OR = 1.166, 95% CI (0.431–3.155), p-value = 0.762]; dominant model [CC + CT vs. TT, OR = 0.825, 95% CI (0.651–1.047), p-value = 0.114]; homozygote comparison [CC vs. TT, OR = 1.108, 95% CI (0.410–2.996), p-value = 0.840]; and heterozygote comparison [CC vs. CT, OR = 1.307, 95% CI (0.478–3.576), p-value = 0.602]; for Italian, allelic model [C allele vs. T allele, OR = 1.144, 95% CI (0.848–1.543), p-value = 0.379]; recessive model [CC vs. CT + TT, OR = 1.296, 95% CI (0.585–2.872), p-value = 0.523]; dominant model [CC + CT vs. TT, OR = 1.112, 95% CI (0.944–1.310), p-value = 0.202]; homozygote comparison [CC vs. TT, OR = 1.364, 95% CI (0.614–3.032), p-value = 0.446]; and heterozygote comparison [CC vs. CT, OR = 1.228, 95% CI (0.561–2.691), p-value = 0.607]; for Belgian, allelic model [C allele vs. T allele, OR = 1.225, 95% CI (0.985–1.523), p-value = 0.069]; recessive model [CC vs. CT + TT, OR = 1.329, 95% CI (0.866–2.039), p-value = 0.193]; dominant model [CC + CT vs. TT, OR = 1.290, 95% CI (0.948–1.755), p-value = 0.105]; homozygote comparison [CC vs. TT, OR = 1.491, 95% CI (0.937–2.373), p-value = 0.092]; and heterozygote comparison [CC vs. CT, OR = 1.207, 95% CI (0.767–1.898), p-value = 0.416]; and for Polish, allelic model [C allele vs. T allele, OR = 0.841, 95% CI (0.656–1.080), p-value = 0.174]; recessive model [CC vs. CT + TT, OR = 0.775, 95% CI (0.465–1.292), p-value = 0.328]; dominant model [CC + CT vs. TT, OR = 0.810, 95% CI (0.575–1.141), p-value = 0.228]; homozygote comparison [CC vs. TT, OR = 0.707, 95% CI (0.410–1.220), p-value = 0.213]; and heterozygote comparison [CC vs. CT, OR = 0.840, 95% CI (0.490–1.441), p-value = 0.527] (Table 2).
Subgroup analyses performed by race/ethnicity
The scrutinized data for the DPP6 (rs10260404) polymorphism showed that there was no significant impact on the Asian population under different genetic models. Interestingly, the subgroup analyses among Caucasians demonstrated that the DPP6 (rs10260404) polymorphism was appreciably associated with increasing sALS risk in the allelic model [C allele vs. T allele, OR = 1.185, 95% CI (1.044–1.344), p-value = 0.009]; dominant model [CC + CT vs. TT, OR = 1.224, 95% CI (1.082–1.384), p-value = 0.010]; homozygote comparison [CC vs. TT, OR = 1.413, 95% CI (1.051–1.898), p-value = 0.022]. Moreover, the subgroup analysis results among Americans suggested that the DPP6 (rs10260404) polymorphism had significant interference with increasing sALS risk in the allelic model [C allele vs. T allele, OR = 1.411, 95% CI (1.104–1.803), p-value = 0.006]; recessive model [CC vs. CT + TT, OR = 1.671, 95% CI (1.029–2.714), p-value = 0.038]; dominant model [CC + CT vs. TT, OR = 1.518, 95% CI (1.073–2.146), p-value = 0.018]; and homozygote comparison [CC vs. TT, OR = 2.006, 95% CI (1.186–3.392), p-value = 0.009] (Table 2).
Subgroup analyses performed based on sources of controls
The investigated data revealed an increased sALS risk among hospital-based studies under four genetic models. For hospital-based studies, the allelic model [C allele vs. T allele, OR = 1.199, 95% CI (1.059–1.357), p-value = 0.004]; recessive model [CC vs CT + TT, OR = 1.405, 95% CI (1.090–1.811), p-value = 0.009]; dominant model [CC + CT vs. TT, OR = 1.205, 95% CI (1.018–1.427), p-value = 0.030]; homozygote comparison [CC vs. TT, OR = 1.600, 95% CI (1.214–2.108), p-value = 0.001]. On the contrary, three models among population-based studies showed an increased sALS risk including the allelic model [C allele vs. T allele, OR = 1.156, 95% CI (1.014–1.318), p-value = 0.030]; dominant model [CC + CT vs. TT, OR = 1.165, 95% CI (1.021–1.331), p-value = 0.024]; homozygote comparison [CC vs. TT, OR = 1.436, 95% CI (1.039–1.985), p-value = 0.029] (Table 2).
Heterogeneity analysis
Between studies, there was significant heterogeneity noticed in terms of DPP6 (rs10260404) polymorphism for the overall sALS allelic model [C allele vs. T allele, Q-test = 39.009, p-value = 0.000, I2 = 66.674%]; recessive model [CC vs. CT + TT, Q-test = 34.798, p-value = 0.001, I2 = 62.641%]; dominant model [CC + CT vs. TT, Q-test = 26.351, p-value = 0.015, I2 = 50.666%]; homozygote comparison [CC vs. TT, Q-test = 36.616, p-value = 0.000, I2 = 64.497%]; and heterozygote comparison [CC vs. CT, Q-test = 27.392, p-value = 0.011, I2 = 52.542%]. To identify the root of heterogeneity subgroup analyses by nationality, ethnicity and sources of controls were executed. In the subgroup investigations, heterogeneity was appreciably reduced in terms of nationality. The findings also suggested that the studies in Chinese, Italian, Asian ethnicity Caucasian ethnicity, population-based sources of controls, and hospital-based sources of controls were the main causes of heterogeneity (Table 2).
Publication bias
The probability of the presence of publication bias within the studies was examined by Begg’s funnel plots. The configurations of these funnel plots exhibited a corroboration of uniformity and represented the lack of publication bias within overall pooled subjects under different genetics models (Fig. 5). Moreover, Egger’s linear regression test estimated no evidence of appreciable publication bias in the allelic model [C allele vs. T allele, p-value = 0.683], recessive model [CC vs. CT + TT, p-value = 0.329], dominant model [CC + CT vs. TT, p-value = 0.918], homozygote comparison [CC vs. TT, p-value = 0.462], and heterozygote comparison [CC vs. CT, p-value = 0.200] (Table 2). Additionally, no evidence of publication bias was detected in the case of subgroup analyses of nationality, ethnicity, and sources of controls (Table 2). However, a clear publication bias was observed among population-based studies for the DPP6 (rs10260404) polymorphism under the heterozygote model [Egger’s regression: p = 0.073] (Table 2).

Funnel plots by Log odds ratio of the meta-analysis on the association between DPP6 (rs10260404) polymorphism and sALS risk for the overall population. The plots under different genetic models show no evidence of significant publication bias
Trial sequential analysis (TSA)
TSA test showed that the available specimens was 17,192 subjects under an allelic model and 8646 subjects under a dominant model for the DPP6 (rs10260404) polymorphism (Fig. 6). TSA test also exhibited that the “Z-curve” crossed the trial sequential supervising circumference before reaching the required sample size, suggesting that the cumulative outcome was satisfactory and no further tests were required to certify the results.

Trial sequential analysis for DPP6 (rs10260404) polymorphism under the allelic model and dominant model
False-positive report probability
We executed FPRP to estimate whether associations described earlier were false positives. We specified FPRP at 0.2 to define biological significance and a prior probability of 0.01 to identify the remarkable OR [42]. The odds ratio fixed at 1.5 to calculate statistical power and FPRP [43]. Fixation of OR at 1.5 is considered as a rational value to detect important biological effects [44, 45]. The association containing the FPRP value less than 0.2 was considered as significant [46]. According to the above discussion, the DPP6 (rs10260404) polymorphism remarkably increased the overall risk of sALS. Also, the DPP6 (rs10260404) polymorphism notably increased the risk of sALS in Caucasian and American patients. Additionally, the rs10260404 variant significantly increased sALS risk among Dutch, Irish, and Swedish patients (Table 3).
Discussion
We conducted a statistical meta-analysis study to evaluate the relation between DPP6 (rs10260404) polymorphism and the risk of sALS more precisely. A total of 14 case–control comparisons for DPP6 (rs10260404) polymorphism (4202 sALS patients and 4444 healthy controls) were analyzed in this meta-analysis study. Surprisingly, a notable relation of DPP6 (rs10260404) polymorphism with the propensity to sALS in overall pooled subjects was noticed among the allelic model (C allele vs. T allele), dominant model (CC + CT vs. TT), and homozygote comparison (CC vs. TT), which signifies that the C allele has significant association with the sALS. Moreover, stratification analysis revealed a sign of association of this variant with increasing sALS risk among Dutch, Irish, American, and Swedish under allelic, dominant, and homozygote models, which reveals that the C allele is expressed significantly among Dutch, Irish, American, and Swedish. Stratification analysis also indicated a remarkable relation of this variant with an increased sALS risk among Dutch and American under the recessive model (CC vs. CT + TT) although C allele is recessive. Additionally, stratification examination explicated a significant association with an increased sALS risk for DPP6 (rs10260404) polymorphism among the ethnicities of Caucasian and American under the allelic model, dominant model, and homozygote comparison, which reveals that the risk C allele expression is more prominent in Caucasians and Americans. Furthermore, stratification analysis in terms of sources of controls elucidated an association of DPP6 (rs10260404) polymorphism with susceptibility to sALS among population-based and hospital-based studies under allelic, dominant, and homozygote models.
One study reported a positive association between DPP6 (rs10260404) and sALS in the Italian population [5], while another study showed inconclusive findings in Italians [1] but the current updated meta-analysis study demonstrated no association of DPP6 (rs10260404) with sALS in Italian population under different genetic models, which illustrates that the C allele is not significantly expressed in Italians to generate sALS. Zhang et al. reported that the rs10260404 in the DPP6 gene was significantly associated with sALS in the Han Ancestry from Mainland China (HACM) [3] which was totally contraposition with other studies among Chinese [38,39,40]. However, our meta-analysis findings suggested no relation of DPP6 (rs10260404) variant with sporadic amyotrophic lateral sclerosis in the Chinese population under allelic, recessive, dominant, homozygote, and heterozygote models, which indicates that the risk C allele is not highly expressed in Chinese. Similarly, consistent with our outcome, one study exhibited that Polish populations were less susceptible to sALS for the DPP6 (rs10260404) polymorphism due to the overrepresentation of the non-risk T allele.
van Es et al. identified that variation in the DPP6 (rs10260404) is highly related to sALS susceptibility in Caucasian populations [21]. Interestingly, our current meta-analysis study found DPP6 (rs10260404) polymorphism with an increased sALS risk in European descent. Perhaps the most unanticipated outcome from our updated meta-analysis is the lack of association between the DPP6 (rs10260404) polymorphism and sALS risk in the Asian descent.
The aforementioned illustration represented an important suggestion that the DPP6 gene is a potential probable factor for the genesis of sALS. Now expression analysis is indispensable to explore the causation of the rs10260404 SNP in the DPP6 gene for the development of sALS. Network analysis or Mendelian randomization procedure was used by some scientists to identify single or multiple disorders regulating genes or SNPs [47,48,49,50,51,52,53]. The identification of direct or indirect effect of the SNPs can be a potential biological marker to detect the disease-causing genes [54]. The cis-acting factors may play a significant role to develop the disease because SNPs alter the gene’s function by occurring within a gene or in a regulatory region. The trans-acting factors that occur in the remote regions of the disease-causing genes are thought as non-causal risk factors for the generation of the disorders [55]. The SNPs remain unexpressed if it occurs within the noncoding areas or may alter the encoded amino acids if it occurs within the coding areas. The SNPs may develop genetic disorders by controlling promoter or enhancer functions, mRNA firmness, and subcellular locations of messenger RNAs and/or proteins. However, a functional disease prognostic model may be established by utilizing an appropriate machine learning approach if SNP records are available for sALS cases and controls. For instance, the SNP-based genetic disorder prognostic models were established by some earlier studies [56, 57].
The current meta-analysis experienced some constraints. First, this study assessed unadjusted estimation for the association of the DPP6 (rs10260404) variant with an increased sALS risk. The unadjusted estimation had been performed due to the lack of information relating to the adjusted estimation described by the published studies. Second, possible covariates bias was revealed within the qualified articles, including inadequate data relating to age, gender, alcohol consumption and smoking, ethnic history, family background, histopathological data, and dietary routines. Moreover, other subjects might associate between the DPP6 (rs10260404) polymorphism and the sensitivity to sALS involving gene–gene and gene-environmental communications. Finally, diverseness and publication bias were noticed in this meta-analysis study.
Conclusion
Results from this meta-analysis study illustrated precise evidence of association for rs10260404 polymorphism in DPP6 gene with increasing sALS risk based on 14 case–control publications. The subgroup analyses by nationality exhibited a clear evidence of association between DPP6 (rs10260404) polymorphism and sALS risk among Dutch, Irish, American, and Swedish under different genetic models. Moreover, stratified analyses based on race showed a strong relation of DPP6 (rs10260404) variant with increasing sALS risk among Caucasians and Americans under allelic, dominant and homozygote models. Surprisingly, none of the comparisons demonstrated any particular association with Asians. In future, further work would be necessary to validate the findings and to disclose the etiopathogenesis of sALS.
Data availability
All data added table, figures, and supplementary file and supplementary tables. In this research work publicly available free mostly online and few offline software/tools were used. Necessary link, reference of the software/tools provided in the method section.
Abbreviations
- ALS:
-
Amyotrophic lateral sclerosis
- ALSFRS:
-
Amyotrophic lateral sclerosis functional rating scale
- CIs:
-
Confidence intervals
- CMA:
-
Comprehensive meta-analysis
- CNS:
-
Central nervous system
- dbSNP:
-
Data base for single-nucleotide polymorphisms
- DPP6:
-
Dipeptidyl-peptidase 6
- DPPX:
-
Dipeptidyl-peptidase like protein
- DPL1:
-
Sphingosine-1-phosphate lyase
- DPPVI:
-
Dipeptidyl-peptidase VI
- DPP6p:
-
Dipeptidyl-peptidase like protein 6
- hDPP6:
-
Human dipeptidyl-peptidase 6
- FPRP:
-
False-positive report probability
- fALS:
-
Familial amyotrophic lateral sclerosis
- GWAS:
-
Genome-wide association study
- HACM:
-
Han Ancestry from Mainland China
- HWE:
-
Hardy-Weinberg equilibrium
- KCND2:
-
Potassium voltage-gated channel subfamily D member 2
- LDF:
-
Fisher’s linear discriminant
- MeSHs:
-
Medical Subjects Headings
- MOOSE:
-
Meta-analysis of Observational Studies in Epidemiology
- mRNA:
-
Messenger ribonucleic acid
- NCBI:
-
National Centre for Biotechnology Information
- PCR-HRMA:
-
Polymerase chain reaction-high resolution melting analysis
- PCR:
-
Polymerase chain reaction
- PRISMA-P:
-
Preferred reporting items for systematic reviews and meta-analysis protocols
- sALS:
-
Sporadic amyotrophic lateral sclerosis
- SNPs:
-
Single-nucleotide polymorphisms
- ORs:
-
Odds ratios
References
Fogh I, D’Alfonso S, Gellera C, Ratti A, Cereda C, Penco S et al (2011) No association of DPP6 with amyotrophic lateral sclerosis in an Italian population. Neurobiol Aging 32:966–967. https://doi.org/10.1016/j.neurobiolaging.2009.05.014
Blauw HM, Al-Chalabi A, Andersen PM, van Vught PWJ, Diekstra FP, van Es MA et al (2010) A large genome scan for rare CNVs in amyotrophic lateral sclerosis. Hum Mol Genet 19:4091–4099. https://doi.org/10.1093/hmg/ddq323
Zhang J, Qiu W, Hu F, Zhang X, Deng Y, Nie H et al (2021) The rs2619566, rs10260404, and rs79609816 polymorphisms are associated with sporadic amyotrophic lateral sclerosis in individuals of Han Ancestry From Mainland China. Front Genet 12:679204. https://doi.org/10.3389/fgene.2021.679204
Krüger S, Battke F, Sprecher A, Munz M, Synofzik M, Schöls L, et al (2016) Rare variants in neurodegeneration associated genes revealed by targeted panel sequencing in a German ALS cohort. Front Mol Neurosci 9. https://doi.org/10.3389/fnmol.2016.00092
Del Bo R, Ghezzi S, Corti S, Santoro D, Prelle A, Mancuso M et al (2008) DPP6 gene variability confers increased risk of developing sporadic amyotrophic lateral sclerosis in Italian patients. J Neurol Neurosurg Psychiatry 79:1085. https://doi.org/10.1136/jnnp.2008.149146
Cronin S, Berger S, Ding J, Schymick JC, Washecka N, Hernandez DG et al (2008) A genome-wide association study of sporadic ALS in a homogenous Irish population. Hum Mol Genet 17:768–774. https://doi.org/10.1093/hmg/ddm361
Garber K (2008) Genetics. The elusive ALS genes. Science 319:20. https://doi.org/10.1126/science.319.5859.20
Schymick JC, Talbot K, Traynor BJ (2007) Genetics of sporadic amyotrophic lateral sclerosis. Hum Mol Genet 16:R233–R242. https://doi.org/10.1093/hmg/ddm215
Ludolph AC, Brettschneider J, Weishaupt JH (2012) Amyotrophic lateral sclerosis. Curr Opin Neurol 25:530–535. https://doi.org/10.1097/WCO.0b013e328356d328
Xue YC, Feuer R, Cashman N, Luo H (2018) Enteroviral infection: the forgotten link to amyotrophic lateral sclerosis? Front Mol Neurosci 11. https://doi.org/10.3389/fnmol.2018.00063
Yu B, Pamphlett R (2017) Environmental insults: critical triggers for amyotrophic lateral sclerosis. Transl Neurodegener 6:15. https://doi.org/10.1186/s40035-017-0087-3
Sher RB (2017) The interaction of genetics and environmental toxicants in amyotrophic lateral sclerosis: results from animal models. Neural Regen Res 12:902–905. https://doi.org/10.4103/1673-5374.208564
Riancho J, Bosque-Varela P, Perez-Pereda S, Povedano M, de Munaín AL, Santurtun A (2018) The increasing importance of environmental conditions in amyotrophic lateral sclerosis. Int J Biometeorol 62:1361–1374. https://doi.org/10.1007/s00484-018-1550-2
Su F-C, Goutman SA, Chernyak S, Mukherjee B, Callaghan BC, Batterman S et al (2016) Association of environmental toxins with amyotrophic lateral sclerosis. JAMA Neurol 73:803–811. https://doi.org/10.1001/jamaneurol.2016.0594
Garzillo EM, Miraglia N, Pedata P, Feola D, Sannolo N (2015) Lamberti M [Amyotrophic lateral sclerosis and exposure to metals and other occupational/environmental hazardous materials: state of the art]. G Ital Med Lav Ergon 37:8–19
Tsitkanou S, Della Gatta P, Foletta V, Russell A (2019) The role of exercise as a non-pharmacological therapeutic approach for amyotrophic lateral sclerosis: beneficial or detrimental? Front Neurol 10:783. https://doi.org/10.3389/fneur.2019.00783
Swash M, Eisen A (2020) Hypothesis: amyotrophic lateral sclerosis and environmental pollutants. Muscle Nerve 62:187–191. https://doi.org/10.1002/mus.26855
Longinetti E, Fang F (2019) Epidemiology of amyotrophic lateral sclerosis: an update of recent literature. Curr Opin Neurol 32
Nowicka N, Juranek J, Juranek JK, Wojtkiewicz J (2019) Risk factors and emerging therapies in amyotrophic lateral sclerosis. Int J Mol Sci 20 https://doi.org/10.3390/ijms20112616
Mathis S, Goizet C, Soulages A, Vallat J-M, Le MG (2019) Genetics of amyotrophic lateral sclerosis: a review. J Neurol Sci 399:217–226. https://doi.org/10.1016/j.jns.2019.02.030
van Es MA, van Vught PWJ, Blauw HM, Franke L, Saris CGJ, Van den Bosch L et al (2008) Genetic variation in DPP6 is associated with susceptibility to amyotrophic lateral sclerosis. Nat Genet 40:29–31. https://doi.org/10.1038/ng.2007.52
Stroup DF, Berlin JA, Morton SC, Olkin I, Williamson GD, Rennie D et al (2000) Meta-analysis of observational studies in epidemiology: a proposal for reporting. Meta-analysis Of Observational Studies in Epidemiology (MOOSE) group. JAMA 283:2008–12. https://doi.org/10.1001/jama.283.15.2008
Moher D, Shamseer L, Clarke M, Ghersi D, Liberati A, Petticrew M et al (2015) Preferred reporting items for systematic review and meta-analysis protocols (PRISMA-P) 2015 statement. Syst Rev 4:1. https://doi.org/10.1186/2046-4053-4-1
Elshazli RM, Toraih EA, Elgaml A, Kandil E, Fawzy MS (2020) Genetic polymorphisms of TP53 (rs1042522) and MDM2 (rs2279744) and colorectal cancer risk: an updated meta-analysis based on 59 case-control studies. Gene 734:144391. https://doi.org/10.1016/j.gene.2020.144391
Qin X, Peng Q, Tang W, Lao X, Chen Z, Lai H et al (2013) An updated meta-analysis on the association of MDM2 SNP309 polymorphism with colorectal cancer risk. PLoS ONE 8:e76031
Thakkinstian A, McEvoy M, Minelli C, Gibson P, Hancox B, Duffy D et al (2005) Systematic review and meta-analysis of the association between β2-adrenoceptor polymorphisms and asthma: a HuGE review. Am J Epidemiol 162:201–211. https://doi.org/10.1093/aje/kwi184
Lan KKG, DeMets DL (1983) Discrete sequential boundaries for clinical trials. Biometrika 70:659–663. https://doi.org/10.2307/2336502
Wetterslev J, Thorlund K, Brok J, Gluud C (2008) Trial sequential analysis may establish when firm evidence is reached in cumulative meta-analysis. J Clin Epidemiol 61:64–75. https://doi.org/10.1016/j.jclinepi.2007.03.013
Wang G, Zhang L, Lou S, Chen Y, Cao Y, Wang R et al (2016) Effect of dexmedetomidine in preventing postoperative side effects for laparoscopic surgery: a meta-analysis of randomized controlled trials and trial sequential analysis (PRISMA). Med (Baltimore) 95:e2927. https://doi.org/10.1097/MD.0000000000002927
Xie S, Shan X-F, Shang K, Xu H, He J, Cai Z-G (2014) Relevance of LIG4 gene polymorphisms with cancer susceptibility: evidence from a meta-analysis. Sci Rep 4:6630. https://doi.org/10.1038/srep06630
Rodriguez S, Gaunt TR, Day INM (2009) Hardy-Weinberg equilibrium testing of biological ascertainment for Mendelian randomization studies. Am J Epidemiol 169:505–514. https://doi.org/10.1093/aje/kwn359
Cochran WG (1954) Some methods for strengthening the common χ2 tests. Biometrics 10:417–451. https://doi.org/10.2307/3001616
Higgins JPT, Thompson SG, Deeks JJ, Altman DG (2003) Measuring inconsistency in meta-analyses. BMJ 327:557–560. https://doi.org/10.1136/bmj.327.7414.557
Mantel N, Haenszel W (1959) Statistical aspects of the analysis of data from retrospective studies of disease. JNCI J Natl Cancer Inst 22:719–748. https://doi.org/10.1093/jnci/22.4.719
DerSimonian R, Laird N (1986) Meta-analysis in clinical trials. Control Clin Trials 7:177–88. https://doi.org/10.1016/0197-2456(86)90046-2
Egger M, Smith GD, Schneider M, Minder C (1997) Bias in meta-analysis detected by a simple, graphical test. BMJ 315:629–634. https://doi.org/10.1136/bmj.315.7109.629
Borenstein M (2022) Comprehensive meta-analysis software. Syst Rev Heal Res 535–48. https://doi.org/10.1002/9781119099369.ch27
Chen Y, Zeng Y, Huang R, Yang Y, Chen K, Song W et al (2012) No association of five candidate genetic variants with amyotrophic lateral sclerosis in a Chinese population. Neurobiol Aging 33(2721):e3-5. https://doi.org/10.1016/j.neurobiolaging.2012.06.004
Li et al (2009) Association between DPP6 polymorphism and the risk of sporadic amyotrophic lateral sclerosis in Chinese patient. Chin Med J (Engl) 122:2989–2992
Wang Y, He Y, Zhu Y, He T, Xu J, Kuang Q et al (2022) Effect of the minor C allele of CNTN4 rs2619566 on medial hypothalamic connectivity in early-stage patients of Chinese Han ancestry with sporadic amyotrophic lateral sclerosis. Neuropsychiatr Dis Treat 18:437–448. https://doi.org/10.2147/NDT.S339456
Cronin S, Tomik B, Bradley DG, Slowik A, Hardiman O (2009) Screening for replication of genome-wide SNP associations in sporadic ALS. Eur J Hum Genet 17:213–218. https://doi.org/10.1038/ejhg.2008.194
Zhou L, Zheng Y, Tian T, Liu K, Wang M, Lin S et al (2018) Associations of interleukin-6 gene polymorphisms with cancer risk: evidence based on 49,408 cancer cases and 61,790 controls. Gene 670:136–47. https://doi.org/10.1016/j.gene.2018.05.104
Wacholder S, Chanock S, Garcia-Closas M, El Ghormli L, Rothman N (2004) Assessing the probability that a positive report is false: an approach for molecular epidemiology studies. J Natl Cancer Inst 96:434–442. https://doi.org/10.1093/jnci/djh075
Marcus PM, Vineis P, Rothman N (2000) NAT2 slow acetylation and bladder cancer risk: a meta-analysis of 22 case-control studies conducted in the general population. Pharmacogenetics 10:115–122. https://doi.org/10.1097/00008571-200003000-00003
Engel LS, Taioli E, Pfeiffer R, Garcia-Closas M, Marcus PM, Lan Q et al (2002) Pooled analysis and meta-analysis of glutathione S-transferase M1 and bladder cancer: a HuGE review. Am J Epidemiol 156:95–109. https://doi.org/10.1093/aje/kwf018
He J, Zou Y, Liu X, Zhu J, Zhang J, Zhang R et al (2018) Association of common genetic variants in pre-microRNAs and neuroblastoma susceptibility: a two-center study in Chinese Children. Mol Ther Nucleic Acids 11:1–8. https://doi.org/10.1016/j.omtn.2018.01.003
Kou N, Zhou W, He Y, Ying X, Chai S, Fei T et al (2020) A Mendelian randomization analysis to expose the causal effect of IL-18 on osteoporosis based on genome-wide association study data. Front Bioeng Biotechnol 8:201. https://doi.org/10.3389/fbioe.2020.00201
Hu P, Jiao R, Jin L, Xiong M (2018) Application of causal inference to genomic analysis: advances in methodology. Front Genet 9:238. https://doi.org/10.3389/fgene.2018.00238
Zhang F, Baranova A (2022) Smoking quantitatively increases risk for COVID-19. Eur Respir J 60. https://doi.org/10.1183/13993003.01273-2021
Hou L, Xu M, Yu Y, Sun X, Liu X, Liu L et al (2020) Exploring the causal pathway from ischemic stroke to atrial fibrillation: a network Mendelian randomization study. Mol Med 26:7. https://doi.org/10.1186/s10020-019-0133-y
Wang X, Fang X, Zheng W, Zhou J, Song Z, Xu M et al (2021) Genetic support of a causal relationship between iron status and type 2 diabetes: a Mendelian randomization study. J Clin Endocrinol Metab 106:e4641–e4651. https://doi.org/10.1210/clinem/dgab454
Zhang F, Rao S, Cao H, Zhang X, Wang Q, Xu Y, et al (2022) Genetic evidence suggests posttraumatic stress disorder as a subtype of major depressive disorder. J Clin Invest 132. https://doi.org/10.1172/JCI145942
Zhang F, Baranova A, Zhou C, Cao H, Chen J, Zhang X et al (2021) Causal influences of neuroticism on mental health and cardiovascular disease. Hum Genet 140:1267–1281. https://doi.org/10.1007/s00439-021-02288-x
Gray IC, Campbell DA, Spurr NK (2000) Single nucleotide polymorphisms as tools in human genetics. Hum Mol Genet 9:2403–2408. https://doi.org/10.1093/hmg/9.16.2403
Harun-Or-Roshid M, Ali MB, Jesmin MMNH (2022) Association of hypoxia inducible factor 1-Alpha gene polymorphisms with multiple disease risks: a comprehensive meta-analysis. PLoS One 17:e0273042
Yu H, Pan R, Qi Y, Zheng Z, Li J, Li H et al (2020) LEPR hypomethylation is significantly associated with gastric cancer in males. Exp Mol Pathol 116:104493. https://doi.org/10.1016/j.yexmp.2020.104493
Liu M, Li F, Yan H, Wang K, Ma Y, Shen L et al (2020) A multi-model deep convolutional neural network for automatic hippocampus segmentation and classification in Alzheimer’s disease. Neuroimage 208:116459. https://doi.org/10.1016/j.neuroimage.2019.116459
Acknowledgements
We acknowledge high performance computing facility support from Centre for Bioinformatics Learning Advancement and Systematics Training (cBLAST) and Texas Advanced Computing Center (TACC) for data analysis. We also acknowledge support of Biomolecular Research Foundation (BMRF), Dhaka, Bangladesh.
Author information
Authors and Affiliations
Contributions
ABMMKI: conceptualization, methodology, software, writing—original draft preparation, visualization, investigation, supervision, validation, writing—reviewing and editing; MMM: data curation, methodology, software, writing—original draft preparation, visualization, validation, writing—reviewing and editing; MAZ: data curation, methodology, software, writing—original draft preparation, visualization, validation, writing—reviewing and editing; NT: data curation, methodology, software, writing—original draft preparation, visualization, investigation, validation, writing—reviewing and editing.
Corresponding author
Ethics declarations
Patient and public involvement
There is no patient’s involvement in the development of the study design, research question, and outcome measure.
Informed consent
There are no human subjects in this article and informed consent is not applicable.
Ethics approval
Ethical approval will not be required because this study will retrieve and synthesize data from already published articles.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Miah, M.M., Zinnia, M.A., Tabassum, N. et al. Association between DPP6 gene rs10260404 polymorphism and increased risk of sporadic amyotrophic lateral sclerosis (sALS): a meta-analysis. Neurol Sci 45, 3225–3243 (2024). https://doi.org/10.1007/s10072-024-07401-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10072-024-07401-2