
Abstract
Leukopenia is a serious, frequent side effect associated with azathioprine use. Currently, we use thiopurine methyltransferase (TPMT) testing to predict leukopenia in patients taking azathioprine. We hypothesized that a risk score incorporating additional clinical and genetic variables would improve the prediction of azathioprine-associated leukopenia. In the discovery phase, we developed four risk score models: (1) age, sex, and TPMT metabolizer status; (2) model 1 plus additional clinical variables; (3) sixty candidate single nucleotide polymorphisms; and (4) model 2 plus model 3. The area under the receiver-operating-characteristic curve (AUC) of the risk scores was 0.59 (95% CI: 0.54–0.64), 0.75 (0.71–0.80), 0.66 (0.61–0.71), and 0.78 (0.74–0.82) for models 1, 2, 3, and 4, respectively. During the replication phase, models 2 and 4 (AUC = 0.64, 95% CI: 0.59–0.70 and AUC = 0.63, 95% CI: 0.58–0.69, respectively) were significant in an independent group. Compared with TPMT testing alone, additional genetic and clinical variables improve the prediction of azathioprine-associated leukopenia.
Similar content being viewed by others
Introduction
Azathioprine is used to treat patients with autoimmune conditions [1], inflammatory bowel disease (IBD) [2, 3], and postorgan transplantation [4]. It is among the most widely prescribed immunosuppressants in the United States, with more than 2.4 million prescriptions filled in 2016, according to the U.S. Department of Health and Human Services [5, 6]. Azathioprine is considered an essential drug by the World Health Organization, which signals its critical role in addressing health care needs [7]. Unfortunately, approximately half of patients receiving azathioprine develop adverse drug reactions ranging from mild to severe, and many patients discontinue treatment due to serious side effects.
One of the most serious side effects associated with azathioprine use is bone marrow suppression, which manifests as leukopenia in 2.5–10% of azathioprine users. Leukopenia is dose-limiting and potentially life-threatening [8,9,10,11,12]. In current clinical practice, the tools for predicting azathioprine-associated leukopenia are limited. The standard of care to predict azathioprine-associated leukopenia is to evaluate the enzyme thiopurine methyltransferase (TPMT)—either by functional assays or by the identification of variants in the gene TPMT, which encodes it. However, these TPMT-based approaches can predict only one out of four cases of severe bone marrow toxicity [13, 14].
Leukopenia is dose-dependent, and azathioprine has a narrow therapeutic index [15]. Possible approaches to improve the prediction of azathioprine-associated leukopenia include accounting for the following: (1) medications that interact with azathioprine pharmacokinetically or pharmacodynamically; (2) comorbid conditions associated with leukopenia; and (3) genetic variants of enzymes involved in azathioprine metabolism [10].
We hypothesized that a risk score composed of multiple clinical factors and select candidate genes—in addition to TPMT—could improve the prediction of azathioprine-associated leukopenia. To test this hypothesis, we conducted a pilot study in a large clinical practice-based DNA biobank.
Methods
The study was conducted using BioVU, a clinical practice-based biobank at Vanderbilt University Medical Center. This biobank stores DNA samples linked to a de-identified version of a patient’s electronic health record (EHR), with all Health Insurance Portability and Accountability Act identifiers removed. BioVU’s enrollment and biobanking approaches have been described in detail previously [16, 17]. In summary, the biobank incorporates diagnostic and procedure codes, demographics, clinical care notes, patient history, problem lists, laboratory values, and medications from which researchers can extract phenotypes, such as disease diagnoses and treatment outcomes [18]. Using natural language processing, we identified 6625 potential azathioprine users in BioVU. We selected a convenience sample of 425 individuals for our discovery phase; of the remaining potential users, 441 individuals met the criteria for the replication phase.
The study was approved by the Vanderbilt University Medical Center Institutional Review Board.
Discovery phase
From the potential azathioprine users in BioVU, we reviewed charts to build a convenience sample designed for a case-control study with a one:one case:control ratio. We initially identified 750 actual users, 325 cases and 325 controls. As our primary outcome, we defined cases as individuals who developed leukopenia (i.e., leukocyte count of less than 4000 WBC/µL) while receiving azathioprine. Subjects taking azathioprine who did not develop leukopenia were classified as control subjects. We presumed a 90-day window of use for each confirmed prescription of azathioprine and only considered laboratory results within those windows.
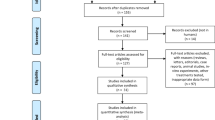
Based on the relatively small size of our sample for this pilot study, we further limited the study to Caucasians (as self-reported) in order to maintain enough power for our analysis. Patients were also required to be at least 18 years old. In order to minimize the effect on clinical care based on TPMT information, we then excluded patients with previous TPMT genotyping testing, as the results have the potential to impact azathioprine dosing. We further excluded patients with a medical history of myeloproliferative disorders or aplastic anemia for the potential impact of stem cell transplants on genetic testing; we also excluded patients with a white blood cell (WBC) count less than 4000 WBC/µL on the date of their first dose in our records to avoid associating existing leukopenia with azathioprine use falsely. Our final study group for the discovery phase included 425 patients, including 216 cases and 209 controls (Fig. 1).

Flowchart of study design: select of cases and controls in discovery and replication.
For these patients, we collected the following demographic and clinical covariates from the EHRs: age at the first azathioprine dose, sex, median weight, azathioprine dose at the time of the low WBC measure, indication for azathioprine, use of concurrent therapies that interact pharmacokinetically with azathioprine (i.e., febuxostat and allopurinol), and concurrent use of other immunosuppressants that have the potential to amplify the risk of leukopenia (i.e., cyclophosphamide, methotrexate, mycophenolate, tacrolimus, and leflunomide).
We selected 71 SNPs in 17 genes for genotyping. The majority of these genes were chosen based on a relevant literature review of prior reported adverse events associated with azathioprine or associations with the metabolic pathway of azathioprine metabolism. We included SNPs in genes TPMT, XDH, MOCOS, ABCC4, ITPA, AOX1, GST, IMPDH1, IL6, and HLADQA1-DRB1 based on previous clinical research [8, 9, 19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49]. We also genotyped an SNP in gene CXCL2, which is associated with benign leukopenia [50,51,52]. Finally, we included additional SNPs from genes TPMT, NUDT15, CMAHP, ST3GAL1, FBLN2, and NRXN3 identified during a preliminary interrogation of the existing genotypes in BioVU during the initial stages of this project. We performed genotyping using the Sequenom (Sequenom Inc., San Diego, CA, USA) and Taqman (Applied Biosystems Inc., Carlsbad, CA, USA) genotyping technologies. We subjected genotyping data to standard quality control (QC) procedures; these measures included removing SNPs with a call rate less than 95%, SNPs that deviated significantly from Hardy–Weinberg equilibrium (p < 0.001), and samples with missing data. From the initial list of 71 SNPs, we removed 5 SNPs from the analysis that did not meet QC standards and 6 that were in perfect linkage disequilibrium (r2 = 1) with 1 of the remaining 60 SNPs. Thus, a total of 60 SNPs were included in our analysis (Supplementary Table 1).
Replication phase
For the replication phase, we built a computer-based algorithm to aid the identification of likely azathioprine users from among the potential users in BioVU. This algorithm used medication and lab data from the EHRs to identify periods of current azathioprine use, defined as a 90-day exposure window following an indicated use of azathioprine, with at least one WBC count during the exposure window. We estimated the algorithm’s ability to predict azathioprine use through chart review of a statistically significant random sample of 134 potential azathioprine users. The algorithm correctly identified 53 of 65 actual users (positive predictive value of 81.54%) and 67 of 69 nonusers (negative predictive value of 97.10%) for an overall accuracy of 89.5% [53]. Among the 6200 individuals not in the final discovery sample, this algorithm identified 2792 additional likely users of azathioprine. For this phase, we limited the study to individuals who have Multi-Ethnic Genotyping Array (MEGA) genotype data available in BioVU and analyzed our SNPs of interest. MEGA is the most frequently used genotyping platform in BioVU, and we could include the largest number of individuals with existing genotyping data in the replication phase. This reduced our replication sample to 1110. We further restricted these patients using parallel guidelines to the discovery phase: self-identified Caucasians, at least 18 years old, without previous TPMT testing, and without leukopenia at their first dose. Similar to the discovery phase, we performed chart review to confirm azathioprine use and extracted relevant clinical variables from the EHRs. Our final replication group included 441 individuals, and they were classified as cases (n = 144) or controls (n = 297) based on the same standard as the discovery phase (Fig. 1). We applied standard QC measures for genome-wide association studies [54] for the results of their MEGA genotyping data, and then used the Michigan imputation for the 1000 Genomes Project [55] to impute genotypes for 10 SNPs included in our analysis that are not available on the MEGA chip. To verify our imputation for the replication phase, we imputed the same SNPs for 265 patients included in the discovery phase, who also had MEGA genotyping available, and we then compared those results to the Sequenom and Taqman testing. Nine of ten were 100% consistent, and the tenth was 99.62% consistent. Results are included in Supplementary Table 1.
TPMT metabolizer status
We determined TPMT metabolizer status using three variant alleles in the TPMT genotype: rs1142345, rs1800460, and rs1800462. We did not include rs1800584 because it was rare (<1% minor allele frequency) among persons of European ancestry—the predominant ancestry of our dataset—according to the HapMap Project and Genome Aggregation Database [56]. In order to replicate the standard of care for patients who receive an azathioprine prescription, we applied the dosing recommendation guidelines provided by the Clinical Pharmacogenetics Implementation Consortium to define TPMT metabolizer status: normal (homozygous wild type); intermediate (heterozygous); and low (homozygous variant) [57]. For the purposes of our analysis, we followed a prespecified classification of intermediate and low metabolizers together.
Statistical analyses
We present demographic and clinical characteristics data as number and percentages for categorical variables and as median and interquartile range (IQR) for continuous variables. We used Fisher’s exact tests to compare binary categorical variables, chi-squared tests to compare polytomous categorical variables, and WilWe next completedcoxon’s rank sum tests to compare continuous variables.
We completed the analysis in two phases. In phase one, we developed risk scores for leukopenia. We developed four models and then used the beta coefficients generated from each model to build weighted risk scores for azathioprine-associated leukopenia for each of the four models. Model 1 used multivariate logistic regression, including age, sex, and TPMT metabolizer status. Model 2 also used multivariate logistic regression and incorporated age, sex, weight, indication, azathioprine dose at low WBC, the concurrent use of xanthine oxidase inhibitors, the concurrent use of other immunosuppressants, and TPMT metabolizer status. Model 3 included candidate gene data. Given that we had multiple SNPs in close proximity and in order to minimize overfitting and alleviate collinearity, we used ridge regression for this model, which employs a standard cross-validated penalty to shrink the coefficients of the covariates. More specifically, we estimated lambda.min, which was the regression penalty associated with the minimum mean cross-validated error, and lambda.1se, which was the largest penalty whose estimated error was within one standard error of this minimum; we then used the regression coefficient estimates associated with lambda.1se to generate the model 3 risk score. Model 4 included age, sex, weight, indication, azathioprine dose at low WBC, the concurrent use of xanthine oxidase inhibitors, and the concurrent use of other immunosuppressants, TPMT metabolizer status, and all candidate gene data. Given the possibility of overfitting and collinearity, as with model 3, we used ridge regression to estimate the beta coefficients for the fourth model risk score.
We next completed receiver-operating-characteristic (ROC) curve analysis to examine whether the risk score could discriminate between those patients who developed azathioprine-associated leukopenia versus those who did not. Results are reported as area under the curve (AUC). We further evaluated model 4 using restricted cubic splines and simple logistic regression, comparing Akaike information criterion (AIC) values. The best-fitted model (with the lowest AIC) was the simple regression, which we used to show the expected proportion of patients who develop leukopenia based on standardized model 4 risk scores.
As a sensitivity analysis, we developed scores and estimated AUCs for the secondary outcome of leukopenia defined as a leukocyte count of less than or equal to 3000 WBC/µL. We calculated results in two ways: (1) including and (2) excluding individuals who had a low leukocyte count between 3000 WBC/µL and 4000 WBC/µL as controls.
In phase two, we used the coefficients estimated from the discovery phase to calculate the risk scores for leukopenia in the replication phase. We then used those scores to calculate their respective AUCs. In addition, we combined the data from both phases and calculated the scores and associated AUCs for each of the four models.
All analyses were conducted using STATA version 16.0 (College Station, TX) or R version 3.4.3 for Windows. Codes will be available to academic researchers upon request.
Results
Table 1 shows the clinical characteristics of the patients included in the discovery and replication phases. In the discovery group, patients who developed leukopenia had statistically significant younger age at the first dose, lower median weight, diagnoses of systemic lupus erythematosus, organ transplants, and concurrent use of immunosuppressants compared with patients who did not develop leukopenia. In the replication group, leukopenia was associated with male sex, organ transplants, and the concurrent use of immunosuppressants.
Risk scores for azathioprine-associated leukopenia
The discriminant capacity of the risk scores for azathioprine-associated leukopenia derived from model 1 (including TPMT metabolizer status, age, and sex) was statistically significant in the discovery phase (AUC = 0.59, 95% CI: 0.54–0.64), but the score was not replicated in a second, independent group (Table 2). Likewise, the AUC for model 3 (candidate SNPs only) was significant in the discovery phase (AUC = 0.66, 95% CI: 0.61–0.71), but not significant in the replication phase. In contrast, the second and fourth models outperformed the first and third models. For the risk scores derived from model 2 (including TPMT metabolizer status, age, sex, weight, indication, azathioprine dose at low WBC, the concurrent use of xanthine oxidase inhibitors, and the concurrent use of other immunosuppressants), the ROC AUC was 0.75 (95% CI: 0.71–0.80) in the discovery phase and 0.64 (95% CI: 0.59–0.70) in the replication phase. The odds ratios generated from the multivariate logistic regression of model 2 in the discovery phase are reported in Supplementary Table 2 and are consistent with expected outcomes. For example, the odds ratio for low/intermediate TPMT metabolizer status in the discovery phase was 2.27 (95% CI: 1.04–4.95, p = 0.04).
Similarly surpassing the first model, the ROC AUC for the risk scores derived from model 4 (including TPMT metabolizer status, age, sex, weight, indication, azathioprine dose at low WBC, the concurrent use of xanthine oxidase inhibitors, the concurrent use of other immunosuppressants, and candidate SNPs) was 0.78 (95% CI: 0.74–0.82) in the discovery phase and 0.63 (95% CI: 0.58–0.69) in the replication phase.
Among patients with azathioprine-associated leukopenia in the discovery phase, the risk scores for model 4 (including all genetic and clinical data) ranged from −0.67 to 0.44 with a median (IQR) score of −0.03 (−0.18 to 0.11) among cases and −0.26 (−0.34 to −0.11) among control subjects. Supplementary Table 3 includes the beta coefficients derived from the discovery phase and applied in the replication phase for each variable in model 4. For accessibility, we then standardized the scores to a scale of 0–100 and then grouped the scores into deciles for the discovery phase. We applied the score ranges from the discovery phase to group the scores in the replication phase. Figure 2 shows the proportion of patients who developed leukopenia for each of these groups in the discovery and replication phases. We have also included the score ranges for each decile, along with the frequency of individuals in decile for both the discovery and replication phases. In order to analyze how well the score could predict the proportion of patients developing azathioprine-associated leukopenia in each score group, we used simple regression to determine expected proportions of patients developing leukopenia based on risk score. Figure 3 shows the expected (red line) proportion of patients developing leukopenia relative to risk score and observed (blue dot) proportion at the median risk score in each decile for the discovery and replication phases. Our results indicate that the risk scores for both models 2 and 4 have discriminating capacity for azathioprine-associated leukopenia (i.e., the percentage of individuals developing azathioprine-associated leukopenia increases in the higher score deciles). In a test of equality between the AUCs generated by models 2 and 4, the p value was not significant (p = 0.088).

Proportion of leukopenia cases per standardized risk score group.

Score performance in predicting azathioprine-associated leukopenia.
Sensitivity analyses
A total of 107 patients developed azathioprine-associated leukopenia defined as leukocytes less than 3000 WBC/µL. When including patients with a low leukocyte count between 3000 and 4000 WBC/µL among controls, in the discovery phase, the ROC AUC for the model 1 risk scores (including TPMT metabolizer status, age, and sex) was 0.55 (95% CI: 0.49–0.62); for the model 2 risk scores (including TPMT metabolizer status and all clinical variables) was 0.76 (95% CI: 0.70–0.81); for the model 3 risk scores (candidate gene data) was 0.68 (95% CI: 0.62–0.74); and for the model 4 risk scores (including all clinical and genetic variables) was 0.79 (0.74–0.84). Results for models 2 and 4 were significant in the replication phase: AUCs of 0.70 (95% CI: 0.63–0.78) and 0.71 (0.64–0.77), respectively (Table 2, Sensitivity analysis 1). Results were similar, if slightly improved, when patients with a low leukocyte count were excluded from controls (Table 2, Sensitivity analysis 2). We also completed an analysis of our primary outcome (WBC < 4000/µL) with data from the two phases combined. As anticipated, the AUCs of the four models were significant and fell between our results in the discovery and replication phases: 0.55 (95% CI: 0.51–0.59), 0.70 (0.69–0.74), 0.60 (0.56–0.64), and 0.72 (0.69–0.76) for models 1, 2, 3, and 4, respectively (Table 2, Combined analysis).
Discussion
To the best of our knowledge this is the first study examining the combined role of clinical variables and genetic variants in models built with data from a wide range of patients, including those with different azathioprine indications. Our results indicate that, compared with TPMT metabolizer status (adjusted for age and sex) alone, a risk score that also incorporates additional important clinical variables, and candidate gene data improve the ability to discriminate those who will present with azathioprine-associated leukopenia and those who will not in routine clinical practice.
While TPMT function and the TPMT genotype are well-established predictors of azathioprine-associated leukopenia, they do not predict the majority of cases of leukopenia [57]. Consequently, adding prespecified clinical variables and a more comprehensive list of candidate genetic variants into a risk score was the logical next step for improving the capacity to discriminate azathioprine-associated leukopenia. There are many methods for building risk scores. We chose an approach that combined clinical and genetic candidates that could account for as much variability as possible. Models including more data (e.g., clinical and genetic risk factors) may help us to clarify the function of common risk loci identified as possible contributors to azathioprine-associated leukopenia, while simultaneously accounting for classical risk factors and allowing for stratification based on clinical data [58]. Indeed, these inclusive models performed better than the current standard of care in our datasets. The use of a penalized regression was intended to limit overfitting and avoid collinearity as the number of variables grew.
While the prespecified primary outcome for this study was leukopenia defined as WBC < 4000/µL, the same set of variables also performed well for our secondary outcome—namely WBC < 3000/µL. This is encouraging for two reasons. First, consistent with the notion that the variables play a mechanistic role in the development of the phenotype, it is reassuring that models incorporating a combination of the same variables improved their performance with a more extreme phenotype. Second, although WBC < 4000/µL is a well-defined cut-off for leukopenia, WBC < 3000/µL is considered more clinically important and actionable.
Prior studies examining leukopenia associated with the use of azathioprine have focused on patients with a specific drug indication, such as IBD or vasculitis [55, 59]. Our approach of including different azathioprine indications, different medication doses, and multiple variables—such as co-administration of medications that interact with azathioprine through pharmacokinetic or pharmacodynamic mechanisms—aimed to improve the generalizability of the score as a potential clinical tool. Nevertheless, many steps are required prior to clinical implementation.
The study was constrained in several ways. First, although we made our best effort in data collection, missing data that were not recorded in the EHRs, lack of information regarding adherence, or use of over-the-counter medications not included in those records could potentially bias the results. Second, we acknowledge that the genetic data contributed less to the improved risk score than clinical data. In a test of equality between the AUCs generated by models 2 and 4, the p value was not significant (p = 0.088). This outcome may be the result of several factors: the number of genetic variants was small; the effect size of individual SNPs was limited; and/or some of our selections may have been suboptimal. We look forward to larger studies with the opportunity the select additional candidate genes and utilize additional unbiased analyses. Third, although we used penalized regressions to overcome overfitting for the more complex model, the sample size was relatively small. The lower AUCs in the replication phase indicate that further refinement and a larger sample size may improve the risk scores’ performance. Fourth, we conducted a replication phase within BioVU from individuals selected for previous genotyping for other studies, which may have influenced the odds ratio for low/intermediate TPMT metabolizer status in the replication phase; thus, an external replication cohort will be desirable. Fifth, this pilot study was limited to Caucasians in order to provide enough power for analysis. We anticipate that risk scores may differ based on ethnicity, and we look forward to larger studies with sufficient power to account for the impact of race in a risk score(s). Lastly, we used a preselected candidate list. Our candidate list was generated prior to the study of the NUDT15 variant rs746071566 reported in Caucasians (minor allele frequency of ~0.3% among Europeans per the 1000 Genomes Project and Genome Aggregation Database) [60,61,62]. Moreover, GWAS analysis may identify alternative SNPs or combinations of SNPs that contribute more to the predictive capacity of the risk score.
Despite these limitations, this study is a proof-of-concept that models with additional variables could perform better than the current standard of care—limited to TPMT function/TPMT variants—in predicting leukopenia associated with the use of azathioprine. Our results clearly demonstrate that increased attention to clinical variables enhance prediction, as evidenced by the increases in the predictive capacity of the risk score for the discovery cohort. Larger cohorts and the use of rich genotype data may further refine the predictive value of complex models.
References
Bertsias G, Ioannidis JP, Boletis J, Bombardieri S, Cervera R, Dostal C, et al. EULAR recommendations for the management of systemic lupus erythematosus. Report of a task force of the EULAR Standing Committee for International Clinical Studies Including Therapeutics. Ann Rheum Dis. 2008;67:195–205.
Rispo A, Testa A, De Palma GD, Donetto S, Diaferia M, Musto D, et al. Different profile of efficacy of thiopurines in ulcerative colitis and Crohn’s disease. Inflamm Bowel Dis. 2015;21:2570–5.
Terdiman JP, Gruss CB, Heidelbaugh JJ, Sultan S, Falck-Ytter YT.AGA Institute Clinical Practice Quality Management Committee American Gastroenterological Association Institute guideline on the use of thiopurines, methotrexate, and anti-TNF-alpha biologic drugs for the induction and maintenance of remission in inflammatory Crohn’s disease. Gastroenterology. 2013;145:1459–63.
van Gelder T, van Schaik RH, Hesselink DA. Pharmacogenetics and immunosuppressive drugs in solid organ transplantation. Nat Rev Nephrol. 2014;10:725–31.
Kane SP. Azathioprine, ClinCalc DrugStats Database, Version 20.0. ClinCalc. 2019. https://clincalc.com/DrugStats/Drugs/Azathioprine. Accessed 10 Feb 2020.
Medical Expenditure Panel Survey (MEPS). Rockville, MD: Agency for Healthcare Research and Quality. 2018. https://www.ahrq.gov/data/meps.html. Accessed 10 Feb 2020.
World Health Organization. WHO model list of essential medicines 2017. 2017. https://www.who.int/medicines/publications/essentialmedicines/en/.
Zabala W, Cruz R, Barreiro-de Acosta M, M C, Panes J, Echarri A, et al. New genetic associations in thiopurine-related bone marrow toxicity among inflammatory bowel disease patients. Pharmacogenomics. 2013;14:631–40.
Roberts RL, Gearry RB. IMPDH1 promoter mutations in a patient exhibiting azathioprine resistance. Pharmacogenom J. 2007;7:312–7.
Matimba A, Li F, Livshits A, Cartwright CS, Scully S, Fridley BL, et al. Thiopurine pharmacogenomics: association of SNPs with clinical response and functional validation of candidate genes. Pharmacogenomics. 2014;15:433–47.
Shih DQ, Nguyen M, Zheng L, Ibanez P, Mei L, Kwan LY, et al. Split-dose administration of thiopurine drugs: a novel and effective strategy for managing preferential 6-MMP metabolism. Aliment Pharmacol Ther. 2012;36:449–58.
Toruner M, Loftus EV Jr., Harmsen WS, Zinsmeister AR, Orenstein R, Sandborn WJ, et al. Risk factors for opportunistic infections in patients with inflammatory bowel disease. Gastroenterology. 2008;134:929–36.
Roberts RL, Barclay ML. Update on thiopurine pharmacogenetics in inflammatory bowel disease. Pharmacogenomics. 2015;16:891–903.
Broekman M, Coenen MJH, Wanten GJ, van Marrewijk CJ, Klungel OH, Verbeek ALM, et al. Risk factors for thiopurine-induced myelosuppression and infections in inflammatory bowel disease patients with a normal TPMT genotype. Aliment Pharmacol Ther. 2017;46:953–63.
Broekman M, Coenen MJH, van Marrewijk CJ, Wanten GJA, Wong DR, Verbeek ALM, et al. More dose-dependent side effects with mercaptopurine over azathioprine in ibd treatment due to relatively higher dosing. Inflamm Bowel Dis. 2017;23:1873–81.
Pulley J, Clayton E, Bernard GR, Roden DM, Masys DR. Principles of human subjects protections applied in an opt-out, de-identified biobank. Clin Transl Sci. 2010;3:42–8.
Roden DM, Pulley JM, Basford MA, Bernard GR, Clayton EW, Balser JR, et al. Development of a large-scale de-identified DNA biobank to enable personalized medicine. Clin Pharmacol Ther. 2008;84:362–9.
Wei WQ, Denny JC. Extracting research-quality phenotypes from electronic health records to support precision medicine. Genome Med. 2015;7:41.
Yang SK, Hong M, Baek J, Choi H, Zhao W, Jung Y, et al. A common missense variant in NUDT15 confers susceptibility to thiopurine-induced leukopenia. Nat Genet. 2014;46:1017–20.
Varnell CD, Fukuda T, Kirby CL, Martin LJ, Warshaw BL, Patel HP, et al. Mycophenolate mofetil-related leukopenia in children and young adults following kidney transplantation: Influence of genes and drugs. Pediatr Transplant. 2017;21:e13033. https://doi.org/10.1111/petr.13033.
Kurzawski M, Dziewanowski K, Lener A, Drozdzik M. TPMT but not ITPA gene polymorphism influences the risk of azathioprine intolerance in renal transplant recipients. Eur J Clin Pharmacol. 2009;65:533–40.
Honda K, Kobayashi A, Niikura T, Hasegawa T, Saito Z, Ito S, et al. Neutropenia related to an azathioprine metabolic disorder induced by an inosine triphosphate pyrophosphohydrolase (ITPA) gene mutation in a patient with PR3-ANCA-positive microscopic polyangiitis. Clin Nephrol. 2018;90:363–9.
Ban H, Andoh A, Imaeda H, Kobori A, Bamba S, Tsujikawa T, et al. The multidrug-resistance protein 4 polymorphism is a new factor accounting for thiopurine sensitivity in Japanese patients with inflammatory bowel disease. J Gastroenterol. 2010;45:1014–21.
Dubois PCA. The risk of azathioprine-induced pancreatitis depends on genetic variants in the HLA gene region. Gut 2011;60:A60.
Abla N, Chinn LW, Nakamura T, Liu L, Huang CC, Johns SJ, et al. The human multidrug resistance protein 4 (MRP4, ABCC4): functional analysis of a highly polymorphic gene. J Pharmacol Exp Ther. 2008;325:859–68.
Hawwa AF, Millership JS, Collier PS, Vandenbroeck K, McCarthy A, Dempsey S, et al. Pharmacogenomic studies of the anticancer and immunosuppressive thiopurines mercaptopurine and azathioprine. Br J Clin Pharmacol. 2008;66:517–28.
Chiabai MA, Lins TC, Pogue R, Pereira RW. Population analysis of pharmacogenetic polymorphisms related to acute lymphoblastic leukemia drug treatment. Dis Markers. 2012;32:247–53.
Zelinkova Z, Derijks LJ, Stokkers PC, Vogels EW, van Kampen AH, Curvers WL, et al. Inosine triphosphate pyrophosphatase and thiopurine s-methyltransferase genotypes relationship to azathioprine-induced myelosuppression. Clin Gastroenterol Hepatol. 2006;4:44–9.
Uchiyama K, Takagi T, Iwamoto Y, Kondo N, Okayama T, Yoshida N, et al. New genetic biomarkers predicting azathioprine blood concentrations in combination therapy with 5-aminosalicylic acid. PLoS ONE. 2014;9:e95080.
Skrzypczak-Zielinska M, Borun P, Bartkowiak-Kaczmarek A, Zakerska-Banaszak O, Walczak M, Dobrowolska A, et al. A simple method for TPMT and ITPA genotyping using multiplex HRMA for patients treated with thiopurine drugs. Mol Diagn Ther. 2016;20:493–9.
Kurzawski M, Dziewanowski K, Safranow K, Drozdzik M. Polymorphism of genes involved in purine metabolism (XDH, AOX1, MOCOS) in kidney transplant recipients receiving azathioprine. Ther Drug Monit. 2012;34:266–74.
Lee MN, Kang B, Choi SY, Kim MJ, Woo SY, Kim JW, et al. Impact of genetic polymorphisms on 6-thioguanine nucleotide levels and toxicity in pediatric patients with IBD treated with azathioprine. Inflamm Bowel Dis. 2015;21:2897–908.
Krishnamurthy P, Schwab M, Takenaka K, Nachagari D, Morgan J, Leslie M, et al. Transporter-mediated protection against thiopurine-induced hematopoietic toxicity. Cancer Res. 2008;68:4983–9.
Steinberg KK, Relling MV, Gallagher ML, Greene CN, Rubin CS, French D, et al. Genetic studies of a cluster of acute lymphoblastic leukemia cases in Churchill County, Nevada. Environ Health Perspect. 2007;115:158–64.
Carroll MB, Smith DM, Shaak TL. Genomic sequencing of uric acid metabolizing and clearing genes in relationship to xanthine oxidase inhibitor dose. Rheumatol Int. 2017;37:445–53.
Eklund BI, Moberg M, Bergquist J, Mannervik B. Divergent activities of human glutathione transferases in the bioactivation of azathioprine. Mol Pharmacol. 2006;70:747–54.
Zhang W, Moden O, Mannervik B. Differences among allelic variants of human glutathione transferase A2-2 in the activation of azathioprine. Chem Biol Interact. 2010;186:110–7.
Stocco G, Pelin M, Franca R, De Iudicibus S, Cuzzoni E, Favretto D, et al. Pharmacogenetics of azathioprine in inflammatory bowel disease: a role for glutathione-S-transferase? World J Gastroenterol. 2014;20:3534–41.
Hollman AL, Tchounwou PB, Huang HC. The association between gene-environment interactions and diseases involving the human GST superfamily with SNP variants. Int J Environ Res Public Health. 2016;13:379.
Silva SN, Azevedo AP, Teixeira V, Pina JE, Rueff J, Gaspar JF. The role of GSTA2 polymorphisms and haplotypes in breast cancer susceptibility: a case-control study in the Portuguese population. Oncol Rep. 2009;22:593–8.
Sylvester RK, Steen P, Tate JM, Mehta M, Petrich RJ, Berg A, et al. Temozolomide-induced severe myelosuppression: analysis of clinically associated polymorphisms in two patients. Anticancer Drugs. 2011;22:104–10.
Andonova IE, Justenhoven C, Winter S, Hamann U, Baisch C, Rabstein S, et al. No evidence for glutathione S-transferases GSTA2, GSTM2, GSTO1, GSTO2, and GSTZ1 in breast cancer risk. Breast Cancer Res Treat. 2010;121:497–502.
Ke HL, Lin J, Ye Y, Wu WJ, Lin HH, Wei H, et al. Genetic variations in glutathione pathway genes predict cancer recurrence in patients treated with transurethral resection and Bacillus Calmette-Guerin instillation for nonmuscle invasive bladder cancer. Ann Surg Oncol. 2015;22:4104–10.
Mitrokhin V, Nikitin A, Brovkina O, Khodyrev D, Zotov A, Vachrushev N, et al. Association between interleukin-6/6R gene polymorphisms and coronary artery disease in Russian population: influence of interleukin-6/6R gene polymorphisms on inflammatory markers. J Inflamm Res. 2017;10:151–60.
Song GG, Choi SJ, Ji JD, Lee YH. Genome-wide pathway analysis of a genome-wide association study on multiple sclerosis. Mol Biol Rep. 2013;40:2557–64.
Heap GA, Weedon MN, Bewshea CM, Singh A, Chen M, Satchwell JB, et al. HLA-DQA1-HLA-DRB1 variants confer susceptibility to pancreatitis induced by thiopurine immunosuppressants. Nat Genet. 2014;46:1131–4.
Zabala-Fernández W, Barreiro-de Acosta M, Echarri A, Carpio D, Lorenzo A, Castro J, et al. A pharmacogenetics study of TPMT and ITPA genes detects a relationship with side effects and clinical response in patients with inflammatory bowel disease receiving azathioprine. J Gastrointest Liver Dis. 2011;20:247–53.
Park SK, Hong M, Ye BD, Kim KJ, Park SH, Yang DH, et al. Influences of XDH genotype by gene-gene interactions with SUCLA2 for thiopurine-induced leukopenia in Korean patients with Crohn’s disease. Scand J Gastroenterol. 2016;51:684–91.
Moriyama T, Nishii R, Perez-Andreu V, Yang W, Klussmann FA, Zhao X, et al. NUDT15 polymorphisms alter thiopurine metabolism and hematopoietic toxicity. Nat Genet. 2016;48:367–73.
Reiner AP, Lettre G, Nalls MA, Ganesh SK, Mathias R, Austin MA, et al. Genome-wide association study of white blood cell count in 16,388 African Americans: the continental origins and genetic epidemiology network (COGENT). PLoS Genet. 2011;7:e1002108.
Ibanez L, Velli PS, Font R, Jaen A, Royo J, Irigoyen D, et al. HIV-infection, atherosclerosis and the inflammatory pathway: candidate gene study in a Spanish HIV-infected population. PLoS ONE. 2014;9:e112279.
Schaefer BA, Flanagan JM, Alvarez OA, Nelson SC, Aygun B, Nottage KA, et al. Genetic modifiers of white blood cell count, albuminuria and glomerular filtration rate in children with sickle cell anemia. PLoS ONE. 2016;11:e0164364.
Dickson A, Stanley B, Anandi P, Kawai V, Birdwell K, Hung A, et al. Development and validation of algorithms using electronic health records to identify azathioprine use and azathioprine-induced leukopenia for pharmacogenetic studies [abstract]. San Diego, CA: American Society of Human Genetics; 2018.
Turner S, Armstrong L, Bradford Y, Carlson C, Crawford D, Crenshaw A, et al. Quality control procedures for genome-wide association studies. Curr Protoc Hum Genet. 2011;1.19.1–1.19.18.
Das S, Forer L, Schonherr S, Sidore C, Locke AE, Kwong A, et al. Next-generation genotype imputation service and methods. Nat Genet. 2016;48:1284–7.
rs1800584 Frequency (updated 1/8/2019). https://useast.ensembl.org/Homo_sapiens/Variation/Population?db=core;r=6:18130281-18131281;v=rs1800584;vdb=variation;vf=104059606.
Relling MV, Gardner EE, Sandborn WJ, Schmiegelow K, Pui CH, Yee SW, et al. Clinical Pharmacogenetics Implementation Consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing. Clin Pharmacol Ther. 2011;89:387–91.
Jostins L, Barrett J. Genetic risk prediction in complex disease. Hum Mol Genet. 2011;20:R182–8.
Hessels AC, Rutgers A, Sanders JSF, Stegeman CA. Thiopurine methyltransferase genotype and activity cannot predict outcomes of azathioprine maintenance therapy for antineutrophil cytoplasmic antibody associated vasculitis: a retrospective cohort study. PloS ONE. 2018;13:e0195524–e.
Walker G, Harrison J, Heap G, Voskuil M, Andersen V, Anderson C, et al. Association of genetic variants in NUDT15 with thiopurine-induced myelosuppression in patients with inflammatory bowel disease. JAMA. 2019;321:773–85.
Project G. rs746071566 Frequency. 2019. https://www.ncbi.nlm.nih.gov/snp/rs746071566#frequency_tab.
Ensembl. rs746071566 Frequency. 2019. https://useast.ensembl.org/Homo_sapiens/Variation/Population?db=core;r=13:48037283-48038288;v=rs746071566;vdb=variation;vf=45794599.
Acknowledgements
This work is supported by grant R01GM126535 R01GM109145, R35GM131770, and by the Rheumatology Research Foundation K-supplement and R01 bridge awards. PA was supported by grants T32AR059039 and T32GM007569. VKK was supported by NIH/NIGMS K23GM117395 and the National Arthritis Research Foundation—All Arthritis Grant Program Award. CPC was also supported by grant AR073764. The dataset(s) used for the analyses described were obtained from Vanderbilt University Medical Center’s BioVU which is supported by numerous sources: institutional funding, private agencies, and federal grants. These include the NIH funded Shared Instrumentation Grant S10RR025141; and CTSA grants UL1TR002243, UL1TR000445, and UL1RR024975. Genomic data are also supported by investigator-led projects that include U01HG004798, R01NS032830, RC2GM092618, P50GM115305, U01HG006378, U19HL065962, R01HD074711; and additional funding sources listed at https://victr.vumc.org/biovu/index.html?sid=229. The funding sources had no role in the collection, analysis, or interpretation of data, writing of the paper, or decision to submit for publication.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Anandi, P., Dickson, A.L., Feng, Q. et al. Combining clinical and candidate gene data into a risk score for azathioprine-associated leukopenia in routine clinical practice. Pharmacogenomics J 20, 736–745 (2020). https://doi.org/10.1038/s41397-020-0163-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41397-020-0163-4
- Springer Nature Limited
We’re sorry, something doesn't seem to be working properly.
Please try refreshing the page. If that doesn't work, please contact support so we can address the problem.