
Abstract
Precision medicine aims to personalize treatment for both effectiveness and safety. As a critical component of this emerging initiative, pharmacogenomics seeks to guide drug treatment based on genetics. In this review article, we give an overview of pharmacogenomics in the setting of an immunosuppressant frequently prescribed by rheumatologists, azathioprine. Azathioprine has a narrow therapeutic index and a high risk of adverse events. By applying candidate gene analysis and unbiased approaches, researchers have identified multiple variants associated with an increased risk for adverse events associated with azathioprine, particularly bone marrow suppression. Variants in two genes, TPMT and NUDT15, are widely recognized, leading drug regulatory agencies and professional organizations to adopt recommendations for testing before initiation of azathioprine therapy. As more gene-drug interactions are discovered, our field will continue to face the challenge of balancing benefits and costs associated with genetic testing. However, novel approaches in genomics and the integration of clinical and genetic factors into risk scores offer unprecedented opportunities for the application of pharmacogenomics in routine practice.
Key Points • Pharmacogenomics can help us understand how individuals’ genetics may impact their response to medications. • Azathioprine is a success story for the clinical implementation of pharmacogenomics, particularly the effects of TPMT and NUDT15 variants on myelosuppression. • As our knowledge advances, testing and dosing recommendations will continue to evolve, with our field striving to balance costs and benefits to patients. • As we aim toward the goals of precision medicine, future research may integrate increasingly individualized traits—including clinical and genetic characteristics—to predict the safety and efficacy of particular medications for individual patients. |
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Precision medicine seeks to identify effective approaches for patients based on a combination of personal factors, including genetics and lifestyle. As part of precision medicine, pharmacogenomics examines how genes impact patients’ responses to drugs and provides that information to clinicians so they can personalize treatments that maximize drug response and minimize adverse drug reactions [1,2,3]. Interpersonal variability in drug response is often unpredictable; therefore, identifying the mechanisms underlying this variability remains one of the most complex therapeutic challenges in internal medicine [4]. In rheumatology, many of our current practices include the prescription of drugs that were efficacious in only ~ 50% of patients during randomized clinical trials (e.g., mycophenolate treatment for lupus nephritis or anti-TNF agents treatment for rheumatoid arthritis) [5,6,7,8,9] or of drugs that have a high rate of adverse events (e.g., azathioprine) [10,11,12,13,14,15,16,17,18].
Genes and drug metabolism
Based on drug metabolism, genetic variation can affect individual drug response in two key steps: activation and elimination. First, a functional variant in a gene which encodes an enzyme responsible for activating a prodrug can affect concentrations of the active drug. If there is a loss-of-function variant or a variant that decreases the function of the activating enzyme, then the drug might have null or reduced efficacy. Conversely, gain-of-function variants can result in excessive drug activation and increase the risks of adverse events [19]. In either case, changes in drug dosing or drug choice might be necessary. The second way genes can have a large effect on drug action is due to loss-of-function variants in enzymes that are responsible for eliminating the active drug from the body. The resulting increased concentration of the active drug can lead to adverse events. This risk for adverse events is particularly concerning for drugs with a narrow therapeutic window (i.e., a small difference between a dose causing side effects and the therapeutic dose).
Pharmacogenomics approaches
The goal of pharmacogenomics is to use an individual’s genetics to guide prescribing decisions. Pharmacogenomics has used two broad methods: (1) the study of candidate gene(s) association studies and (2) unbiased approaches to identify functionally important variants. Candidate gene association studies have focused on variations in genes that encode enzymes, drug transporters, or drug receptors. The genes are chosen based on foreknowledge of drug metabolic pathways. The advantage to this approach is that fewer cases are required to have sufficient power to detect difference [4]. In contrast, unbiased approaches—predominantly, genome-wide association studies (GWAS), exome sequencing, and whole genome sequencing—interrogate millions of variants [20]. GWAS are more costly and complex, and they may obscure causative variants because they adjust the significance threshold for multiple comparisons; however, the unbiased approach offers the opportunity to discover unexpected mechanisms or pathways [21].
Despite pharmacogenomics being a relatively new field, the research derived from using biased and unbiased approaches has yielded substantive information regarding the links between genetic variants and drug response. As of December 2019, the Food and Drug Administration’s (FDA) Table of Pharmacogenomic Biomarkers in Drug Labeling includes 404 pharmacogenomic biomarkers for 282 drugs [22]. Indeed, numerous rheumatological medications and their associated recommendations (e.g., testing and concurrent prescription warnings) appear on this table: azathioprine, celecoxib, flurbiprofen, lesinurad, pegloticase, piroxicam, probenecid, and upadacitinib.
Although these discoveries can and should contribute to therapeutic decision-making, implementing pharmacogenomics into clinical practice still faces many challenges. The first challenge is to identify clinically relevant drug response phenotypes. Technology has brought the resources to manage and interrogate genome data robustly [23], but determining clinically important phenotypes may not be feasible or may be too costly in time or labor, particularly if the standard for clinical relevance demands a randomized controlled trial showing the benefit of genotyping [24]. Consequently, real-world data obtained in routine clinical care are an attractive resource. However, the use of real-world data to define the clinical relevance of pharmacogenomics variants also has challenges. For example, the clinical variables used to define response in clinical trials (e.g., DAS28, SLEDAI) may not be recorded routinely in clinical practice. Moreover, accurate collection of data still requires review of individual medical records in many cases, which is labor intensive. The next challenge is to determine which gene-drug pairs are actionable [23]—in other words, identifying a particular genotype leads to either a change in the recommended dose of a drug or the substitution of an alternative drug, ideally implemented at the point of care. Finally, we must consider the potential costs of incorporating genetic testing in the clinical environment (e.g., financial or privacy-related) and carefully balance them with the potential benefits to patients (e.g. improved disease treatment). Overcoming these challenges is crucial as we move forward into the precision medicine era.
Pharmacogenomics applied in rheumatology: the example of azathioprine
Azathioprine and its associated side effects mark the most successful application of pharmacogenomics to rheumatology clinical practice and one of the most successful cases in internal medicine more broadly. In this review, we describe some of the concepts and history related to azathioprine and present the rationale supporting current guidelines to illustrate the current status and the potential direction of pharmacogenomics in general.
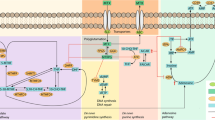
Thiopurine drugs (azathioprine and its metabolite 6-mercaptopurine) are immunosuppressants that are used widely for treating autoimmune conditions (e.g., systemic lupus erythematosus and ulcerative colitis), forms of cancer, and preventing organ transplant rejection. Azathioprine is rapidly converted to 6-mercaptopurine (6-MP) outside of the cell; 6-MP is then transported inside the cell where it is converted into thioinosine monophosphate (TIMP). Through a series of reactions, TIMP is converted, in part, to active metabolites for treatment, 6-thioguanine nucleotides (6-TGNs: deoxy-thioguanosine monophosphate, deoxy-thioguanosine triphosphate, thioguanosine diphosphate, thioguanosine monophosphate, and thioguanosine triphosphate). 6-TGNs suppress the immune system by incorporating into DNA that is replicating and RNA that is translating as well as blocking the de novo pathway of purine synthesis (Fig. 1) [25, 26]. However, as first discovered in 1983, excessive 6-TGNs’ concentrations are toxic and can cause bone marrow suppression [27, 28] and certain types of cancers [29, 30].

Azathioprine metabolism. *6-Mercaptopurine (6-MP), 6-methyl-mercaptopurine (6-MMP), 6-thioinosine monophosphate (TIMP), 6-thiouracil (6-TU), deoxy-thioguanosine monophosphate (TdGMP), deoxy-thioguanosine triphosphate (TdGTP), nudix hydrolase 15 (NUDT15), thioguanosine diphosphate (TGDP), thioguanosine monophosphate (TGMP), thioguanosine triphosphate (TGTP), thiopurine methyltransferase (TPMT), xanthine oxidase (XO)
Variants in TPMT can predict leukopenia
The first link between phenotypic variations in the enzyme thiopurine methyltransferase (TPMT) and variability in 6-TGNs concentrations was observed in 1987 [31]. We now know that TPMT inactivates intermediate compounds during the metabolism of thiopurine drugs, thereby reducing the final concentration of 6-TGNs at a given dose of the drug (Fig. 1). With this knowledge, geneticists used candidate gene association studies to identify variants in TPMT responsible for reduced activity. Now, they have identified dozens of single nucleotide polymorphisms (SNPs) on the TPMT gene that reduce or eliminate its function, which results in an increased likelihood of myelosuppression for patients taking azathioprine. Three of these variants account for 90% of low activity phenotypes in individuals of European descent: *2, *3A, and *3C (found in 0.2%, 3.4%, and 0.4%, respectively) [32,33,34]. Accordingly, due to the high level of risk, potential users are now routinely tested for this genetic variation before receiving a prescription.
Despite this real-world application, preemptive TPMT testing does not eliminate the risk of myelosuppression for most patients who use azathioprine; indeed, known TPMT variants appear to account for only 25% of cases of myelosuppression [35,36,37,38]. As such, researchers have focused on using unbiased approaches to find additional genetic variants causing adverse events with azathioprine use.
Variants in NUDT15 can predict leukopenia
The limitations of testing for known TPMT variants are particularly relevant to Asian populations. Asians have a much lower frequency of TPMT variants identified with azathioprine-associated leukopenia compared to Caucasians (~ 2% among South/Central Asians versus ~ 12% of Caucasians); nevertheless, the rate of leukopenia is higher among Asians (31–40% among Asians with Crohn’s disease versus ~ 5% among Caucasians with inflammatory bowel disease). This points to the limitations of using TPMT alone to predict azathioprine toxicity [39,40,41,42]. Suspecting genes outside TPMT played a role in leukopenia, researchers utilized an unbiased approach to identify additional genetic variants causing leukopenia in individuals of Asian descent.
In 2014, Yang et al. found that a missense SNP, rs116855232, in nudix hydrolase 15 (NUDT15) was strongly associated with increased risk for leukopenia among Koreans (OR = 35.6; p = 4.88 × 10−94) [43]. The same NUDT15 SNP was later found to cause azathioprine-associated leukopenia in individuals of Chinese descent [44,45,46]. While this particular SNP is rare in Caucasians, researchers have found other NUDT15 variants associated with azathioprine-induced myelosuppression in Caucasians [47, 48]. Indeed, Schaeffeler et al. observed that NUDT15 variants contributed to 13% of azathioprine-associated leukopenia among Caucasians; further, they observed that TPMT and NUDT15 variants combined to explain ~ 50% of hematotoxicity among azathioprine users of European descent [47]. Having identified the significance of certain NUDT15 variants, researchers have since been able to identify the mechanisms by which NUDT15 impacts thiopurine metabolism. Similar to TPMT, NUDT15 inactivates thiopurine metabolites, ultimately leading to reduced concentrations of cytotoxic 6-TGNs (Fig. 1). [49]
A variant in HLA can predict pancreatitis
Leukopenia is not the only serious side effect associated with azathioprine use. Approximately 4% of patients develop pancreatitis after the initiation of azathioprine; notably, unlike leukopenia, pancreatitis is not dose related [50]. Heap et al. conducted a GWAS to identify potential genetic predictors of pancreatitis among a cohort of European patients who developed pancreatitis after thiopurine treatment for Crohn’s disease or ulcerative colitis; notably, these patients had no additional risk factors for pancreatitis, including their concurrent medications. The group found a SNP, rs2647087 (occurring in ~ 30% of individuals with European ancestry), within the human leukocyte antigen (HLA) complex (fine-mapping revealed this SNP was associated with HLADQA1*02:01-HLA-DRB1*07:01 haplotype) significant for the development of pancreatitis among azathioprine users; they were able to replicate their results in a group of patients with inflammatory bowel disease (IBD). The study found no association between TPMT variants (*3A, *3C, *2, *4, and *8) and pancreatitis [51]. A separate study confirmed the association between HLA SNP rs2647087 and pancreatitis in a second, independent cohort of patients with IBD taking azathioprine (OR = 0.53% for wild-type, 4.25% for heterozygous, and 14.63% homozygous) [52].
Cost-effectiveness evaluation
Alongside the focus on safety, we must consider cost-effectiveness when considering implementation of genetic testing for patients. Identifying potential side effects and efficacy through genetic testing may ultimately save more money, despite upfront costs. [53]
Analysis of the cost-effectiveness of genetic testing for azathioprine use has brought some controversy, but the results of most studies favor genotyping. Studies in several healthcare systems around the world have demonstrated that the aggregate costs associated with testing tend to be lower compared with the aggregate costs of treatment for patients who are heterozygous (approximately 1 in 10) or homozygous (approximately 1 in 300) for TPMT loss-of-function variants [54, 55]. For example, in the USA, one study compared TPMT testing, metabolite monitoring, and community care. TPMT testing was the least costly strategy at year one ($7142 for community care versus $3861 for testing) [56]. Of course, results are not always so definitive. Several studies in Canada’s single payer system have found only small differences in the costs associated with screening versus not [57, 58]. Despite the lack of “savings,” these results promisingly show that patients at the greatest risk may be identified for relatively neutral costs.
Gene-drug pairs with the strongest evidence to support the necessity of genetic testing and proven cost-effectiveness of preemptive genotyping are more likely to be reimbursed by Medicare (and similar systems in other countries); accordingly, Medicare reimburses testing for TPMT variants before prescribing azathioprine. [59]
Clinical recommendations and guidelines
The success in identifying genetic predictors for drug-associated side effects has led several groups to provide therapeutic guidelines for these drugs. As noted above, the FDA now requires labeling with guidelines for individuals who have variants in TPMT and NUDT15. Additionally, the Clinical Pharmacogenetics Implementation Consortium (CPIC) and Dutch Pharmacogenetics Working Group (DPWG) both provide evidence-based recommendations for dosing or alternative medication options [27, 60]. The most recent CPIC publication includes dosing guidelines based on TPMT and NUDT15 metabolizer status (Table 1) [34]. For example, for patients who are considered TPMT or NUDT15 poor metabolizers (two nonfunctional alleles), CPIC recommends against using azathioprine to treat non-malignant conditions. CPIC also has guidelines for treating patients with various combinations of TPMT and NUDT15 phenotypes. Creating these types of evidence-based recommendations are not only imperative to improving healthcare but also for reducing healthcare costs and encouraging healthcare reimbursement from insurance companies and healthcare programs like Medicare and Medicaid.
Future opportunities: more genes and the potential role of risk scores
Along with the widely recognized genetic variants described above, numerous additional variants may play a role in myelosuppression or other adverse effects associated with azathioprine use. Researchers are continuing to examine the potential links between side effects and proteins known to be involved in the metabolic pathway of azathioprine. For example, xanthine oxidase/dehydrogenase (XDH), aldehyde oxidase (AOX1), and molybdenum cofactor sulfurase (MOCOS) are enzymes involved in the metabolism of azathioprine. MOCOS is an enzyme that sulfurates the molybdenum cofactor for AOX1 and XDH, which compete with TPMT to inactivate certain azathioprine metabolites [61, 62]. Initial studies have shown that variants in AOX1 and MOCOS can impact effective dosage among transplant candidates; however, additional studies are required to determine if these variants in these genes play a role in azathioprine-associated side effects. Additional studies have noted the potential for variants in the genes ABCC4, ITPA, GST, IMPDH1, IL6, and FTO [43, 49, 51, 62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92] to impact azathioprine metabolism and its side effects. Nevertheless, more research is needed to determine whether they should be tested in clinical practice.
Because of the interaction between genetics and environment, the most useful information for clinicians and patients may lie in risk scores that consider genetic, clinical, and environmental characteristics. As a proof-of-concept, we recently published a pilot study that generated such a score for azathioprine-associated leukopenia [93]. As discussed above, genetic testing to determine TPMT metabolizer status has become the standard of care for patients initiating azathioprine; however, even with the addition of NUDT15 testing, almost half of leukopenia cases remain unexplained [47, 94, 95]. We hypothesized that a risk score composed of multiple clinical factors and additional candidate genes could improve the prediction of azathioprine-associated leukopenia. The risk score included demographic characteristics, medications that interact pharmacokinetically or pharmacodynamically with azathioprine, leukopenia-associated comorbid conditions, and genetic variants coding for enzymes involved in azathioprine metabolism (including TPMT and NUDT15). We generated multiple models using information from electronic health records, including patients with a wide range of indications, and/or the results of genetic testing. The model that included all clinical variables and genetic information performed the best in both the discovery and replication phases of the study. These results indicate that a multivariable score that incorporates important clinical variables and genetic data outperforms scores based on traditional genetic testing (e.g., TPMT metabolizer status).
Many steps are required before implementing risk scores in clinical practice; these include larger studies and GWAS analysis for additional genetic information. Also, while we have determined some of the genetic variants that lead to azathioprine adverse events, further research is needed to examine whether additional genetic information, as well as proteomic and metabolomic approaches, could improve prediction of adverse events in real-world clinical practice. Although these efforts require an investment of time and resources, we can look forward to risks scores—based on complex clinical and genetic information—that can more effectively determine rheumatology patients’ risk for serious side effects.
Conclusion and future directions
Along with this focus on safety, we also anticipate more studies regarding the impact of genetics on the efficacy of medications. As we work toward the goal of precision medicine, future research ultimately may combine genetic and clinical data into a generalized recommendation score that accounts for the efficacy and risk of individual drugs for individual patients. In closing, we see promise, in particular for the multiple drugs with complex metabolism and narrow therapeutic indices that are strong candidates for applied pharmacogenomics in rheumatology.
References
National Institutes of Health NLoM What is pharmacogenomics? https://ghr.nlm.nih.gov/primer/genomicresearch/pharmacogenomics. Accessed April 17, 2020 2020
Roses AD (2004) Pharmacogenetics and drug development: the path to safer and more effective drugs. Nat Rev Genet 5(9):645–656. https://doi.org/10.1038/nrg1432
Roden DM, McLeod HL, Relling MV, Williams MS, Mensah GA, Peterson JF, Van Driest SL (2019) Pharmacogenomics. Lancet 394(10197):521–532. https://doi.org/10.1016/s0140-6736(19)31276-0
Roden DM, Altman RB, Benowitz NL, Flockhart DA, Giacomini KM, Johnson JA, Krauss RM, HL ML, Ratain MJ, Relling MV, Ring HZ, Shuldiner AR, Weinshilboum RM, Weiss ST, Pharmacogenetics Research N (2006) Pharmacogenomics: challenges and opportunities. Ann Intern Med 145(10):749–757
Appel GB, Contreras G, Dooley MA, Ginzler EM, Isenberg D, Jayne D, Li L-S, Mysler E, Sánchez-Guerrero J, Solomons N, Wofsy D, Aspreva Lupus Management Study G (2009) Mycophenolate mofetil versus cyclophosphamide for induction treatment of lupus nephritis. J Am Soc Nephrol 20(5):1103–1112. https://doi.org/10.1681/ASN.2008101028
Rubbert-Roth A, Finckh A (2009) Treatment options in patients with rheumatoid arthritis failing initial TNF inhibitor therapy: a critical review. Arthritis Res Ther 11 Suppl 1(Suppl 1):S1. https://doi.org/10.1186/ar2666
Ma X, Xu S (2013) TNF inhibitor therapy for rheumatoid arthritis. Biomed Rep 1(2):177–184. https://doi.org/10.3892/br.2012.42
Johnson KJ, Sanchez HN, Schoenbrunner N (2019) Defining response to TNF-inhibitors in rheumatoid arthritis: the negative impact of anti-TNF cycling and the need for a personalized medicine approach to identify primary non-responders. Clin Rheumatol 38(11):2967–2976. https://doi.org/10.1007/s10067-019-04684-1
Dooley MA, Jayne D, Ginzler EM, Isenberg D, Olsen NJ, Wofsy D, Eitner F, Appel GB, Contreras G, Lisk L, Solomons N (2011) Mycophenolate versus azathioprine as maintenance therapy for lupus nephritis. N Engl J Med 365(20):1886–1895. https://doi.org/10.1056/NEJMoa1014460
Avallone EV, Pica R, Cassieri C, Zippi M, Paoluzi P, Vernia P (2014) Azathioprine treatment in inflammatory bowel disease patients: type and time of onset of side effects. Eur Rev Med Pharmacol Sci 18(2):165–170
Singh G, Fries JF, Spitz P, Williams CA (1989) Toxic effects of azathioprine in rheumatoid arthritis. A national post-marketing perspective. Arthritis Rheum 32(7):837–843
Currey HL, Harris J, Mason RM, Woodland J, Beveridge T, Roberts CJ, Vere DW, Dixon AS, Davies J, Owen-Smith B (1974) Comparison of azathioprine, cyclophosphamide, and gold in treatment of rheumatoid arthritis. Br Med J 3(5934):763–766. https://doi.org/10.1136/bmj.3.5934.763
Sahasranaman S, Howard D, Roy S (2008) Clinical pharmacology and pharmacogenetics of thiopurines. Eur J Clin Pharmacol 64(8):753–767. https://doi.org/10.1007/s00228-008-0478-6
Kirk AP, Lennard-Jones JE (1982) Controlled trial of azathioprine in chronic ulcerative colitis. Br Med J (Clin Res Ed) 284(6325):1291–1292. https://doi.org/10.1136/bmj.284.6325.1291
Pruijt JFM, Haanen JBAG, Hollander AAMJ, den Ottolander GJ (1996) Azathioprine-induced pure red-cell aplasia. Nephrol Dial Transplant 11(7):1371–1373. https://doi.org/10.1093/ndt/11.7.1371
Gilissen LPL, Derijks LJJ, Bos LP, Bus PJ, Hooymans PM, Engels LGJB (2004) Therapeutic drug monitoring in patients with inflammatory bowel disease and established azathioprine therapy. Clin Drug Investig 24(8):479–486. https://doi.org/10.2165/00044011-200424080-00006
Kaskas BA, Louis E, Hindorf U, Schaeffeler E, Deflandre J, Graepler F, Schmiegelow K, Gregor M, Zanger UM, Eichelbaum M, Schwab M (2003) Safe treatment of thiopurine S-methyltransferase deficient Crohn’s disease patients with azathioprine. Gut 52(1):140–142. https://doi.org/10.1136/gut.52.1.140
Adam L, Phulukdaree A, Soma P (2018) Effective long-term solution to therapeutic remission in inflammatory bowel disease: role of azathioprine. Biomed Pharmacother 100:8–14. https://doi.org/10.1016/j.biopha.2018.01.152
Westervelt P, Cho K, Bright DR, Kisor DF (2014) Drug-gene interactions: inherent variability in drug maintenance dose requirements. P T 39(9):630–637
Crews KR, Hicks JK, Pui CH, Relling MV, Evans WE (2012) Pharmacogenomics and individualized medicine: translating science into practice. Clin Pharmacol Ther 92(4):467–475. https://doi.org/10.1038/clpt.2012.120
Manolio TA (2010) Genomewide association studies and assessment of the risk of disease. N Engl J Med 363(2):166–176. https://doi.org/10.1056/NEJMra0905980
Administration. FaD table of pharmacogenomic biomarkers in drug labeling. https://www.fda.gov/media/124784/download. Accessed April 24, 2020 2020
Relling MV, Evans WE (2015) Pharmacogenomics in the clinic. Nature 526(7573):343–350. https://doi.org/10.1038/nature15817
Wei WQ, Denny JC (2015) Extracting research-quality phenotypes from electronic health records to support precision medicine. Genome Med 7(1):41. https://doi.org/10.1186/s13073-015-0166-y
Lennard L, Van Loon JA, Weinshilboum RM (1989) Pharmacogenetics of acute azathioprine toxicity: relationship to thiopurine methyltransferase genetic polymorphism. Clin Pharmacol Ther 46(2):149–154. https://doi.org/10.1038/clpt.1989.119
Zaza G, Cheok M, Krynetskaia N, et al. (2010) Thiopurine pathway. Pharmacogenet Genomics 20(9):573–574. https://doi.org/10.1097/FPC.0b013e328334338f
Relling MV, Gardner EE, Sandborn WJ, Schmiegelow K, Pui CH, Yee SW, Stein CM, Carrillo M, Evans WE, Hicks JK, Schwab M, Klein TE (2013) Clinical pharmacogenetics implementation consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing: 2013 update. Clin Pharmacol Ther 93(4):324–325. https://doi.org/10.1038/clpt.2013.4
Lennard L, Rees CA, Lilleyman JS, Maddocks JL (1983) Childhood leukaemia: a relationship between intracellular 6-mercaptopurine metabolites and neutropenia. Br J Clin Pharmacol 16(4):359–363. https://doi.org/10.1111/j.1365-2125.1983.tb02178.x
Lennard L, Thomas S, Harrington CI, Maddocks JL (1985) Skin cancer in renal transplant recipients is associated with increased concentrations of 6-thioguanine nucleotide in red blood cells. Br J Dermatol 113(6):723–729. https://doi.org/10.1111/j.1365-2133.1985.tb02408.x
Bo J, Schrøder H, Kristinsson J, Madsen B, Szumlanski C, Weinshilboum R, Andersen JB, Schmiegelow K (1999) Possible carcinogenic effect of 6-mercaptopurine on bone marrow stem cells: relation to thiopurine metabolism. Cancer 86(6):1080–1086. https://doi.org/10.1002/(sici)1097-0142(19990915)86:6<1080::aid-cncr26>3.0.co;2-5
Lennard L, Van Loon JA, Lilleyman JS, Weinshilboum RM (1987) Thiopurine pharmacogenetics in leukemia: correlation of erythrocyte thiopurine methyltransferase activity and 6-thioguanine nucleotide concentrations. Clin Pharmacol Ther 41(1):18–25. https://doi.org/10.1038/clpt.1987.4
Yates CR, Krynetski EY, Loennechen T, Fessing MY, Tai HL, Pui CH, Relling MV, Evans WE (1997) Molecular diagnosis of thiopurine S-methyltransferase deficiency: genetic basis for azathioprine and mercaptopurine intolerance. Ann Intern Med 126(8):608–614. https://doi.org/10.7326/0003-4819-126-8-199704150-00003
Schaeffeler E, Fischer C, Brockmeier D, Wernet D, Moerike K, Eichelbaum M, Zanger UM, Schwab M (2004) Comprehensive analysis of thiopurine S-methyltransferase phenotype-genotype correlation in a large population of German-Caucasians and identification of novel TPMT variants. Pharmacogenetics 14(7):407–417. https://doi.org/10.1097/01.fpc.0000114745.08559.db
Consortium CPI CPIC guideline for thiopurines and TPMT and NUDT15. https://cpicpgx.org/guidelines/guideline-for-thiopurines-and-tpmt/. Accessed April 17, 2020 2020
Roberts RL, Barclay ML (2015) Update on thiopurine pharmacogenetics in inflammatory bowel disease. Pharmacogenomics 16(8):891–903. https://doi.org/10.2217/pgs.15.29
Broekman M, Coenen MJH, Wanten GJ, van Marrewijk CJ, Klungel OH, Verbeek ALM, Hooymans PM, Guchelaar HJ, Scheffer H, Derijks LJJ, Wong DR, de Jong DJ (2017) Risk factors for thiopurine-induced myelosuppression and infections in inflammatory bowel disease patients with a normal TPMT genotype. Aliment Pharmacol Ther 46(10):953–963. https://doi.org/10.1111/apt.14323
Colombel JF, Ferrari N, Debuysere H, Marteau P, Gendre JP, Bonaz B, Soule JC, Modigliani R, Touze Y, Catala P, Libersa C, Broly F (2000) Genotypic analysis of thiopurine S-methyltransferase in patients with Crohn’s disease and severe myelosuppression during azathioprine therapy. Gastroenterology 118(6):1025–1030
Goldberg R, Irving PM (2015) Toxicity and response to thiopurines in patients with inflammatory bowel disease. Expert Rev Gastroenterol Hepatol 9(7):891–900. https://doi.org/10.1586/17474124.2015.1039987
Kumagai K, Hiyama K, Ishioka S, et al. (2001) Allelotype frequency of the thiopurine methyltransferase (TPMT) gene in Japanese. Pharmacogenetics 11(3):275–278.
Cao Q, Zhu Q, Shang Y, Gao M, Si J (2009) Thiopurine methyltransferase gene polymorphisms in Chinese patients with inflammatory bowel disease. Digestion 79(1):58–63. https://doi.org/10.1159/000205268
Takatsu N, Matsui T, Murakami Y, Ishihara H, Hisabe T, Nagahama T, Maki S, Beppu T, Takaki Y, Hirai F, Yao K (2009) Adverse reactions to azathioprine cannot be predicted by thiopurine S-methyltransferase genotype in Japanese patients with inflammatory bowel disease. J Gastroenterol Hepatol 24(7):1258–1264. https://doi.org/10.1111/j.1440-1746.2009.05917.x
Collie-Duguid ESR, Pritchard P, Sludden J, Collier LT, McLeod HL (1999) The frequency and distribution of thiopurine methyltransferase alleles in Caucasian and Asian populations. Pharmacogenetics 9(1):37–42
Yang SK, Hong M, Baek J, Choi H, Zhao W, Jung Y, Haritunians T, Ye BD, Kim KJ, Park SH, Park SK, Yang DH, Dubinsky M, Lee I, McGovern DP, Liu J, Song K (2014) A common missense variant in NUDT15 confers susceptibility to thiopurine-induced leukopenia. Nat Genet 46(9):1017–1020. https://doi.org/10.1038/ng.3060
Yin D, Xia X, Zhang J, Zhang S, Liao F, Zhang G, Zhang Y, Hou Q, Yang X, Wang H, Ma Z, Wang H, Zhu Y, Zhang W, Wang Y, Liu B, Wang L, Xu H, Shu Y (2017) Impact of NUDT15 polymorphisms on thiopurines-induced myelotoxicity and thiopurines tolerance dose. Oncotarget 8(8):13575–13585. https://doi.org/10.18632/oncotarget.14594
Liu Y, Meng Y, Wang L, Liu Z, Li J, Dong W (2018) Associations between the NUDT15 R139C polymorphism and susceptibility to thiopurine-induced leukopenia in Asians: a meta-analysis. Onco Targets Ther 11:8309–8317. https://doi.org/10.2147/OTT.S177007
Fan X, Yin D, Men R, Xu H, Yang L (2019) NUDT15 Polymorphism confer increased susceptibility to thiopurine-induced leukopenia in patients with autoimmune hepatitis and related cirrhosis. Front Pharmacol 10:346
Schaeffeler E, Jaeger SU, Klumpp V, Yang JJ, Igel S, Hinze L, Stanulla M, Schwab M (2019) Impact of NUDT15 genetics on severe thiopurine-related hematotoxicity in patients with European ancestry. Genet Med 21(9):2145–2150. https://doi.org/10.1038/s41436-019-0448-7
Walker GJ, Harrison JW, Heap GA, Voskuil MD, Andersen V, Anderson CA, Ananthakrishnan AN, Barrett JC, Beaugerie L, Bewshea CM, Cole AT, Cummings FR, Daly MJ, Ellul P, Fedorak RN, Festen EAM, Florin TH, Gaya DR, Halfvarson J, Hart AL, Heerasing NM, Hendy P, Irving PM, Jones SE, Koskela J, Lindsay JO, Mansfield JC, McGovern D, Parkes M, Pollok RCG, Ramakrishnan S, Rampton DS, Rivas MA, Russell RK, Schultz M, Sebastian S, Seksik P, Singh A, So K, Sokol H, Subramaniam K, Todd A, Annese V, Weersma RK, Xavier R, Ward R, Weedon MN, Goodhand JR, Kennedy NA, Ahmad T (2019) Association of genetic variants in NUDT15 with thiopurine-induced myelosuppression in patients with inflammatory bowel disease. JAMA 321(8):773–785. https://doi.org/10.1001/jama.2019.0709
Moriyama T, Nishii R, Perez-Andreu V, Yang W, Klussmann FA, Zhao X, Lin TN, Hoshitsuki K, Nersting J, Kihira K, Hofmann U, Komada Y, Kato M, McCorkle R, Li L, Koh K, Najera CR, Kham SK, Isobe T, Chen Z, Chiew EK, Bhojwani D, Jeffries C, Lu Y, Schwab M, Inaba H, Pui CH, Relling MV, Manabe A, Hori H, Schmiegelow K, Yeoh AE, Evans WE, Yang JJ (2016) NUDT15 polymorphisms alter thiopurine metabolism and hematopoietic toxicity. Nat Genet 48(4):367–373. https://doi.org/10.1038/ng.3508
Chaparro M, Ordás I, Cabré E, Garcia-Sanchez V, Bastida G, Peñalva M, Gomollón F, García-Planella E, Merino O, Gutiérrez A, Esteve M, Márquez L, Garcia-Sepulcre M, Hinojosa J, Vera I, Muñoz F, Mendoza JL, Cabriada JL, Montoro MA, Barreiro-de Acosta M, Ceña G, Saro C, Aldeguer X, Barrio J, Maté J, Gisbert JP (2013) Safety of thiopurine therapy in inflammatory bowel disease: long-term follow-up study of 3931 patients. Inflamm Bowel Dis 19(7):1404–1410. https://doi.org/10.1097/MIB.0b013e318281f28f
Heap GA, Weedon MN, Bewshea CM, Singh A, Chen M, Satchwell JB, Vivian JP, So K, Dubois PC, Andrews JM, Annese V, Bampton P, Barnardo M, Bell S, Cole A, Connor SJ, Creed T, Cummings FR, D'Amato M, Daneshmend TK, Fedorak RN, Florin TH, Gaya DR, Greig E, Halfvarson J, Hart A, Irving PM, Jones G, Karban A, Lawrance IC, Lee JC, Lees C, Lev-Tzion R, Lindsay JO, Mansfield J, Mawdsley J, Mazhar Z, Parkes M, Parnell K, Orchard TR, Radford-Smith G, Russell RK, Reffitt D, Satsangi J, Silverberg MS, Sturniolo GC, Tremelling M, Tsianos EV, van Heel DA, Walsh A, Watermeyer G, Weersma RK, Zeissig S, Rossjohn J, Holden AL, International Serious Adverse Events Consortium, I. B. D. Pharmacogenetics Study Group et al (2014) HLA-DQA1-HLA-DRB1 variants confer susceptibility to pancreatitis induced by thiopurine immunosuppressants. Nat Genet 46(10):1131–1134. https://doi.org/10.1038/ng.3093
Wilson A, Jansen LE, Rose RV, Gregor JC, Ponich T, Chande N, Khanna R, Yan B, Jairath V, Khanna N, Sey M, Beaton M, McIntosh K, Teft WA, Kim RB (2018) HLA-DQA1-HLA-DRB1 polymorphism is a major predictor of azathioprine-induced pancreatitis in patients with inflammatory bowel disease. Aliment Pharmacol Ther 47(5):615–620. https://doi.org/10.1111/apt.14483
Verbelen M, Weale ME, Lewis CM (2017) Cost-effectiveness of pharmacogenetic-guided treatment: are we there yet? Pharmacogenomics J 17(5):395–402. https://doi.org/10.1038/tpj.2017.21
Krynetski EY, Evans WE (2000) Genetic polymorphism of thiopurine S-methyltransferase: molecular mechanisms and clinical importance. Pharmacology 61(3):136–146
McLeod HL, Lin JS, Scott EP, Pui CH, Evans WE (1994) Thiopurine methyltransferase activity in American white subjects and black subjects. Clin Pharmacol Ther 55(1):15–20. https://doi.org/10.1038/clpt.1994.4
Dubinsky MC, Reyes E, Ofman J, Chiou CF, Wade S, Sandborn WJ (2005) A cost-effectiveness analysis of alternative disease management strategies in patients with Crohn’s disease treated with azathioprine or 6-mercaptopurine. Am J Gastroenterol 100(10):2239–2247. https://doi.org/10.1111/j.1572-0241.2005.41900.x
Tavadia SM, Mydlarski PR, Reis MD, Mittmann N, Pinkerton PH, Shear N, Sauder DN (2000) Screening for azathioprine toxicity: a pharmacoeconomic analysis based on a target case. J Am Acad Dermatol 42(4):628–632
Marra CA, Esdaile JM, Anis AH (2002) Practical pharmacogenetics: the cost effectiveness of screening for thiopurine s-methyltransferase polymorphisms in patients with rheumatological conditions treated with azathioprine. J Rheumatol 29(12):2507–2512
Simeonidis S, Koutsilieri S, Vozikis A, Cooper DN, Mitropoulou C, Patrinos GP (2019) Application of economic evaluation to assess feasibility for reimbursement of genomic testing as part of personalized medicine interventions. Front Pharmacol 10:830. https://doi.org/10.3389/fphar.2019.00830
Swen JJ, Nijenhuis M, van Rhenen M, de Boer-Veger NJ, Buunk AM, Houwink EJF, Mulder H, Rongen GA, van Schaik RHN, van der Weide J, Wilffert B, Deneer VHM, Guchelaar HJ (2018) Pharmacogenetic information in clinical guidelines: the European perspective. Clin Pharmacol Ther 103(5):795–801. https://doi.org/10.1002/cpt.1049
Smith MA, Marinaki AM, Arenas M, Shobowale-Bakre M, Lewis CM, Ansari A, Duley J, Sanderson JD (2009) Novel pharmacogenetic markers for treatment outcome in azathioprine-treated inflammatory bowel disease. Aliment Pharmacol Ther 30(4):375–384. https://doi.org/10.1111/j.1365-2036.2009.04057.x
Kurzawski M, Dziewanowski K, Safranow K, Drozdzik M (2012) Polymorphism of genes involved in purine metabolism (XDH, AOX1, MOCOS) in kidney transplant recipients receiving azathioprine. Ther Drug Monit 34(3):266–274. https://doi.org/10.1097/FTD.0b013e31824aa681
Varnell CD, Fukuda T, Kirby CL, Martin LJ, Warshaw BL, Patel HP, Chand DH, Barletta GM, Van Why SK, VanDeVoorde RG, Weaver DJ, Wilson A, Verghese PS, Vinks AA, Greenbaum LA, Goebel J, Hooper DK (2017) Mycophenolate mofetil-related leukopenia in children and young adults following kidney transplantation: Influence of genes and drugs. Pediatr Transplant 21(7). https://doi.org/10.1111/petr.13033
Kurzawski M, Dziewanowski K, Lener A, Drozdzik M (2009) TPMT but not ITPA gene polymorphism influences the risk of azathioprine intolerance in renal transplant recipients. Eur J Clin Pharmacol 65(5):533–540. https://doi.org/10.1007/s00228-009-0630-y
Honda K, Kobayashi A, Niikura T, Hasegawa T, Saito Z, Ito S, Sasaki T, Komine K, Ishizuka S, Motoi Y, Kubota T, Yamamoto H, Yokoo T (2018) Neutropenia related to an azathioprine metabolic disorder induced by an inosine triphosphate pyrophosphohydrolase (ITPA) gene mutation in a patient with PR3-ANCA-positive microscopic polyangiitis. Clin Nephrol 90(5):363–369. https://doi.org/10.5414/CN109383
Ban H, Andoh A, Imaeda H, Kobori A, Bamba S, Tsujikawa T, Sasaki M, Saito Y, Fujiyama Y (2010) The multidrug-resistance protein 4 polymorphism is a new factor accounting for thiopurine sensitivity in Japanese patients with inflammatory bowel disease. J Gastroenterol 45(10):1014–1021. https://doi.org/10.1007/s00535-010-0248-y
Dubois PCA (2011) The risk of azathioprine-induced pancreatitis depends on genetic variants in the HLA gene region. Gut 60(Suppl 1):A60–A60. https://doi.org/10.1136/gut.2011.239301.120
Abla N, Chinn LW, Nakamura T, Liu L, Huang CC, Johns SJ, Kawamoto M, Stryke D, Taylor TR, Ferrin TE, Giacomini KM, Kroetz DL (2008) The human multidrug resistance protein 4 (MRP4, ABCC4): functional analysis of a highly polymorphic gene. J Pharmacol Exp Ther 325(3):859–868. https://doi.org/10.1124/jpet.108.136523
Hawwa AF, Millership JS, Collier PS, Vandenbroeck K, McCarthy A, Dempsey S, Cairns C, Collins J, Rodgers C, McElnay JC (2008) Pharmacogenomic studies of the anticancer and immunosuppressive thiopurines mercaptopurine and azathioprine. Br J Clin Pharmacol 66(4):517–528. https://doi.org/10.1111/j.1365-2125.2008.03248.x
Chiabai MA, Lins TC, Pogue R, Pereira RW (2012) Population analysis of pharmacogenetic polymorphisms related to acute lymphoblastic leukemia drug treatment. Dis Markers 32(4):247–253. https://doi.org/10.3233/DMA-2011-0884
Zelinkova Z, Derijks LJ, Stokkers PC, Vogels EW, van Kampen AH, Curvers WL, Cohn D, van Deventer SJ, Hommes DW (2006) Inosine triphosphate pyrophosphatase and thiopurine s-methyltransferase genotypes relationship to azathioprine-induced myelosuppression. Clin Gastroenterol Hepatol 4(1):44–49. https://doi.org/10.1016/j.cgh.2005.10.019
Uchiyama K, Takagi T, Iwamoto Y, Kondo N, Okayama T, Yoshida N, Kamada K, Katada K, Handa O, Ishikawa T, Yasuda H, Sakagami J, Konishi H, Yagi N, Naito Y, Itoh Y (2014) New genetic biomarkers predicting azathioprine blood concentrations in combination therapy with 5-aminosalicylic acid. PLoS One 9(4):e95080. https://doi.org/10.1371/journal.pone.0095080
Skrzypczak-Zielinska M, Borun P, Bartkowiak-Kaczmarek A, Zakerska-Banaszak O, Walczak M, Dobrowolska A, Kurzawski M, Waszak M, Lipinski D, Plawski A, Slomski R (2016) A simple method for TPMT and ITPA genotyping using multiplex HRMA for patients treated with thiopurine drugs. Mol Diagn Ther 20(5):493–499. https://doi.org/10.1007/s40291-016-0217-0
Lee MN, Kang B, Choi SY, Kim MJ, Woo SY, Kim JW, Choe YH, Lee SY (2015) Impact of genetic polymorphisms on 6-thioguanine nucleotide levels and toxicity in pediatric patients with ibd treated with azathioprine. Inflamm Bowel Dis 21(12):2897–2908. https://doi.org/10.1097/MIB.0000000000000570
Krishnamurthy P, Schwab M, Takenaka K, Nachagari D, Morgan J, Leslie M, Du W, Boyd K, Cheok M, Nakauchi H, Marzolini C, Kim RB, Poonkuzhali B, Schuetz E, Evans W, Relling M, Schuetz JD (2008) Transporter-mediated protection against thiopurine-induced hematopoietic toxicity. Cancer Res 68(13):4983–4989. https://doi.org/10.1016/j.jsbmb.2011.07.002.Identification
Steinberg KK, Relling MV, Gallagher ML, Greene CN, Rubin CS, French D, Holmes AK, Carroll WL, Koontz DA, Sampson EJ, Satten GA (2007) Genetic studies of a cluster of acute lymphoblastic leukemia cases in Churchill County, Nevada. Environ Health Perspect 115(1):158–164. https://doi.org/10.1289/ehp.9025
Carroll MB, Smith DM, Shaak TL (2017) Genomic sequencing of uric acid metabolizing and clearing genes in relationship to xanthine oxidase inhibitor dose. Rheumatol Int 37(3):445–453. https://doi.org/10.1007/s00296-016-3592-2
Eklund BI, Moberg M, Bergquist J, Mannervik B (2006) Divergent activities of human glutathione transferases in the bioactivation of azathioprine. Mol Pharmacol 70(2):747–754. https://doi.org/10.1124/mol.106.025288
Zhang W, Moden O, Mannervik B (2010) Differences among allelic variants of human glutathione transferase A2-2 in the activation of azathioprine. Chem Biol Interact 186(2):110–117. https://doi.org/10.1016/j.cbi.2010.04.028
Stocco G, Pelin M, Franca R, De Iudicibus S, Cuzzoni E, Favretto D, Martelossi S, Ventura A, Decorti G (2014) Pharmacogenetics of azathioprine in inflammatory bowel disease: a role for glutathione-S-transferase? World J Gastroenterol 20(13):3534–3541. https://doi.org/10.3748/wjg.v20.i13.3534
Hollman AL, Tchounwou PB, Huang HC (2016) The association between gene-environment interactions and diseases involving the human GST superfamily with SNP variants. Int J Environ Res Public Health 13(4):379. https://doi.org/10.3390/ijerph13040379
Silva SN, Azevedo AP, Teixeira V, Pina JE, Rueff J, Gaspar JF (2009) The role of GSTA2 polymorphisms and haplotypes in breast cancer susceptibility: a case-control study in the Portuguese population. Oncol Rep 22(3):593–598. https://doi.org/10.3892/or_00000477
Sylvester RK, Steen P, Tate JM, Mehta M, Petrich RJ, Berg A, Kolesar J (2011) Temozolomide-induced severe myelosuppression: analysis of clinically associated polymorphisms in two patients. Anti-Cancer Drugs 22(1):104–110. https://doi.org/10.1097/CAD.0b013e3283407e9f
Andonova IE, Justenhoven C, Winter S, Hamann U, Baisch C, Rabstein S, Spickenheuer A, Harth V, Pesch B, Bruning T, Ko YD, Ganev V, Brauch H (2010) No evidence for glutathione S-transferases GSTA2, GSTM2, GSTO1, GSTO2, and GSTZ1 in breast cancer risk. Breast Cancer Res Treat 121(2):497–502. https://doi.org/10.1007/s10549-009-0589-5
Ke HL, Lin J, Ye Y, Wu WJ, Lin HH, Wei H, Huang M, Chang DW, Dinney CP, Wu X (2015) Genetic variations in glutathione pathway genes predict cancer recurrence in patients treated with transurethral resection and bacillus calmette-guerin instillation for non-muscle invasive bladder cancer. Ann Surg Oncol 22(12):4104–4110. https://doi.org/10.1245/s10434-015-4431-5
Mitrokhin V, Nikitin A, Brovkina O, Khodyrev D, Zotov A, Vachrushev N, Dragunov D, Shim A, Mladenov M, Kamkin A (2017) Association between interleukin-6/6R gene polymorphisms and coronary artery disease in Russian population: influence of interleukin-6/6R gene polymorphisms on inflammatory markers. J Inflamm Res 10:151–160. https://doi.org/10.2147/jir.S141682
Song GG, Choi SJ, Ji JD, Lee YH (2013) Genome-wide pathway analysis of a genome-wide association study on multiple sclerosis. Mol Biol Rep 40(3):2557–2564. https://doi.org/10.1007/s11033-012-2341-1
Roberts RL, Gearry RB (2007) IMPDH1 promoter mutations in a patient exhibiting azathioprine resistance. Pharmacogenomics J 7(5):312–317. https://doi.org/10.1038/sj.tpj.6500421
Zabala W, Cruz R, Barreiro-de Acosta M, Chaparro M, Panes J, Echarri A, Esteve M, Carpio D, Andreu M, García-Planella E, Domenech E, Carracedo A, Gisbert JP, Barros F, Eiga Eneida investigators (2013) New genetic associations in thiopurine-related bone marrow toxicity among inflammatory bowel disease patients. Pharmacogenomics 14(6):631–640
Zabala-Fernández W, Barreiro-de Acosta M, Echarri A, Carpio D, Lorenzo A, Castro J, Martínez-Ares D, Pereira S, Martin-Granizo I, Corton M, Carracedo A, Barros F (2011) A pharmacogenetics study of TPMT and ITPA genes detects a relationship with side effects and clinical response in patients with inflammatory bowel disease receiving azathioprine. J Gastrointestin Liver Dis 20(3):247–253. https://doi.org/10.1002/cncr.26110
Park SK, Hong M, Ye BD, Kim KJ, Park SH, Yang DH, Hwang SW, Kwak MS, Lee HS, Song K, Yang SK (2016) Influences of XDH genotype by gene-gene interactions with SUCLA2 for thiopurine-induced leukopenia in Korean patients with Crohn's disease. Scand J Gastroenterol 51(6):684–691. https://doi.org/10.3109/00365521.2015.1133698
Chang JY, Park SJ, Jung ES, Jung SA, Moon CM, Chun J, Park JJ, Kim ES, Park Y, Kim TI, Kim WH, Cheon JH (2019) Genotype-based treatment with thiopurine reduces incidence of myelosuppression in patients with inflammatory bowel diseases. Clin Gastroenterol Hepatol. https://doi.org/10.1016/j.cgh.2019.08.034
Anandi P, Dickson AL, Feng Q, Wei WQ, Dupont WD, Plummer D, Liu G, Octaria R, Barker KA, Kawai VK, Birdwell K, Cox NJ, Hung A, Stein CM, Chung CP (2020) Combining clinical and candidate gene data into a risk score for azathioprine-associated leukopenia in routine clinical practice. Pharmacogenomics J. https://doi.org/10.1038/s41397-020-0163-4
Payne K, Newman W, Fargher E, Tricker K, Bruce IN, Ollier WE (2007) TPMT testing in rheumatology: any better than routine monitoring? Rheumatology (Oxford) 46(5):727–729. https://doi.org/10.1093/rheumatology/kel427
van Gennep S, Konté K, Meijer B, Heymans MW, D'Haens GR, Löwenberg M, de Boer NKH (2019) Systematic review with meta-analysis: risk factors for thiopurine-induced leukopenia in IBD. Aliment Pharmacol Ther 50(5):484–506. https://doi.org/10.1111/apt.15403
Acknowledgments
The authors would like to thank Dr. C. Michael Stein for his review of the manuscript.
Funding
Supported by grant R01GM126535. Dr. Chung is also funded by the Veterans Health Administration Merit Award 1I01CX001741.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosures
None.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Laura L. Daniel and Alyson L. Dickson are considered co-first authors
Rights and permissions
About this article

Cite this article
Daniel, L.L., Dickson, A.L. & Chung, C.P. Precision medicine for rheumatologists: lessons from the pharmacogenomics of azathioprine. Clin Rheumatol 40, 65–73 (2021). https://doi.org/10.1007/s10067-020-05258-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10067-020-05258-2