
Abstract
A novel low-temperature Escherichia coli phage vB_EcoS_NBD2 was isolated in Lithuania from agricultural soil. With an optimum temperature for plating around 20 °C, vB_EcoS_NBD2 efficiently produced plaques on Escherichia coli NovaBlue (DE3) at a temperature range of 10–30 °C, yet failed to plate at temperatures above 35 °C. Phage vB_EcoS_NBD2 virions have a siphoviral morphology with an isometric head (65 nm in diameter), and a non-contractile flexible tail (170 nm). The 51,802-bp genome of vB_EcoS_NBD2 has a G + C content of 49.8%, and contains 87 probable protein-encoding genes as well as 1 gene for tRNASer. Comparative sequence analysis revealed that 22 vB_EcoS_NBD2 ORFs encode unique proteins that have no reliable identity to database entries. Based on homology to biologically defined proteins and/or proteomics analysis, 36 vB_EcoS_NBD2 ORFs were given a putative functional annotation, including 20 genes coding for morphogenesis-related proteins and 13 associated with DNA metabolism. Phylogenetic analysis revealed that vB_EcoS_NBD2 belongs to the subfamily Tunavirinae, but cannot be assigned to any genus currently recognized by ICTV.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Ever since their discovery in the early twentieth century, bacteriophages (phages) have attracted the interest of scientists from many fields. From the identification of DNA as the genetic material to the discovery of CRISPR-Cas, phage research has had an enormous impact on our understanding of core biological processes and has provided many reagents and techniques routinely used in molecular biology [1,2,3]. Moreover, the rise of antibiotic-resistant bacteria has led to a renewed interest in phage-based antimicrobials [4]. Based on this, one may conclude that bacterial viruses are very well researched. However, recent evidence suggests that phages represent the most numerous and biologically diverse viral population, of which we have only a very limited understanding [5, 6].
Approximately 95% of known phages are double-stranded DNA (dsDNA)-containing tailed viruses that comprise the order Caudovirales, which is divided into three families: Podoviridae (phages with short tails), Siphoviridae (those with long noncontractile tails), and Myoviridae (phages with contractile tails) [7, 8]. More than three decades ago, three physiological types of phages were recognized: high-temperature (HT) phages plating at or above 25 °C, low-temperature (LT) phages, plating at or below 30 °C, and mid-temperature (MT) phages, plating in the range of 15 to 42 °C [9]. The vast majority of characterized coliphages, those that are active against the enterobacterium Escherichia coli, are MT phages [5, 10]. Coliphages belonging to the MT group have an optimum temperature for plating about 37-40 °C, which correlates closely with the optimum growth temperature of the host [11]. The ability of coliphages to lyse their hosts at low temperatures is affected at various stages of phage infection. In the case of phage T4, a typical representative of MT coliphages, certain steps in virion assembly are inhibited even at 19 °C, leading to an accumulation of capsids, preheads, partially sheathed tails, and naked cores [12]. In E. coli phage λ, at temperatures below 22 °C, the DNA injection process is ineffective [13]. Therefore, unsurprisingly, only seven E. coli-specific phages, all belonging to the family Myoviridae, have been assigned to the LT group to date [5, 14].
In this paper, we present the molecular characterization of E. coli-specific siphovirus vB_EcoS_NBD2 (subsequently referred to by its shorter common laboratory name, NBD2), which shows a low-temperature plating profile and has an optimum temperature for plating of ~20 °C.
MATERIALS AND METHODS
Phage techniques
Bacteriophage NBD2 was isolated from agricultural soil (strawberry patch, Alytus, Lithuania), using E. coli strain NovaBlue (DE3) (Novagen) as the host for phage propagation. Phage isolation was carried out as described previously [15]. Since NBD2 failed to reproduce after inoculation into liquid bacterial culture, a modified soft agar overlay method was employed for phage multiplication. Briefly, after the addition of 5 ml of LB broth to the surface of the plate, the top agar was scraped off and the suspension recovered. After 30 min of incubation at 4 °C with mild stirring, the mixture was then centrifuged at 6000 rpm for 15 min. The phage-containing supernatant was decanted and the phage was concentrated by high-speed centrifugation at 30000 rpm for 3 h. The resulting pellets were suspended in PB buffer (70 mM NaCl, 10 mM MgSO4, 50 mM Na2HPO4, 30 mM KH2PO4). To avoid bacterial DNA contamination, DNase I was added to the phage suspension, and the sample was incubated 1 h at 37 °C. Further purification was performed using a CsCl step gradient as described in [15]. For all phage experiments, bacteria were cultivated in Luria-Bertani broth (LB) or LB agar. Determination of the efficiency of plating (e.o.p.) was performed as described by Seeley and Primrose [9]. Briefly, high-titer phage stocks were diluted and plated in duplicate. Plates incubated at 15-36 °C were read after 24 h, while those incubated at 10 °C were read after 48 h. The temperature at which the largest number of plaques formed was taken as the standard for the e.o.p. calculation.
The host range of NBD2 was investigated using a quantitative spot dilution test as described in [8]; the list of E. coli strains used is presented in Table 1.
Viral DNA isolation and restriction analysis
For the isolation of phage DNA, aliquots of phage suspension (1011–1012 PFU/ml) were subjected to phenol/chloroform extraction and ethanol precipitation as described by Carlson and Miller [16]. Isolated phage DNA was subsequently subjected to genome sequencing and restriction digestion analysis.
Restriction digestion was performed with EcoRI, ScaI, and Bsp14071 restriction endonucleases (Thermo Fisher Scientific) according to the supplier’s recommendations. DNA fragments were separated by electrophoresis (7V/cm) in a 0.8% agarose gel containing ethidium bromide. Restriction digestion was repeated at least three times to confirm the results.
Transmission electron microscopy (TEM)
CsCl density gradient-purified phage particles were diluted to approximately 1011 PFU/ml with distilled water. In all TEM analyses, 5 µl of the sample was directly applied on the carbon-coated nitrocellulose grid, excess liquid was drained with filter paper before staining with two successive drops of 2% uranyl acetate (pH 4.5). Samples were then dried and examined in a Tecnai G2 F20 X-TWIN (FEI) transmission electron microscope operating at 200 kV with an 11 MPix ORIUS SC1000B (Gatan) CCD camera. Magnification was calibrated using T4 phage particles and dimensions established by measurement of at least 20 intact phage particles.
Genome sequencing and analysis
The isolated genomic DNA of NBD2 (~100 μg) was sent to BaseClear (Netherlands) for sequencing. Paired-end sequence reads were generated using the Illumina HiSeq2500 system. FASTQ sequence files were generated using the Illumina Casava pipeline version 1.8.3. Initial quality assessment was based on data passing the Illumina Chastity filtering. The second quality assessment was based on the remaining reads using the FASTQC quality control tool version 0.10.0.
The quality of the FASTQ sequences was enhanced by trimming off low-quality bases using the “Trim sequences” option of the CLC Genomics Workbench version 8.5.1. The quality filtered sequence reads were puzzled into a number of contig sequences. The analysis was performed using the “De novo assembly” option of the CLC Genomics Workbench version 8.5.1. Misassemblies and nucleotide disagreement between the Illumina data and the contig sequences were corrected with Pilon version 1.11. The contigs were linked and placed into scaffolds or supercontigs. The orientation, order and distance between the contigs was estimated using the insert size between the paired-end and/or matepair reads. The analysis was performed using the SSPACE Premium scaffolder version 2.3. The gapped regions within the scaffolds were (partially) closed in an automated manner using GapFiller version 1.10. Thus, reads were assembled into a single linear contig of 51,802 bp (2,661,576 reads; 6,306 av. coverage).
Open reading frames (ORFs) were predicted with Glimmer v3.02 (https://www.ncbi.nlm.nih.gov/genomes/MICROBES/glimmer_3.cgi) and Geneious Pro v5.5.6. (http://www.geneious.com ), using a minimum ORF size of 75 nt. Analysis of the genome sequence was performed using BLAST, PSI-BLAST, Megablast (https://blast.ncbi.nlm.nih.gov/Blast.cgi) as well as Transeq, Clustal Omega (http://www.ebi.ac.uk), and HHPred, HHblits, HHsenser [17, 18]. Also, tRNAscan-SE 1.21 (http://lowelab.ucsc.edu/tRNAscan-SE/) was used to search for tRNAs. Phylogenetic analysis was conducted using MEGA version 5 [19]. Whole genome alignment was performed using mVista [20, http://genome.lbl.gov/vista/], and VIRFAM [21, http://biodev.cea.fr/virfam/] was used for total proteome comparisons.
Analysis of structural proteins
Analysis of NBD2 virion structural proteins was performed following a modified filter-aided sample preparation (FASP) protocol as described in [15].
SDS-PAGE of NBD2 virion proteins
CsCl-purified phage particles (~1010 PFU/ml) resuspended in a buffer containing 60 mM Tris-HCl (pH 6.8), 1% SDS (w/v), 1% 2-mercaptoethanol (v/v), 10% glycerol (v/v) and 0.01% bromophenol blue (w/v) were boiled for 3 min and separated on a 12% SDS PAGE gel. Protein bands were visualized by staining with PageBlue Protein Staining Solution (Thermo Fisher Scientific).
RESULTS
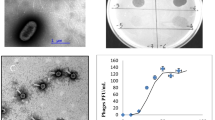
Virion morphology. Based on morphological characteristics, phage NBD2 belongs to the family Siphoviridae (Fig. 1); characterized by having an isometric head (~65 nm in diameter) and an apparently non-contractile flexible tail (~170 nm in length, ~12 nm in width). Although neither the baseplate nor the tail fibers are clearly visible by TEM, several ORFs coding for putative tail fiber proteins have been detected during bioinformatics analysis and/or by proteomics (see below).

Electron micrographs of CsCl-purified NBD2 virions
Basic physiological characteristics and host range. To determine the optimal conditions for phage propagation, the effect of temperature on the efficiency of plating (e.o.p.) was tested. The e.o.p. of NBD2 was examined at a temperature range of 7–36 °C, with the test revealing that NBD2 is a low-temperature virus: the phage forms plaques on E. coli NovaBlue (DE3) lawns at a temperature range of 10–34 °C, and has an optimum temperature for plating of ~20 °C (Fig. S1). Notably, the same low-temperature plating profile was observed when NBD2 was plated on the lawns of other E. coli strains, listed in Table 1. After 24 h of incubation at an optimum temperature, bacteriophage NBD2 formed circular smooth plaques of about 6 mm in diameter with a clear center and turbid edge (data not shown). Notably, since bacteriophage NBD2 failed to reproduce after inoculation into liquid bacterial culture, the one-step growth experiment was not performed.
To investigate the host range of NBD2, the phage was tested for its ability to infect different laboratory strains of E. coli. As seen in Table 1, bacteriophage NBD2 has a broad host range towards laboratory strains of E. coli, and shows the capacity to infect both E. coli K-12 and B derivatives. According to the literature, although genetically closely related, strains of E. coli B and K-12 differ in the structure of the outer membrane [25], particularly in the composition of the lipopolysaccharide (LPS) core and expression of outer membrane proteins [26]. In wild-type enterobacteria, a major component of the outer membrane, LPS, consists of an O-antigen, the core-region, and lipid A [27, 28]. Many common laboratory strains of E. coli, such as K-12 strains, are devoid of the O-antigen, while B strains lack not only O-antigen but the distal part of the core oligosaccharide as well [29]. Although quite a number of E. coli phages described in the literature use outer membrane LPS as a receptor during adsorption [30], the results presented in Table 1 (section I) indicate that the ability of bacteriophage NBD2 to adsorb to the host cell is not influenced by the structure of LPS. To confirm this, bacteriophage NBD2 was tested against E. coli K-12 strains that are mutated in their LPS biosynthesis pathway (Table 1, II). Also, since bacterial viruses are known to recognize outer membrane proteins as well [30], a set of E. coli K-12 strains with deletions in genes encoding outer membrane proteins (Table 1, III), most commonly targeted by known tailed phages, was included in the test. The test revealed that NBD2 is indeed insensitive to alterations in LPS structure because this phage most likely either recognizes more than one host receptor (one of which may just as well be LPS) or adsorbs solely to the outer membrane proteins, other than those listed in Table 1. However, further studies are required to confirm this hypothesis.
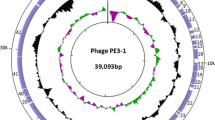
Overview of the phage NBD2 genome. Bacteriophage NBD2 has a linear, circularly permuted dsDNA genome consisting of 51,802 bp with a G+C content of 49.8%. As observed in other tailed phages, the genome of NBD2 is rather compact, with ~93% of the DNA sequence representing coding sequences (Fig. 2). A total of 87 probable protein-encoding genes and one tRNASer have been identified in the genome of NBD2. As seen in Fig. 2, there is a certain asymmetry in the distribution of the genes on the two NBD2 DNA strands. In total, 65 NBD2 open reading frames (ORFs) have been predicted to be transcribed from the same DNA strand, whereas 22 ORFs have been found on the opposite strand.

Functional genome map of bacteriophage NBD2. The coding capacity of the NBD2 genome is shown. Functions are assigned according to characterized ORFs in the NCBI database and/or MS/MS analysis. The colour code for the online version of the article is as follows: yellow – DNA replication, recombination, repair; green – transcription, translation, nucleotide metabolism; blue – structural proteins; light blue – lysis; grey – ORFs of unknown function; red – NBD2-specific ORFs that encode unique proteins with no reliable identity to database entries
The results of BLASTP analysis revealed that 51 NBD2 ORFs (~59% of NBD2 genes) code for hypothetical proteins of unknown function, and 22 of these ORFs have no reliable identity (E values of ≥0.001) to database entries. Using bioinformatics approaches, a putative function was assigned to 35 NBD2 ORFs, including 19 genes coding for virion morphogenesis-related proteins and packaging, as well as 8 genes associated with DNA replication (Table S1), recombination, and repair. None of the predicted NBD2 proteins showed sequence homology with integration-related proteins, antibiotic resistance determinants, or virulence factors.
DNA replication, recombination and repair. The genome of bacteriophage NBD2 contains no homologues to characterized DNA polymerase genes, suggesting that this phage relies quite heavily on the replication machinery of the host cell. However, the presence of a DNA primase (gp58; HHpred probability, 100.0%; E value, 4.1e-36), helicase (gp60; HHpred probability, 100.0%; E value, 9.4E-31), replication licensing factor (gp74; HHpred probability, 97.99%; E value, 1.2e-06), and SSB protein (gp56; HHpred probability, 100.0%; E value, 4.8E-33) suggests that NBD2 DNA replication depends not only on host factors but on phage-encoded proteins as well.
Homology searches also allowed the identification of the phage-encoded RecET recombination system in the genome of NBD2. The product of NBD2 ORF54 shows homology to the RecE protein of Escherichia virus TLS (56% aa identity; E value, 3e-133) and is homologous to E.coli exonuclease RecE (HHpred probability, 99.95%; E value, 1.8e-27). NBD2 ORF54 is followed by NBD2 ORF55, which codes for an ERF superfamily protein that is homologous to gp26 from Klebsiella phage Sushi (60% aa identity; E value, 1e-73). The ERF superfamily (pfam04404) includes the single-strand annealing proteins such as RecT, Redbeta, ERF, and Rad52, that all function in RecA-dependent and RecA-independent DNA recombination pathways [31]. Other NBD2 gene products possibly involved in DNA recombination, replication and repair include a TopA domain-containing (COG0550) protein, NBD2 Gp79, and a putative DNA repair nuclease, Gp61, which shows homology to a VRR-NUC domain-containing protein from Escherichia phage ADB-2 (73% aa identity; E value, 1e-68).
Nucleotide metabolism and DNA modification. Based on the amino acid sequence homology, four gene products of NBD2 were assigned as nucleotide metabolism and modification enzymes, including two nucleoside kinases, a polynucleotide kinase and dNMP kinase, encoded by two adjacent genes, NBD2 ORF14 and NBD2 ORF15, respectively. To escape from the digestion of restriction endonucleases and replicate successfully, phages often encode their own enzymes for DNA modification [32]. The results of bioinformatics analysis indicate that two genes for methylases are present in the genome of NBD2. The gene NBD2 ORF62 codes for a putative N-6-adenine-methyltransferase that has been predicted to belong to the Dam superfamily of methylases (pfam05869), whereas the product of NBD2 ORF76 is a Dcm superfamily (cl21501) methylase that is homologous to that from Klebsiella phage Sushi (64% aa identity; E value, 9e-107).
Cell lysis. It has been reported recently that the lysis of Gram-negative hosts by phages is a three-step process [33]. The infection cycle terminates when a small phage encoded protein (known as a holin) permeabilizes the membrane at a programmed time. Then, a muralytic enzyme (endolysin) escapes through the holes to attack the peptidoglycan. Finally, the fusion of the inner and outer membranes is accomplished by spanins [33, 34]. As seen in Fig. 2, the host-cell lysis cassette of NBD2 comprises three genes: ORF69, ORF70, and ORF71. The gene product of ORF70 is a Lysozyme-like superfamily (cl00222) protein that shows homology to different endolysins and lysozymes, whereas ORF69 and ORF70 code for the holin and u-spanin, respectively.
Structural proteins. As mentioned above, using bioinformatics approaches, 16 NBD2 structural genes were identified, including those coding for head (ORF32, ORF33, ORF35), neck (ORF36, ORF37), tail (ORF38, ORF39, ORF42, ORF43, ORF44), and tail fiber (ORF47, ORF57) proteins, as well as 3 ORFs coding for virion morphogenesis-related proteins, namely, ORF31 (putative prohead protease), ORF30 (head morphogenesis protein), and ORF46 (putative tail assembly protein). FASP followed by LC-MS/MS led to the experimental identification of 18 NBD2 structural proteins, including two, gp26 and gp49, which have no reliable homologues to annotated structural proteins in other organisms (Table 2). Therefore, in total, 20 NBD2 gene products were assigned as proteins involved in virion morphogenesis and DNA packaging (Figure 2).
In tailed phages, the packaging machine consists of two essential components: a portal ring and a terminase complex. Most characterized terminases are heteroligomers that consist of a small subunit (TerS) involved in DNA recognition and a large terminase subunit (TerL) containing the ATPase and the endonuclease activities [35, 36]. The GP3_package domain-containing (pfam16677) protein TerS of NBD2 is encoded by ORF27, while NBD2 ORF29 codes for a portal protein that shows homology to the corresponding proteins from various phages and has been detected by proteomics approaches as well. Based on the results of bioinformatics analysis, the gene product of NBD2 ORF28 is a TerL, since it has been predicted to belong to the terminase-like family (pfam03237) and shows detectable homology to the TerL proteins of a number of different bacteriophages, including those that have been reported to use a headful DNA packaging mechanism (e.g., Escherichia coli phage Rtp [37]). Notably, restriction digestion profiles of phage NBD2 DNA matched in silico predictions of a circular DNA molecule and, in addition to the expected fragments of the circular map, a discrete submolar pac fragment was observed in each digest (Fig. S2). According to the literature, this type of restriction pattern is considered to be diagnostic of headful packaging [38].
As seen in Fig. 1, neither the baseplate nor the tail fibers of NBD2 are clearly visible by TEM. Nevertheless, two genes potentially coding for baseplate components (ORF44 and ORF45) as well as two ORFs coding for tail fiber proteins (ORF47 and ORF57) have been identified in the genome of NBD2, and verified by proteomics analysis. NBD2 gp44 has been predicted to belong to the Phage-tail-L superfamily (cl01908) and is homologous to minor tail proteins from a wide range of diverse phages and bacteria. Meanwhile NBD2 gp45, which has an N-terminal MPN_NLC_P60 family (cd08059) domain and a C-terminal NLPC_P60 (pfam00877) family domain, is homologous to phage-encoded minor tail proteins and tail tip assembly proteins. Phage-tail-3 family (pfam13550) domain-containing protein NBD2 gp47 (1200 aa) has two conserved C-terminal domains of unknown function (DUF1983, pfam09327, and DUF1640, pfam07798) and is homologous to a putative tail fiber protein, T1p33 (1172 aa) of Escherichia virus T1 (64% aa ident; E value, 0.0). Another tail fiber protein of NBD2, gp57 (765 aa), has a C-terminal Peptidase_S74 domain (pfam13884), a conserved chaperone domain that is commonly found in endosialidases, and shares two regions of similarity (aa 1-398 and aa 585-756; 51% and 57% aa identity, respectively) with the tail fiber protein (622 aa) of Cronobacter virus ESP29491. Notably, phage endosialidases are usually present on the virus particle in the form of tail spikes or tail fibers and are responsible for host polysialic acid capsule recognition, binding and degrading [39, 40].
In the case of siphoviruses, two proteins, the major capsid protein and the major tail protein, are the main building blocks of the virion. A BLASTP search against the non-redundant NCBI protein database revealed that the gene product of NBD2 ORF33 shows homology to the major capsid proteins of Escherichia, Klebsiella, Citrobacter, Vibrio, Lactobacillus, and Edwardsiella siphoviruses. Also, HHPred returned a high-probability hit to the major capsid protein of Escherichia phage HK97 (HHPred probability, 99.9%; E value, 2.2E-26). NBD2 gp39 has been predicted to belong to the Phage-tail-3 (pfam08813) superfamily and is an orthologue of the major tail protein V of bacteriophage lambda (HHPred probability, 99.84%; E value, 6.4e-21).
Phylogenetic analysis. The VIRFAM analysis, a novel “head-neck-tail”-based classification method proposed by Lopes and coauthors [21] classified NBD2 as a siphovirus of Type 1 suggesting that this phage adopts the structural organization of the Siphoviridae phage SPP1 neck, which is formed by a portal protein, an Ad1 (Adaptor of type 1), a Hc1 (Head-closure of type 1), a Ne1 (Neck protein of type 1) and a Tc1 (Tail-completion of type 1) proteins [21]. However, as seen in Fig. S3, VIRFAM failed to assign NBD2 to a specific cluster and grouped it with viruses belonging to a subfamily, Tunavirinae.
Since the results of homology searches also hinted at similarity between NBD2 and tunaviruses, four phylogenetic trees based on the alignment of the NBD2 portal, tape measure, major capsid, and major tail protein sequences with those returned by BLASTP homology searches were constructed. As seen in Fig. 3, bacteriophage NBD2, while clearly related to tunaviruses, occupies a distinct branch on all four phylogenetic trees.

Phylogenetic analysis. Relationships of the: (a)- major capsid protein; (b)- major tail protein; (c)- tape-measure protein; and (d)- portal protein are analysed across diverse phages. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (500 replicates) is shown next to the branches
For many years, bacteriophages related to E. coli phage T1 were called T1-like viruses. In 2014 a new subfamily Tunavirinae, comprising all T1-like viruses characterized at the time, was established [41]. According to the 2016 ICTV taxonomy release (https://talk.ictvonline.org/taxonomy/), Tunavirinae is further classified into five genera, namely T1virus, Tlsvirus, Kp36virus, Rogue1virus, and Rtpvirus, each named after the prototype phages Escherichia virus T1, Escherichia virus TLS, Klebsiella virus KP36, Escherichia virus Rogue1, and Escherichia virus Rtp, respectively. Within each genus, phages share 64–82% nucleotide sequence identity, whereas nucleotide sequence identity shared between genera is 49–56% [41].
To obtain a more detailed picture of the phylogenetic relationships between viruses classified within the subfamily Tunavirinae and NBD2, the genome sequences of 17 tunaviruses recognized by ICTV were downloaded from the NCBI database and compared to that of NBD2 using mVISTA (Fig. S4) and Progressive Mauve (data not shown). Comparative whole-genome sequence alignment revealed that the genomes of NBD2 and tunaviruses shared several regions of nucleotide sequence similarity that, in NBD2, covered the virion morphogenesis as well as DNA metabolism and modification genes. Nevertheless, the overall nucleotide sequence similarity shared between NBD2 and phages from the subfamily Tunavirinae was relatively low and ranged from 24% (NBD2 vs Rogue1) to 33% (NBD2 vs T1). Thus, based on the criteria mentioned above, NBD2 cannot be assigned to any genus currently recognized by ICTV and is a singleton phage classifiable in the subfamily Tunavirinae.
DISCUSSION
Foodborne illnesses resulting from the consumption of agricultural commodities contaminated with enteric pathogens are an increasing problem around the world [42, 43]. While various possibilities for the contamination of produce with pathogens exist, global warming combined with the widespread use of animal manure in agriculture likely contributes to the increased number of such outbreaks [44 ]. In this regard, phages and their derived products may provide a safe and effective intervention against bacterial contamination [42]. However, while a number of reports in the literature have recently described encouraging results, using phages to reduce foodborne pathogens on a variety of fresh and fresh-cut produce [45], one of the main barriers to the success of such applications is the restrictive temperature used for the storage of many fresh and minimally processed produce, since at low temperatures, most pathogenic bacteria will not be metabolically active and the cycle of phage infection cannot be completed [42, 45]. To overcome this limitation, bacteriophages that are adapted to replicate at lower temperatures may be used. However, to date, only a limited number of LT enterobacteria phages have been described in the literature, and little is known about the molecular mechanisms underlying cold-adaptation of bacterial viruses.
Here we present the results of the molecular characterization of the LT siphophage NBD2 that is active against E. coli. Both comparative sequence and phylogenetic analyses indicate that this phage is related to a group of siphoviruses that are classifiable within the subfamily Tunavirinae. Since all E. coli-specific tunaviruses described in the literature are MT phages, one may expect that cold-adaptation of coliphages can be elucidated from the genome sequence comparisons. However, the results of the molecular analysis of NBD2 presented here, in conjunction with our previously published findings [14], indicate that it is not possible to demonstrate cold-adaptation of LT phages solely from sequence data. Nevertheless, while for now, we have come no closer to unraveling the mystery of phage cold-adaptation, low-temperature coliphages are not only extremely valuable for studying this phenomenon but also represent an attractive tool for application in the selective detection and/or the control of bacterial contamination in areas where adaptation to grow at low temperatures is desirable.
Nucleotide sequence accession numbers
The complete genome sequence of E. coli bacteriophage NBD2 was deposited in the NCBI nucleotide sequence database under accession number KX130668. The accession numbers of phage genomes used in this study are as follows: Shigella virus Shf1 (NC_015456), Shigella virus Psf2 (NC_026010), Escherichia virus ADB-2 (NC_019725), Escherichia virus vB_EcoS_ACG-M12 (NC_019404), Escherichia virus bV_EcoS_AHP42 (NC_024793), Escherichia virus bV_EcoS_AHS24 (NC_024784), Escherichia virus bV_EcoS_AKS96 (NC_024789), Escherichia virus e4/1c (NC_024210), Escherichia virus phiKP26 (KC579452), Enterobacter virus F20 (JN672684), Klebsiella virus 1513 (NC_028786), Citrobacter virus Stevie (KM236241), Escherichia virus TLS (NC_009540), Klebsiella virus KP36 (NC_029099), Escherichia virus vB_EcoS_Rogue1 (JQ182736), Escherichia virus RTP (NC_007603), Escherichia virus T1 (NC_005833).
References
Salmond GP, Fineran PC (2015) A century of the phage: past, present and future. Nat Rev Microbiol 13:777–786
Clokie MR, Millard AD, Letarov AV, Heaphy S (2011) Phages in nature. Bacteriophage 1:31–45
Keen EC (2015) A century of phage research: Bacteriophages and the shaping of modern biology. Bioessays 37:6–9
Klumpp J, Fouts DE, Sozhamannan S (2013) Bacteriophage functional genomics and its role in bacterial pathogen detection. Brief Funct Genomics 12:354–365
Jurczak-Kurek A, Gąsior T, Nejman-Faleńczyk B, Bloch S, Dydecka A et al (2016) Biodiversity of bacteriophages: morphological and biological properties of a large group of phages isolated from urban sewage. Sci Rep 6:34338
Hatfull GF (2015) Dark matter of the biosphere: the amazing world of bacteriophage diversity. Goodrum F, ed. J Virol 89:8107–8110
Ackermann HW (2001) Frequency of morphological phage descriptions in the year 2000. Arch Virol 146:843–857
Kutter E, Sulakvelidze A (2004) Bacteriophages: biology and applications. CRC Press, Boca Raton
Seeley ND, Primrose SB (1980) The effect of temperature on the ecology of aquatic bacteriophages. J Gen Virol 46:87–95
Klausa V, Piešinienė L, Staniulis J, Nivinskas R (2003) Abundance of T4-type bacteriophages in municipal wastewater and sewage. Ekologija 1:47–50
Leclerc H, Mossel DAA, Edberg SC, Struijk CB (2001) Advances in the bacteriology of the coliform group: their suitability as markers of microbial water safety. Annu Rev Microbiol 55:201–234
Yanagida M, Suzuki Y, Toda T (1984) Molecular organization of the head of bacteriophage T-even: underlying design principles. Adv Biophys 17:97–146
Wegrzyn G, Wegrzyn A (2005) Genetic switches during bacteriophage lambda development. Prog Nucleic Acid Res Mol Biol 79:1–48
Kaliniene L, Zajančkauskaitė A, Šimoliūnas E, Truncaitė L, Meškys R (2015) Low-temperature bacterial viruses VR—a small but diverse group of E. coli phages. Arch Virol 160:1367–1370
Šimoliūnas E, Kaliniene L, Stasilo M, Truncaitė L, Zajančkauskaitė A et al (2014) Isolation and characterization of vB_ArS-ArV2—first Arthrobacter sp. infecting bacteriophage with completely sequenced genome. PLoS One 9:e111230
Carlson K, Miller E (1994) Experiments in T4 genetics. In: Karam JD (ed) Bacteriophage T4. ASM Press, Washington DC, pp 419–483
Alva V, Nam SZ, Söding J, Lupas AN (2016) The MPI bioinformatics toolkit as an integrative platform for advanced protein sequence and structure analysis. Nucleic Acids Res. doi:10.1093/nar/gkw348
Söding J, Biegert A, Lupas AN (2005) The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res 33(suppl 2):W244–W248
Tamura K, Peterson D, Peterson N, Stecher G, Nei M et al (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Frazer KA, Pachter L, Poliakov A, Rubin EM, Dubchak I (2004) VISTA: computational tools for comparative genomics. Nucleic Acids Res 32(suppl 2):W273–W279
Lopes A, Tavares P, Petit MA, Guérois R, Zinn-Justin S (2014) Automated identification of tailed bacteriophages and classification according to their neck organization. BMC Genomics 15:1027
Selick HE, Kreuzer KN, Alberts BM (1988) The bacteriophage T4 insertion/substitution vector system. A method for introducing site-specific mutations into the virus chromosome. J Biol Chem 263:11336–11347
Grenier F, Matteau D, Baby V, Rodrigue S (2014) Complete genome sequence of Escherichia coli BW25113. Genome Announc 2:e01038-14
Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H (2006) Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2(2006):0008
Yoon SH, Han MJ, Jeong H, Lee CH, Xia XX, Lee DH, Shim JH, Lee SY, Oh TK, Kim JF (2012) Comparative multi-omics systems analysis of Escherichia coli strains B and K-12. Genome Biol 13:R37
Han MJ, Lee SY, Hong SH (2012) Comparative analysis of envelope proteomes in Escherichia coli B and K-12 strains. J Microbiol Biotechnol 22:470–478
Silhavy TJ, Kahne D, Walker S (2010) The bacterial cell envelope. Cold Spring Harb Perspect Biol 2:a000414
Müller-Loennies S, Lindner B, Brade H (2003) Structural analysis of oligosaccharides from lipopolysaccharide (LPS) of Escherichia coli K12 strain W3100 reveals a link between inner and outer core LPS biosynthesis. J Biol Chem 278:34090–34101
Jansson PE, Lindberg AA, Lindberg B, Wollin R (1981) Structural studies on the hexose region of the core in lipopolysaccharides from Enterobacteriaceae. Eur J Biochem 115:571–577
Bertozzi Silva J, Storms Z, Sauvageau D (2016) Host receptors for bacteriophage adsorption. FEMS Microbiol Lett 363(4):fnw002
Iyer LM, Koonin EV, Aravind L (2002) Classification and evolutionary history of the single-strand annealing proteins, RecT, Redbeta, ERF and RAD52. BMC Genomics 3:8
Warren RA (1980) Modified bases in bacteriophage DNAs. Annu Rev Microbiol 34:137–158
Young R (2014) Phage lysis: three steps, three choices, one outcome. J Microbiol 52:243–258
Rajaure M, Berry J, Kongari R, Cahill J, Young R (2015) Membrane fusion during phage lysis. Proc Natl Acad Sci USA 112:5497–5502
Rao VB, Feiss M (2008) The bacteriophage DNA packaging motor. Annu Rev Genet 42:647–681
Rao VB, Feiss M (2015) Mechanisms of DNA packaging by large double-stranded DNA viruses. Annu Rev Virol 2:351–378
Wietzorrek A, Schwarz H, Herrmann C, Braun V (2006) The genome of the novel phage Rtp, with a rosette-like tail tip, is homologous to the genome of phage T1. J Bacteriol 188:1419–1436
Casjens SR, Gilcrease EB (2009) Determining DNA packaging strategy by analysis of the termini of the chromosomes in tailed-bacteriophage virions. Methods Mol Biol 502:91–111
Stirm S, Bessler W, Fehmel F, Freund-Mölbert E (1971) Bacteriophage particles with endo-glycosidase activity. J Virol 8:343–346
Stummeyer K, Dickmanns A, Mühlenhoff M, Gerardy-Schahn R, Ficner R (2005) Crystal structure of the polysialic acid-degrading endosialidase of bacteriophage K1F. Nat Struct Mol Biol 12:90–96
Niu YD, McAllister TA, Nash JH, Kropinski AM, Stanford K (2014) Four Escherichia coli O157:H7 phages: a new bacteriophage genus and taxonomic classification of T1-like phages. PLoS One 9(6):e100426
Sharma M (2013) Lytic bacteriophages: potential interventions against enteric bacterial pathogens on produce. Bacteriophage 3:e25518
Painter JA, Hoekstra RM, Ayers T, Tauxe RV, Braden CR et al (2013) Attribution of foodborne illnesses, hospitalizations, and deaths to food commodities by using outbreak data, United States, 1998–2008. Emerg Infect Dis 19:407–415
Ongeng D, Geeraerd AH, Springael D, Ryckeboer J, Muyanja C et al (2015) Fate of Escherichia coli O157:H7 and Salmonella enterica in the manure-amended soil-plant ecosystem of fresh vegetable crops: a review. Crit Rev Microbiol 41:273–294
Pérez Pulido R, Grande Burgos MJ, Gálvez A, Lucas López R (2016) Application of bacteriophages in post-harvest control of human pathogenic and food spoiling bacteria. Crit Rev Biotechnol 36:851–861
Acknowledgments
This work was supported by the Research Council of Lithuania (Project No. SIT-7/2015).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interests.
Human and animal rights statement
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Handling Editor: Horst Neve.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kaliniene, L., Truncaitė, L., Šimoliūnas, E. et al. Molecular analysis of the low-temperature Escherichia coli phage vB_EcoS_NBD2. Arch Virol 163, 105–114 (2018). https://doi.org/10.1007/s00705-017-3589-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-017-3589-5