
Abstract
Pantoea bacteria species cause human animal infections, and contribute to soil and aquatic environmental pollution. A novel bacteriophage, vB_Pd_C23 was isolated from a Tunisian wastewater system and represents the first new phage infecting P. dispersa. Lysis kinetics, electron microscopy, and genomic analyses revealed that the vB_Pd_C23 phage has a head diameter of 50 nm and contractile tail dimensions of 100 nm by 23 nm; vB_Pd_C23 has a linear double-stranded DNA genome consisting of 44,714-bp and 49.66% GC-content. Predicted functions were assigned to 75 open reading frames (ORFs) encoding proteins and one tRNA, the annotation revealed that 21 ORFs encode for unique proteins of yet unknown function with no reliable homologies. This indicates that the new species vB_Pd_C23 exhibits novel viral genes. Phylogenetic analysis along with comparative analyses generating nucleotide identity and similarity of vB_Pd_C23 whole genome suggests that the phage is a candidate for a new genus within the Caudoviricetes Class. The characteristics of this phage could not be attributed to any previous genera recognized by the International Committee on Taxonomy of Viruses (ICTV).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Pantoea was described in the previous decade and has been proposed to include the genus Erwinia and some species of Enterobacter. Pantoea, a Gram-negative, rod-shaped bacterium, is non-encapsulated and produces dark yellow colonies; this bacterium is mobile and has been widely identified and isolated from animals, soils, aquatic environments [1]. Pantoea species grow at an optimum temperature range of 28–30 °C; the DNA GC-content ranges from 52.7 to 60.6% [2]. Pantoea identification is primarily based on phenotypic characters that include microanatomy as well as biochemical and genotypic assays [3]. Among the twenty-four species of Pantoea described, five species have been reported as phytopathogens. These phytopathogens impact at least 31 crop plants that could result in substantial global agricultural losses [4]. In addition, Pantoea species causes human infections [5], mainly in immunocompromised individuals, resulting in wound, blood or urinary-tract infections [6, 7]. Pantoea susceptibility to first-line antibiotics is variable and exhibits a high prevalence of multidrug resistance [8]. Growing bacterial anti-microbial resistance is a serious public health problem worldwide [9]. This pathogen resistance to antibiotics has motivated researchers to examine alternative treatments for bacterial infections.
Bacteriophages represent an effective therapy to overcome antibiotic-resistant bacteria; customized phages are now used not only for medical purposes [10], clinical trials [11], but also in aquaculture [12], the food processing industry, and agriculture [13]. Alternative therapies to antibiotics aim to find new tools with fewer risks in human, animals, and plants.
Bacteriophages can play an important role in plant and animal pathogens management. Phages have been shown effective in reducing pathogenic bacteria in wastewater effluents [14]. The efficiency of phage therapy is due to high specificity for targeted bacterial pathogens.
This research focused on the isolation, genome characterization, and comparative genomic analysis of a virulent phage Pd_C23, the first isolated bacteriophage infecting the bacteria P. dispersa.
Materials and Methods
Bacterial Strain Identification
P. dispersa was isolated in January 2014 from the wastewater treatment plant (WWTP) of Charguia, Tunis City (Tunisia). A 5-L sample of wastewater was collected in sterilized glass bottles. The physicochemical characteristics of the strain sample of P. dispersa are as follows: COD (185 ± 8.1 mg O2/L), BOD (62 ± 4.2 mg O2/L) and MES (14 ± 2.4 mg/L), PO4–P (16 ± 2.3 mg/L), NH4–N (1.51 ± 0. 2 mg/L), NO3–N (3.4 ± 1.1 mg/L) pH (7.3 ± 0.4). A monoclonal culture was produced from each colony after plating and incubating the wastewater sample for 24 h, at 37 °C. An observation with a JEOL transmission electron microscope allowed further characterization for the isolated strain to determine size. The "Boiling" method was adopted to extract the chromosomal DNA [15]. The obtained sequences were compared to the nucleotide sequences available on the National Center for Biotechnology Information (NCBI) databases (http://www.ncbi.nlm.nih.gov/) using the Basic Local Alignment Search Tool (BLAST) and Ribosomal Database Project (RDP): https://rdp.cme.msu.edu/seqmatch/seqmatch.
Phage Isolation and Purification
P. dispersa phage was isolated in 2014, from a sample of 5 L of secondary wastewater collected from the WWTP. The isolation of the phages followed the protocols recommended by [16]. 50 mL of wastewater and 1 mL of an overnight culture of the P. dispersa host strains were added to 50 mL of tryptic soy broth (TSB, Difco) and were incubated at 37 °C for 4 h to allow phage expression in its host cells, then 500 µL of chloroform was added, afterward the lysate was centrifuged at 5 000×g for 15 min, and the supernatant was filtered through 0.22 µm pore filters to extract the vB_Pd_C23 phage from the suspension. A volume of 100 μL of suspension was added to 5 mL of TSB medium previously inoculated with 100 μL of P. dsipersa overnight culture.
The phage was purified by soft agar overlay method [17]. The phage titer (i.e., infectious dose) was determined by plaque assay expressed as plaque-forming units PFU/mL, using spread plate method [18].
The method used for characterization was based on host cell interaction, plaque morphology, electron microscopy, and genome sequencing [19].
Characterization of Phage Pd_C23
Transmission Electron Microscopy
The methods used are described by [19]. Ten microliters of uranyl acetate (2%) were deposited on a Formvar carbon-coated grid (200 meshes, Pelco International) mixed with a drop of the phage particles. The grids were then examined using JEOL1230 transmission electron microscope with high contrast, 40–120 kV transmission electron microscope operated at 80 kV at the Platform of Molecular Imaging and Microscopy of the Laval University.
One Step Growth Curve
One-step growth curve was performed to determine the phage latent period and burst size as described by [20].
Phage DNA Extraction and Restriction of vB_Pd_C23
Phage DNA was extracted from 500 mL of lysate using the Qiagen Lambda Maxi DNA purification kit with modifications as described by [21]. The phage DNA was digested with 3 enzymes restriction (Sma1: recognizes the sequence 5'---CCCGGG---3'; Nae I: recognizes the sequence 5 '---GCCGGC---3'; Nar IV: recognizes the sequence 5'---GGCGCC---3'). The DNA fragments were separated by agarose gel (0.8%) electrophoresis with size marker of 1 kb suspended in 1X Tris-Acetate-EDTA buffer. A voltage of 100 V was considered during one hour (Thermo Fisher Scientific). The obtained restriction profiles were compared to those predicted in silico using GeneiousPrime2020 (www.geneious.com).
Sequencing and Genome Analysis of P. dispersa Phage vB_Pd_C23
DNA extraction was performed using titer vB_Pd_C23 phage of 9 × 107 performed by plasmid Kit Maxi (Qiagen) as described by [21]. The sequencing library was first set up with the Nextera XT DNA sample preparation kit (Illumina) according to the manufacturer’s instructions. The library was sequenced using a MiSeq reagent kit (v2) on a MiSeq system (Illumina) [21]. De novo assembly was achieved with the Ray assembler 2.2.0. [17, 22]. Coverage was calculated with Samtools. PCR product was sequenced with an ABI 3730xl sequencer. Each vB_Pd_C23 gene sequence was submitted to a homology test using Blastx on NCBI. The whole genome characteristics were compared by length, GC%, ORF number, and presence of genes. The open reading frames (ORFs) were identified using Geneious Prime 2020; NCBI ORF finder (http://www.ncbi.nlm.nih.gov/gorf/orfig.cgi), and gene prediction was carried out with GeneMark.hmm prokaryotic64 on http://exon.gatech. edu/GeneMark/gmhmmp.cgi. Genome annotation involving functions and products was performed using the FastaProtein, FastaNucleotide and the non-redundant protein database https://www.ncbi.nlm.nih.gov/protein.
Transeq and HHpred were used for further results on http://www.ebi.ac.uk/Tools/st/emboss_transeq, the tRNA placements were determined with http://lowelab.ucsc.edu/tRNAscan-SE/ [23].
Phylogenetic Analysis and Average Nucleotide Identity (ANI)
Whole vB_Pd_C23 genome was aligned in MUSCULE with five best matches Caudovirales complete genomes. Sequence alignments, phylogenetic tree reconstruction and visualization were performed using Molecular Evolutionary Genetics Analyses (MEGA.7) [24] via the Maximum Likelihood method (ML). In addition, the pairwise alignment with ClustalW of the whole genome was used to compare differences between vB_Pd_C23 and the closest 500 replicates Myovirus. Whole vB_Pd_C23 was furthermore aligned with ViPTree analysis, 2.0version developed by [25, 26]. The dendrogram of phage proteomic tree was generated from selected best fit genomes and gene markers to obtain a viral proteomic tree of vB_Pd_C23 examining the whole genomic nucleotide of 11 sequences of double-stranded DNA (dsDNA) viruses belonging to the family of Caudovirales by ML.
At last, to determine the phylogenetic relationship of vB_Pd_C23 with its close relatives, a phylogenetic relationship analysis between large terminase subunit and major capsid individual genes were identified via BLASTn. Sequences were aligned using MUSCLE [27, 28]; phylogenetic trees were generated using neighbor-joining tree analysis based on MLM alignment through PhyML software [29].
Nucleotide and amino acid identity between vB_Pd_C23 whole genome and best homologs were assessed using a Software package (RRID: SCR_001591), as multiple sequence alignment tool between multiple sequences performed in: http://www.ebi.ac.uk/Tools/msa/clustalo/. A Heatmap of the nucleotide and amino acid identities between the complete vB_Pd_C23 genome and Caudovirales sharing the highest genetic homologies and marker genes was constructed to detect the evolutionary relationship of the bacteriophage with the rest of the homologous selected phages.
Accession Number
Bacteriophage vB_Pd_C23 genome sequences were deposited to GenBank, under accession number: OL396571.1.
Results
Bacterial Identification
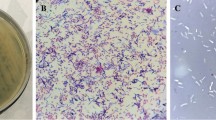
Transmission electron microscopy (TEM) showed a bacterium of 2 μm in size characterized by long peritrichous cilia (Fig. 1A). Sequence comparison of the isolated strain with existing data in the genomic database indicated that the strain is classified as strain Pant_SSU27 of bacteria P. dispersa; LMG2603; Genus Pantoea. The isolated complete genome strain is registered on Genbank, NCBI, under the accession Number: MN725743.

A Electron micrographs of P. dispersa strain seen by JEOL Transmission electron microscopy (MET). B Plaques formed by vB_Pd_C23 phage after 24 h of incubation at 37 °C on a lawn of Pantoea dispersa strain by Spot testing increasing dilutions estimated by plate-forming units (PFU)/mL. C Electron micrographs of vB_Pd_C23 phage by JEOL transmission electron microscopy (MET). D One-step growth curve of vB_Pd_C23 phage each data point is the mean of three independent experiments, and error bars indicate the means ± standard deviations
Phage Isolation and Purification
The phage suspension titer was estimated at 9×107 PFU/mL, as illustrated in Fig. 1B. Transmission electron microscopy revealed that the isolated phage is Myovirus presenting a long straight contractile tail with total size of 150 nm; a head size of 50 nm and a contractile tail of 100 nm×23 nm (Fig. 1C).
Characterization of Phage Pd_C23
One Step Growth Curve
One-step growth curve of the vB_Pd_C23 Myovirus was performed. The growth curve showed a 20-min latent period and a rise period estimated at 10 min, with the peak growth at 50 min with a total load of 136 PFU/mL (Fig. 1D).
Phage DNA Extraction and Restriction of vB_Pd_C23 DNA
Gel electrophoresis of vB_Pd_C23 DNA, revealed the presence of distinct bands suggesting the presence of putative dsDNA. vB_Pd_C23 DNA hydrolyzed by the enzymes NaeI, NarIV, and SmaI was cleaved into fragments, as presented in Fig. 2A; Results showed that vB_Pd_C23 genome was sensitive to all considered restriction enzymes NaeI, NarIV, and SmaI, as predicted in silico. Expected positions, and fragments sizes are shown in Supplementary Table 2. Data analyses were performed using Geneious software are summarized in Fig. 2B.

DNA electrophoresis of vB_Pd_C23 genome profile (REN) by restriction enzymes; NaeI, NarIV, and SmaI. A On the right is the in silico restriction profile and on the left is the in vitro restriction profile, molecular mass (MT indicates the size marker 1 Kb +) is indicated on the left of each profile (Bio-Rad). B Circular Genome map of vB_Pd_C23 phage indicating the positions and fragments size of the restriction enzymes NaeI, NarIV, and SmaI (Generated par Geneious 8.0 softwar)
Genome Analysis of P. dispersa Phage vB_Pd_C23
The restriction-digestion and analyses of the generated consensus sequence used for the analysis and mapped by denovo assembly of the new Tunisian phage vB_Pd_C23 revealed a linear double-stranded DNA, with a genome size of 44,714 bp. vB_Pd_C23 has 49.7% G-C content similar to other reported Pantoea phages infecting species than P. dispersa. Coding regions represent approximately 49% of the genome, and the longest noncoding region is 795 bp, occurring between ORF 40303pb and ORF41097pb (Fig. 3) (Supplementary Table 1). Transcription starts codon ATG for ribosome binding site (RBS) represent 80.3% of the total phage vB_Pd_C23 genes, and GTG represent only 12.5% of phage genes. Moreover, 19 ORFs out of 75 ORFs were reversed consecutively from the genomic position of 24735pb to the position 32429pb. From ORF32 to ORF50, the predicted proteins in this region are represented by 15 hypothetical proteins (ORF32-ORF40; ORF44; ORF46-ORF50), ORF41 was predicted to be a resistance-protein which function as a trigger to the antibiotic resistance response. HNH endonuclease (ORF42) function as an essential recombination protein trigger (ORF43) and zf-TFIIB was predicted to be a domain-containing protein (ORF45). An asymmetrical gene distribution was observed on the two DNA strands; 56 ORFs were predicted to be transcribed from the forward DNA strand, while 19 ORFs are located on the reverse complementary DNA strand.

Functional genome map of bacteriophage vB_Pd_C23. Numbers indicate open reading frame (ORF) position on the genome, functions are assigned according to the color code as follows: yellow—Proteins with no reliable identity; green— transcription, translation, nucleotide metabolism; pink ‐Bacterial origin; gray— DNA replication, recombination, repair, and packaging conserved; blue—structural proteins; red—Lysis, Orange: ORFs with unknown function (Color figure online)
Supplementary Table 1 grouped the annotation of predicted genes with putative functions. Only ORFs with a significant protein “best-match” with Myovirus sp. and Siphoviral sp. are presented in the Supplementary Table 1.
Fifty-three gene products of vB_Pd_C23 phage shared similar protein functions found in Siphoviral partial genomes phages. 46 ORFs had known functions, and the hypothetical proteins are similar to partial genomes of viruses isolated from human metagenomes revealing potential associations with chronic diseases [30] (Supplementary Table 1).
DNA Packaging Genes
The replication process requires genes for DNA packaging in the capsid, responsible for DNA recognition and initiation of DNA packaging. vB_Pd_C23 phage has a large terminase unit (ORF1) predicted to be located at the beginning of the genome and sharing 83% identity with the Klebsiella phage vB_Kpn_Chronis large unit terminase.
Structural Proteins
Four groups ORFs were determined to encode phage head and tail genes: (i) major capsid protein (ORF 6) shared 79% identity with Klebsiella phage vB_Kpn_Chronis, (ii) a putative baseplate J-like family protein (ORF21) also sharing 64% identity with Klebsiella phage, (iii) two tail fiber proteins (ORF23; ORF24), and (iv) one tail needle protein (ORF 25). All four ORFs were involved in triggering the cell infection by host recognition and promoting phage adsorption.
vB_Pd_C23 Phage Comparison with Klebsiella Phage vB_Kpn_Chronis
Pairwise alignment genomes of P. dispersa phage vB_Pd_C23 and K. pneumoniae phage (vB_KPn-chronis) were performed using Visual Interactive Simulation Tools and Applications for comparative analysis of genomic sequences (VISTA. a pairwise alignment program). Synteny test between Genomic organization of P. dispersa phage vB_Pd_C23 (OL396571) and vB_Kpn_Chronis (MN013086) was performed with blast alignment of the phages [31]. VISTA revealed the presence of similar 23 ORFs, respecting the same gene orientation from ORF 1 to ORF 23 (Supplementary Table 1).
Phylogeny and Average Nucleotide Identity (ANI)
Phylogeny was first constructed with the whole genome sequence of the vB_Pd_C23 (Fig. 4A).

vB_Pd_C23 phylogeny based on maximum likelihood method (MLM) of whole genomic nucleotide sequences of different species and genomic similarity relationships between all dsDNA viruses belonging to Caudoviricetes Class, Myoviruses, Siphoviruses, and Podoviruses mainly, with Gammaproteobacteria as host on the database. A Nucleotide sequences were aligned using Geneious.8 with 500 bootstrap replicates; model is WAG with gamma and invariant sites Bootstrap values (> 50%) are shown at each node; B vB_Pd_C23 Viral proteomic tree; Branch lengths are logarithmically scaled, the log scaled is represented by the numbers on top representing the SG values (normalized scores by blastx)
A second viral proteomic tree was constructed using VipTree, based on the ML method of the entire genomic nucleotide sequences of 11 selected dsDNA viruses among Caudovirales (Supplementary Table 3), the tree was generated based on the chronology of appearance between the closest related relationships (Fig. 4B), it revealed that vB_Pd_C23 genome embodies a distant branch, which according to the International Taxonomy Committee of the Bacterial Viruses Subcommittee and Archaeal indicates that the Monophyletic grouping generated meets the criteria for a new genus’.
A third phylogenetic tree based of individual genes (Fig. 5). The vB_Pd_C23 phylogenetic tree based on the major capsid homologous protein sequences included five phages (Fig. 5A). The phylogenetic tree, based on the large terminase unit protein sequence, included 19 phages protein sequences sharing the highest genetic similarities (Fig. 5B). (Supplementary Table 4).

Bioinformatic analyses of vB_Pd_C23 based on individual genes A Phylogenetic analyses based on large terminase large unit genes B Phylogenetic analysis based on major capsid genes. Sequences were aligned using MUSCLE, phylogenetic trees were generated using Neighbor-joining tree analysis based on MLM alignment through PhyML software (Tree percentage replicate in the bootstrap test is the value indicated next to the branches)
Average Nucleotide Identity (ANI)
Nucleotide and amino acid identity heat map constructed between vB_Pd_C23 whole genome and best hits whole genome homologs is presented on Supplementary Table 5. Overall, phylogenetic analysis revealed that vB_Pd_C23 did not significantly cluster with any known phage, and therefore represents a novel genus.
Discussion
TEM identification of the isolated strain along with the sequence comparison with existing genomic databases showed that the strain is classified as P. dispersa, LMG2603. Transmission electron microscopy of the isolated phage revealed that it belongs to the Myovirus phages. vB_Pd_C23 has a linear double-stranded DNA; with a genome size of 44,714 bp, and has a 49.7% G-C content similar to other Pantoea phages. For example, [32] showed that the phage Pantoea had a 49% G-C content. Similarly, vB_PagM_AAM22 phages (AAM22) were characterized by a 48.4% G C content [58]. However, related phages can display a GC content that can range from 52 to 55% [1].
Sequence analysis of the entire vB_Pd_C23 genome revealed the existence of 75 predicted ORF-encoding for proteins and identified one tRNA detected between the position 40706 and 40830pb, with a length of 124pb. The presence of tRNA gene in vB_Pd_C23 phage could be interpreted as a compensation for the disproportionate amount of amino acid codon between the virus and its hosts. This characteristic may offer an effective mechanism to extend the host range, as shown in research on related viruses [33]. Also, the presence of tRNA gene in vB_Pd_C23 phage confirms its lytic character as this gene is missing in lysogenic phages [34]. Bioinformatic analysis uncovered the lack of any integrase or other lysogeny-associated genes [35]. The genome start site is likely a large terminase unit gene. Predicted protein functions for vB_Pd_C23 were verified by searching for sequence similarity in the NCBI database by BLASTp. Bioinformatics analyses were based on identifying regions with similarities to gene products of tailed Caudovirales homologs. Only one protein DUF1367 family protein (ORF65) was identified to encode for a protein of Salmonella typhimurium. 35ORFs were similar with known genes of Myovirus and/or Siphoviruses associated proteins. The 40 remaining ORFs were divided between 19 ORFs that encode for hypothetical proteins of Myovirus and Siphoviruses with no reliable identity, and the other 21 ORFs had no reliable homologies of phages or non-phages genes in the NCBI database. The results suggest that there are 21 new viral proteins were associated with the phage vB_Pd_C23.
Among the vB_Pd_C23 gene products compared with detectable homologs, a total of 49 vB_Pd_C23 ORFs shared similarities with Myovirus proteins, with 22 of these ORFs sharing similarities with the complete genome of Klebsiella pneumoniae phage vB_Kpn_Chronis (MN013086.1). Similarities were verified from ORF1 to ORF 23 except for the ORF7. The best gene similarity match (83%) was registered for ORF1 with a large unit terminase of vB_Kpn_Chronis phage. The remaining ORFs matched with a Myovirus from various isolates of a human metagenome, with partial genome submitted from the Cellular Oncology Laboratory, National Cancer Institute, USA [30].
As observed for the Myoviruses genome organization, phage vB_Pd_C23 genome contains an early genomic region necessary for the phage life cycles; these genes are involved in the initiation of bacterial infection, which then triggers intermediate and late genes [36]. In addition, vB_Pd_C23 also has a small unit (ORF75) for DNA binding predicted to be at the genome end, sharing 54% of its identity with a Pseudomonas phage (DAQ82909.1). DNA packaging is known to engage the portal protein (ORF2) during viral packaging in the capsid and tail assembly. DNA packaging produces the pre-formed proheads that are needed in interactions of the portal protein with the terminase protein; this packaging is critical in the translocation of dsDNA for the final viral capsid assembly [37].
DNA Replication, Recombination, and Repair genes
vB_Pd_C23 replication process required the genes ORF 4 that produce Nudix hydrolase; this enzyme is found in viruses, bacteria and humans and plays an important role in breaking down toxic intracellular compounds as well as in creating the bacterial and viral pathogenesis [38, 39]. Additional roles may include the regulating the lytic transglycosylase (ORF15) which is associated with the initiation of bacterial cell infection. In the absence of the transglycosylase, the internalization of the phage genome is significantly delayed during infection [40]. Three ORFs (ORF42, ORF71, and ORF73) were predicted to be homing endonucleases, thus would promote DNA packaging, and act as mobile elements in genetic exchange and horizontal transfer and play a part in the dissemination of tRNA gene clusters among related organisms [41]. Restriction alleviation protein (ORF62) has endonuclease activity was predicted as the gene responsible for hindering the cleavage of the bacterial DNA restriction-modification system. The repair process for the vB_Pd_C23 genome is likely accomplished by the SOS-system (ORF56) which responds to DNA damage by increasing the gene expression required for DNA repair. The RecA (ORF66) is used to repair breaks into the bacterial chromosome [42].
Phage-Host Interaction and Lysis Genes
Six ORFs were associated with Phage-host interaction and lysis genes, involving both Holin (ORF28) and lysozyme (ORF27) along with adhesin (ORF30) and cytadhesin (ORF29) P30/P32. The lysis occurs by encoding endolysin and the protein holin. These peptidoglycan-degrading enzymes are synthesized during the late phase of gene expression [43], causing the degradation of the bacterial cell wall [17]. Holin creates small pores in the membrane facilitating the phage access to the host cell peptidoglycan, and allows the endolysins to reach their substrates. Endolysins have a high degree of biochemical variability, with higher bactericidal function than antibiotics; their associated antimicrobial activity can be exploited to reduce bacterial pathogenicity [44].
Annexin (ORF10) is known as a protein with immunomodulatory functions in viral infections and host responses [13], endosialidase (ORF5) works during the viral passage into cells and also in their release from infected cells, [45], PVL ORF-50-like family (ORF63) play a role in the pathogenesis of necrotizing pneumoniae [46] and is found in both bacteria and bacteriophages, encoded as a member of the leukocidin group of bacterial toxins that kill leukocytes by creating pores in the cell membrane [47]. The vB_Pd_C23 consensus genome is 50.6% similar to that of Klesbiella phage. The genome of vB_Pd_C23 is shorter by 988 bp, sequences encoding putative proteins made up 92.1%, this is compared to 96.7% for vB_KPn-chronis, The number of intragenic regions was 32 for vB_Pd_C23 compared to 26 for vB_KPn-chronis; the mean distances for these intragenic regions was 115 for vB_Pd_C23 and 65 bp Klesbiella phage. The genome organization of vB_Pd_C23 clearly confirms its relatedness to the Klesbiella phage.
vB_Pd_C23 phylogeny was grouped with pairwise alignment of phage vB_Kpn_Chronis along with four best matches blasted Caudovirales complete genomes. The genetic distances between the two Myovirus showed a genetic link between the two isolates, sharing a distant relationship. Also results indicate that the large terminase unit designated as the utmost commonly conserved gene sequence in phages thus exploited to build viral evolutionary relationships phylogeny between different phages is along with major Capsid protein carrying highly conserved domains [48]. As shown on Neighbor-joining tree analysis of the large unit and the major capsid protein, vB_Pd_C23 appears to embody a distinct branch in a position of aproximity with the vB_Kpn Chronis phage, confirming the previous results. This reveals vB_Pd_C23 represents a new genus, which shares a distant relationship with the vB_Kpn_Chronis phage [49]. vB_Pd_C23 demonstrated an exclusive organization regarding its clusters, supporting Hendrix theory promoting those dsDNA bacteriophages adapts to their living environment through horizontal gene exchanges, giving them the ability to recognize specifically host cells granting them the mosaic genome architecture. This suggests that vB_Pd_C23 could potentially be a mosaic of Klebsiella phage. Multiple sequence analysis revealed that although a connection was found between homologous phages, (the highest identity observed was estimated at 30.37% for Salmonella phage vB_Se_STGO_35_1 (NC_054648), proceed by 16.48% identity with vB_Kpn_Chronis phage percentage values remained lower than the nucleotide identity required between same genera, described as a cohesive group of viruses sharing a high degree of greater than 70% nucleotide identity over the entire length of the genome [50]. The new vB_Pd_C23 is associated within a new genus, since it cannot be classified in any of the currently phage recognized by the ICTV.
A submission of a taxonomic proposal for the new genus of vB_Pd_C23 (OL396571.1) as the first lytic phage specific to the P. dispersa strain was submitted to the Caudoviral Study Group. ICTV link to our proposal: https://talk.ictvonline.org/files/proposals/taxonomy_proposals_prokaryote1/m/bact01/13637
Conclusion
Myovirus phage vB_Pd_C23 was isolated from Tunisia wastewater treatment facilities. The fully sequenced genome revealed 21 ORFs with no reliable homology, indicating that vB_Pd_C23 has 21 novel viral proteins Analyses of the results indicated that the lytic phage may share a common ancestor with the unclassified K. pneumonia phage. Overall vB_Pd_C23 represents a novel bacteriophage within a new genus and presents the first lytic phage with a high specificity for its host P. dispersa.
Data Availability
Data and material are Available and transparent.
Code Availability
Not applicable.
References
Walterson AM, Stavrinides J (2015) Pantoea: insights into a highly versatile and diverse genus within the Enterobacteriaceae. FEMS Microbiol Rev 39(6):968–984. https://doi.org/10.1093/femsre/fuv027
Mergaert J, Verdonck L, Kersters K (1993) Transfers of Erwinia ananas (synonym, Erwinia uredovora) and Erwinia stewartii to the Genus Pantoea emend. As Pantoea ananas (Serrano 1928) comb. Nov. and Pantoea stewartii (Smith 1898) comb, nov., respectively, and description of Pantoea stewartii subsp. indologenes subsp. nov. Int J Syst Microbiol 43:162–173. https://doi.org/10.1099/00207713-43-1-162
Wan MLY, Forsythe SJ, El-Nezami H (2019) Probiotics interaction with foodborne pathogens: a potential alternative to antibiotics and future challenges. Crit Rev Food Sci Nutr 59(20):3320–3333. https://doi.org/10.1080/10408398.2018.1490885
Kini K (2018) Thesis: Pantoea spp: a new bacterial threat to rice production in sub-Saharan Africa (Doctoral dissertation, Université Montpellier; AfricaRice (Abidjan).
Büyükcam A, Tuncer Ö, Gür D, Sancak B, Ceyhan M, Cengiz AB, Kara A (2018) Clinical and microbiological characteristics of Pantoea agglomerans infection in children. J Infect Public Health 11(3):304–309. https://doi.org/10.1016/j.jiph.2017.07.020
Asai N, Koizumi Y, Yamada A, Sakanashi D, Watanabe H, Kato H, Shiota A, Hagihara M, Suematsu H, Yamagishi Y, Mikamo H (2019) Pantoea dispersa bacteremia in an immunocompetent patient: a case report and review of the literature. J Med Case Reports 13(1):33. https://doi.org/10.1186/s13256-019-1969-z
Cruz AT, Cazacu AC, Allen CH (2007) Pantoea agglomerans, a plant pathogen causing human disease. J Clin Microbiol 45(6):1989–1992. https://doi.org/10.1128/JCM.00632-07
Mani S, Nair J (2021) Pantoea Infections in the neonatal intensive care unit. Cureus 13(2):e13103. https://doi.org/10.7759/cureus.13103
Callaway TR, Lillehoj H, Chuanchuen R, Gay CG (2021) Alternatives to antibiotics: a symposium on the challenges and solutions for animal health and production. Antibiotics 10(5):471. https://doi.org/10.3390/antibiotics10050471
Biswas B (2002) Bacteriophage therapy rescues mice bacteremic from a clinical isolate ofvancomycin-resistant Enteroccocus faecium. Infect Immun 70(1):204–210. https://doi.org/10.1128/IAI.70.1.204-210.2002
Thiyagarajan S, Chrisolite B, Alavandi SV (2021) Degenerate primed randomly amplified polymorphic DNA (DP-RAPD) fingerprinting of bacteriophages of Vibrio harveyi from shrimp hatcheries in Southern India. Cold Spring Harbor Laboratory. https://doi.org/10.1101/2021.08.10.455891
Ninawe AS, Sivasankari S, Ramasamy P et al (2020) Bacteriophages for aquaculture disease control. Aquacult Int 28:1925–1938. https://doi.org/10.1007/s10499-020-00567-4
Sieiro C, Areal-Hermida L, Pichardo-Gallardo Á, Almuiña-González R, de Miguel T, Sánchez S, Sánchez-Pérez Á, Villa TG (2020) A hundred years of bacteriophages: can phages replace antibiotics in agriculture and aquaculture? Antibiotics 9(8):493. https://doi.org/10.3390/antibiotics9080493
Grami E, Salhi N, Sealey KS, Hafiane A, Ouzari HI, Saidi N (2022) Siphoviridae bacteriophage treatment to reduce abundance and antibiotic resistance of Pseudomonas aeruginosa in wastewater. Int J Environ Sci Technol 19(4):3145–3154. https://doi.org/10.1007/s13762-021-03366-3
Sambrook J, Fritschi EF, Maniatis T (1989) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, New York
Petrovski S, Seviour RJ, Tillett D (2011) Genome sequence and characterization of the Tsukamurella Bacteriophage TPA2. Appl Environ Microbiol 77(4):1389–1398. https://doi.org/10.1128/AEM.01938-10
Kropinski AM, Mazzocco A, Waddell TE, Lingohr E, Johnson RP (2009) Enumeration of bacteriophages by double agar overlay plaque assay in bacteriophages. Humana Press. https://doi.org/10.1007/978-1-60327-164-6_7
Armon R, Kott Y (1993) A simple, rapid and sensitive presence/absence detection test for bacteriophage in drinking water. J Appl Bacteriol 744:490–496. https://doi.org/10.1111/j.1365-2672.1993.tb05159.x
Ackermann HW (2001) Frequency of morphological phage descriptions in the year 2000. Brief review Arch Virol 146:843–857. https://doi.org/10.1007/s007050170120
Bolger-Munro M, Cheung K, Fang A, Wang L (2013) T4 bacteriophage average burst size varies with Escherichia coli B23 cell culture age. J Exp Microbiol Immunol 17:115–119
Deveau H, Van Calsteren MR, Moineau S (2002) Effect of exopolysaccharides on phage-host interactions in Lactococcus lactis. Appl Environ Microbiol 68:4364–4369. https://doi.org/10.1128/AEM.68.9.4364-4369.2002
Boisvert S, Laviolette F, Corbei IJ (2010) Ray: simultaneous assembly of reads from a mix of high-throughput sequencing technologies. J Comput Biol 17:1519–1533. https://doi.org/10.1089/cmb.2009.0238
Delesalle VA, Tanke NT, Vill AC, Krukonis GP (2016) Testing hypotheses for the presence of tRNA genes in mycobacteriophage genomes. Bacteriophage 6(3):e1219441. https://doi.org/10.1080/21597081.2016.1219441
Kumar S, Stecher G, Tamura K (2016) MEGA7 molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33(7):1870–1874. https://doi.org/10.1093/molbev/msw054
Nishimura Y, Yoshida T, Kuronishi M, Uehara H, Ogata H, Goto S (2017) ViPTree: the viral proteomic tree server. Bioinformatics 33(15):2379–2380. https://doi.org/10.1093/bioinformatics/btx157
Rohwer FE, dwards R (2002) The phage proteomic tree: a genome-based taxonomy for phage. ASM J 184:16. https://doi.org/10.1128/JB.184.16.4529-4535.2002
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32(5):1792–1797. https://doi.org/10.1093/nar/gkh340
Madeira F, Park YM, Lee J (2019) The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids. https://doi.org/10.1093/nar/gkz268
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 30. Syst Biol 59(3):307–321
Tisza MJ, Buck CBA (2021) Catalog of tens of thousands of viruses from human metagenomes reveals hidden associations with chronic diseases. Proc Natl Acad Sci USA. https://doi.org/10.1073/pnas.2023202118
Frazer KA, Pachter L, Poliakov A, Rubin EM, Dubchak I (2004) VISTA: computational tools for comparative genomics. Nucleic Acids Res 1(32):W273-9. https://doi.org/10.1093/nar/gkh458
Šimoliūnas E, Šimoliūnienė M, Kaliniene L, Zajančkauskaitė A, Skapas M, Meškys R et al (2018) Pantoea bacteriophage vB_PagS_Vid5: A low-temperature Siphovirus that harbors a cluster of genes involved in the biosynthesis of archaeosine. Viruses 10(11):583
Šimoliūnienė M, Truncaitė L, Petrauskaitė E, Zajančkauskaitė A, Meškys R, Skapas M, Kaupinis A, Valius M, Šimoliūnas E (2020) Pantoea agglomerans-infecting bacteriophage vB_PagS_AAS21: a cold-adapted virus representing a novel genus within the family siphoviridae. Viruses 12(4):479. https://doi.org/10.3390/v10110583
McCullor K, Postoak B, Rahman M, King C, McSha WM (2018) Genomic sequencing of high-efficiency transducing streptococcal bacteriophage A25: consequences of escape from lysogeny. ASM J 200:23. https://doi.org/10.1128/JB.00358-18
Varona HC, Hargreaves K, Abedon S (2017) Lysogeny in nature: mechanisms, impact and ecology of temperate phages. ISME J 11:1511–1520. https://doi.org/10.1038/ismej.2017.16
Low DA, Weyand NJ, Mahan M (2001) Roles of DNA adenine methylation in regulating bacterial gene expression and virulence. Infect Immun 69:7197–7204
Casjens SR (2011) The DNA-packaging nanomotor of tailed bacteriophages. Nat Rev Microbiol 9:647–657. https://doi.org/10.1038/nrmicro2632
Bessman MJ, Walsh J-D, Dunn CA, Swaminathan J, Weldon JE, Shen J (2001) The gene ygdP, associated with the invasiveness of Escherichia coli K1, designates a Nudix hydrolase, Orf176, active on adenosine (5′)-pentaphospho-(5′)-adenosine (Ap5A). J Biol Chem 276:37834–37838. https://doi.org/10.1074/jbc.M107032200
Cartwright J-L, Safrany L-K, Dixon E, Darzynkiewicz J, Stepinski R, McLennan B (2002) The g5R (D250) gene of African swine fever virus encodes a Nudix hydrolase that preferentially degrades diphosphoinositol polyphosphates. J Virol 76:1415–1421. https://doi.org/10.1128/jvi.76.3.1415-1421.2002
Moak M, Molineux IJ (2000) Role of the Gp16 lytic transglycosylase motif in bacteriophage T7 virions at the initiation of infection. Mol Microbiol 37(2):345–355. https://doi.org/10.1046/j.1365-2958.2000.01995.x
Morgado S, Vicente AC (2019) Global in-silico scenario of tRNA genes and their organization in virus genomes. Viruses 11:180. https://doi.org/10.3390/v11020180
Lesterlin C, Ball G, Schermelleh L, Sherratt DJ (2014) RecA bundles mediate homology pairing between distant sisters during DNA break repair. Nature 506:249–253
Abedon ST (2011) Lysis from without. Bacteriophage 1:46–49. https://doi.org/10.4161/bact.1.1.13980
Saier MH, Reddy B (2015) Holins in bacteria, eukaryotes, and archaea: multifunctional xenologues with potential biotechnological and biomedical applications. J Bacteriol 197:7–17. https://doi.org/10.1128/JB.02046-14
Ampomah PB, Kong WT, Zharkova O, Chua SC, Perumal Samy RAMAR, Lim LH (2018) Annexins in influenza virus replication and pathogenesis. Front Pharmacol 9:1282. https://doi.org/10.3389/fphar.2018.01282
Crémieux AC, Dumitrescu O, Lina G, Vallee C, Muffat-Joly CJF, M, et al (2009) Panton–Valentine leukocidin enhances the severity of community-associated methicillin-resistant Staphylococcus aureus rabbit osteomyelitis. PLoS ONE 4(9):e7204. https://doi.org/10.1371/journal.pone.0007204
Lina G, Piémont Y, Godail-Gamot F, Bes M, Peter MO, Gauduchon V, Vandenesch F, Etienne J (1999) Involvement of Panton–Valentine leukocidin-producing Staphylococcus aureus in primary skin infections and pneumoniae. Clin Infect Dis 29(5):1128–1132. https://doi.org/10.1086/313461
Casjens SR, Gilcrease EB, Winn-Stapley DA, Schicklmaier P, Schmieger H, Pedulla ML, Ford ME, Houtz JM, Hatfull GF, Hendrix RW (2005) The generalized transducing Salmonella bacteriophage ES18: complete genome sequence and DNA packaging strategy. J Bacteriol 87(3):1091–1104. https://doi.org/10.1128/JB.187.3.1091-1104
Thurgood TL, Sharma R, Call JJ et al (2020) Genome sequences of 12 phages that infect Klebsiella pneumoniae. Microbiol Resour Announc 9(16):e00024-e120. https://doi.org/10.1128/MRA.00024-20
Turner D, Kropinski AM, Adriaenssens EM (2021) A roadmap for genome-based phage taxonomy. Viruses 13:506. https://doi.org/10.3390/v13030506
Acknowledgements
This study was supported by a grant from Tunisian Ministry of Higher Education and Scientific Research LR18CERTE04 (2019_2022 Programs). Authors are grateful for technical team of CERTE. We thank Pr. Sylvain Moineau and Pr Josée Harel and members of their teams for discussion at the beginning stages of the project and for phage morphological identification.
Funding
This study was supported by a grant from Tunisian Ministry of Higher Education and Scientific Research LR18CERTE04 (2019_2022 Programs).
Author information
Authors and Affiliations
Contributions
EG performed the experiments, analyzed the data, performed bioinformatics analysis and drafted the manuscript. IL contributed in data analysis. NS is the supervisor of the work and participated in the continuous revision of the manuscript. KSS contributed in genius figures, participated in the annotation and edited English Language. DT and GR conducted the sequencing assembly and the electron microscopy. SBT designed the genius figures and participated in the annotation. AH the Head of LMBE laboratory contribute in the article revision. All authors read and approved the manuscript. The phage described here under the name of vB_Pd_C23 Phage and host are available at the Félix d'Hérelle Reference Center for Bacterial Viruses of the University Laval (Québec, Canada; www.phage.Ulaval.ca), as well as at CERTE Tunisia.
Corresponding author
Ethics declarations
Conflict of interest
Authors disclose that there are no conflicts of interest.
Ethical Approval
Not applicable.
Consent for Publication
Authors Consent for publication.
Consent to Participate
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Grami, E., Laadouze, I., Ben Tiba, S. et al. Isolation, Characterization, and Comparative Genomic Analysis of vB_Pd_C23, a Novel Bacteriophage of Pantoea dispersa. Curr Microbiol 80, 52 (2023). https://doi.org/10.1007/s00284-022-03152-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00284-022-03152-5