
Abstract
The genome sequence of a novel avian cyclovirus is described in this study. The genome size and orientation of predicted genes was similar to those described in other vertebrate and insect origin cycloviruses. The greatest genome sequence identity was shared with a dragonfly cyclovirus (nt, 60.6%). Phylogenetic analysis showed marginal relatedness with another avian cyclovirus, the chicken associated cyclovirus 1. In contrast, along a short fragment of the replication-associated protein coding gene (rep) (spanning nt 1240-1710) the duck origin cyclovirus was very similar to human origin and honey bee origin rep sequences (human – TN4, 98%; honey bee – hb10, 100%). Related cyclovirus strains existing amongst various animal species living in diverse ecosystems and separated by large geographic distances show the need for additional studies to better understand the ecology and epidemiology of cycloviruses.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The Circoviridae family classifies viruses comprising small, icosahedral, non-enveloped particles infecting eukaryote organisms including both invertebrate and vertebrate animals. The family is classified into two genera, Circovirus and Cyclovirus [16]. The single-stranded circular DNA (ssDNA) genome of circoviruses and cycloviruses measures 1.7 to 2.1 kb in both genera with two open reading frames (ORFs) which encode the replication-associated protein (Rep) and the capsid protein (Cap). The orientation and strand-specificity of these ORFs are key features that distinguish members of the two genera [3, 16]. Of interest, fossil circoviral elements, mainly restricted to the rep gene (or its non-functional derivative), have been identified integrated into eukaryotic host genomes [5, 8]. Differentiating these integrated fossil genetic traits from replication competent circoviruses and circovirus-like agents is critical for developing a better understanding of viral genome biology, ecology, possible disease associations, and finally virus taxonomy.
Circoviruses have been identified, with or without disease, in several bird species including parrots, pigeons, ravens, ducks, finches, and chickens [11, 12, 15, 17,18,19,20]. In contrast, chickens are the only bird species, in which cycloviruses have been identified [11, 12]. Recently, we have conducted an ecological survey among wild birds to identify potential reservoirs of circoviruses pathogenic to domestic poultry. One virus whose genome sequence we determined could be classified in the Cyclovirus genus and showed close genetic relatedness to cyclovirus-like partial rep sequences amplified from feces of a healthy Tunisian child and to cyclovirus-like partial rep sequence detected in honey bees in Hungary [11, 13]. These earlier reports did not clarify whether the novel cyclovirus-like sequences are integrated genomic elements, or, are parts of the genome of replication competent exogenous viruses. Thus, we made an attempt to perform whole genome sequencing of a wild bird-origin cyclovirus-like agent.
For this study cloacal swab specimens were resuspended in 1 ml of PBS buffer. Nucleic acid was extracted using the Direct-zol RNA MiniPrep Kit (Zymo Research) omitting DNase treatment. A pan viral degenerated primer set targeting the circoviral rep gene was utilized in a screening PCR assay [11, 13]. PCR mixtures (25 μl final volume) contained 1x DreamTaq Green buffer, 200 μM dNTP mix, 200 nM primers, 0.625 U DreamTaq DNA Polymerase (Thermo Fisher Scientific), and 1 μl of nucleic acid templates. The cycling protocols used for the nested PCR were as follows: denaturation at 95 °C for 3 min, 40 cycles of 95 °C for 30 s, 52 °C (first round of nested PCR) and 56 °C (second round of nested PCR) for 30 s and extension at 72 °C 1 min; a final extension step (72 °C for 10 min) was also included [13]. The second round PCR product was extracted from the gel slice by the Geneaid Gel/PCR DNA Fragments Extraction Kit and was directly sequenced using the BigDye Terminator v1.1 Cycle sequencing Kit (Thermo Fisher Scientific) on an ABI PRISM 3100-Avant Genetic Analyzer.
A total of 16 cloacal swab samples collected from waterfowl species, including seven samples from mallard duck (Anas platyrhynchos), seven samples from lesser white-fronted goose (Anser erythropus) and two samples from great crested grebe (Podiceps cristatus) tested positive for circoviral rep gene by PCR. All samples were collected during December 2013, around the town of Mezőberény. The settlement is located in south-east Hungary. Within and near the settlement there are over a dozen ponds and lakes and a mid-size river (Kőrös) is located about 1-1.5 km north from the town; favorable conditions for many waterfowl species to inhabit the neighborhood.
One specimen collected from mallard duck was selected for whole genome sequencing. Amplification of the whole virus genome was performed by back-to-back PCR primers (forward primer 5’ TCATCTCTTGAACTGGTGTGCC-3’ and reverse primer 5’-CTGTGACGCAATAACGAGGTC-3’) designed based on the sequence of the nested PCR product. The PCR mixtures (25 μl final volume) contained 1x Phusion Green HF buffer, 200 μM dNTP mix, 200 nM primers and 0.25 U Phusion DNA Polymerase (Thermo Fisher Scientific), and 1 μl of nucleic acid templates. The cycling protocol used for the back-to-back PCR was as follows: denaturation at 98 °C for 30 s, 45 cycles of 98 °C for 10 s, annealing at 57 °C for 30 s and extension at 72 °C 1 min; a final extension step at 72 °C for 10 min was added to the protocol [13].
The approximately 2 kb long amplicon generated by the back-to-back primers was initially processed for next-generation sequencing (NGS) using the Ion Torrent PGM instrument. We have previously shared procedures concerning library preparation, emulsion PCR, templated bead enrichment and sequencing of amplified PCR products and we applied the same strategy in this study as well [6]. De novo assembly, which was carried out by using the Geneious software [9], gave a sequence scaffold for additional primer design. To confirm sequence data obtained by semiconductor sequencing we used these additional primers (data not shown) in a primer walking sequencing strategy using the same amplicon that served as template for NGS library preparation. Sequence reads were subsequently assembled into a single consensus genomic sequence by AliView [10] that was deposited in GenBank under the accession number KY851116.
The genome of the suspect cyclovirus strain was 1,902 nt in length. Genome annotation [9, https://www.ncbi.nlm.nih.gov/orffinder/] identified the ORFs coding for the rep and the cap with lengths of 981 nt and 759 nt, respectively. Introns were not found in any ORFs. The two ORFs were predicted to localize on complementary DNA strands (Fig. 1). The non-coding region between the 5’ end of rep and cap gene measured 158 nt, whereas the non-coding region between the 3’ end of rep and cap was 4 nt long. The sequence of the nonanucleotide motif located upstream of the start codon of the cap gene was TAGTATTAC (Fig. 1). Collectively, these genomic features suggested that the duck-origin ssDNA virus can be classified within the newly proposed Cyclovirus genus.

Genomic organization of the novel duck associated cyclovirus 1 representing the rep and cap genes and the nonanucleotide motif
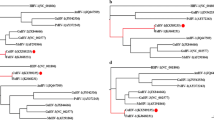
This finding was confirmed by phylogenetic analysis. The PhyML software was used to infer cyclovirus phylogeny [7]. The maximum likelihood algorithm using the GTR+G+I+F substitution model was selected and SH-like support was chosen to validate tree topology (Fig. 2). Pairwise distances from the whole genome alignment were calculated by using the Sequence Demarcation Tool v1.2 [14] using the Muscle alignment algorithm [4], which showed a range of sequence identities between 54.5% (FeACyV-1) and 60.6% (DfACyV-3) (Fig. 3). These values fell below the species demarcation threshold at 80% [16]. Further sequence analyses showed a close genetic relationship for the duck origin cyclovirus sequence with the human origin TN4 (nt, 98%) and the honey bee origin hb10 (nt, 100%) rep sequences along a ~470 nt fragment within the ORF (nt 1240-1710) encoding the rep protein (data not shown). These data together with current classification criteria strongly suggest that this duck origin cyclovirus isolate represents a novel species within the genus. Seeing the putative broad host range of the identified cyclovirus(es), however, assigning a host species may be challenging at this moment. It is unclear whether the duck cyclovirus related sequences detected in other host species might represent (i) viral genetic elements integrated into the respective animal genomic DNA, (ii) exogenous multi-host virus strains, (iii) exogenous viruses of yet unidentified organisms that are capable of colonizing various invertebrate and vertebrate animals, or (iv) these viruses are swallowed and pass through the intestine of animals by consumption of water or food where many newly described viruses possessing circular ssDNA genome can be effectively accumulated [1, 2]. The recent classification proposal for the Circoviridae which provides new taxonomic criteria for cycloviruses, does not satisfactorily discuss issues relating to the host origin of characterized strains. Nonetheless, given that the first representative full genome sequence originated from a wild duck specimen, we propose to introduce the name duck associated cyclovirus 1 for this isolate (DuACyV-1) and “Duck associated cyclovirus 1” for the species.

A maximum likelihood phylogenetic tree of representative cycloviruses’ whole genome sequences. Branches with SH-like support < 80% are not shown. Scale bar represents nucleotide substitutions per site. Sequence names include GenBank accession numbers followed by viral species using the acronyms introduced by Rosario et al. [16]; the novel duck origin cyclovirus is highlighted

A genome-wide pairwise identity matrix for representative cyclovirus strains. Sequence names include GenBank accession numbers followed by viral species using the acronyms introduced by Rosario et al. [16]; the novel duck origin cyclovirus is highlighted
In summary, this paper is the first to report a cyclovirus in wild bird species. Our study illustrates how little is known about the ecology and epidemiology of cycloviruses, a paradigm that needs to be addressed in future research and taxonomy proposals.
References
Dayaram A, Goldstien S, Argüello-Astorga GR, Zawar-Reza P, Gomez C, Harding JS, Varsani A (2015) Diverse small circular DNA viruses circulating amongst estuarine molluscs. Infect Genet Evol 31:284–295
Dayaram A, Galatowitsch ML, Argüello-Astorga GR, van Bysterveldt K, Kraberger S, Stainton D, Harding JS, Roumagnac P, Martin DP, Lefeuvre P, Varsani A (2016) Diverse circular replication-associated protein encoding viruses circulating in invertebrates within a lake ecosystem. Infect Genet Evol 39:304–316
Delwart E, Li L (2012) Rapidly expanding genetic diversity and host range of the Circoviridae viral family and other Rep encoding small circular ssDNA genomes. Virus Res 164:114–121
Edgar RC (2004) Muscle: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797
Fehér E, Székely C, Lőrincz M, Cech G, Tuboly T, Singh HS, Bányai K, Farkas SL (2013) Integrated circoviral rep-like sequences in the genome of cyprinid fish. Virus Genes 47:374–377
Fehér E, Pazár P, Kovács E, Farkas SL, Lengyel G, Jakab F, Martella V, Bányai K (2014) Molecular detection and characterization of human gyroviruses identified in the ferret fecal virome. Arch Virol 159:3401–3406
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59:307–321
Katzourakis A, Gifford RJ (2010) Endogenous viral elements in animal genomes. PLoS Genet 6:e1001191
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Meintjes P, Drummond A (2012) Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649
Larsson A (2014) AliView: a fast and lightweight alignment viewer and editor for large data sets. Bioinformatics 30:3276–3278
Li L, Kapoor A, Slikas B, Bamidele OS, Wang C, Shaukat S, Masroor MA, Wilson ML, Ndjango JB, Peeters M, Gross-Camp ND, Muller MN, Hahn BH, Wolfe ND, Triki H, Bartkus J, Zaidi SZ, Delwart E (2010) Multiple diverse circoviruses infect farm animals and are commonly found in human and chimpanzee feces. J Virol 84:1674–1682
Li L, Shan T, Soji OB, Alam MM, Kunz TH, Zaidi SZ, Delwart E (2011) Possible cross-species transmission of circoviruses and cycloviruses among farm animals. J Gen Virol 92:768–772
Marton S, Ihász K, Lengyel G, Farkas SL, Dán Á, Paulus P, Bányai K, Fehér E (2015) Ubiquiter circovirus sequences raise challenges in laboratory diagnosis: the case of honey bee and bee mite, reptiles, and free living amoebae. Acta Microbiol Immunol Hung 62:57–73
Muhire BM, Varsani A, Martin DP (2014) SDT: a virus classification tool based on pairwise sequence alignment and identity calculation. PLoS One 9:e108277
Ritchie BW, Niagro FD, Lukert PD, Steffens WL 3rd, Latimer KS (1989) Characterization of a new virus from cockatoos with psittacine beak and feather disease. Virology 171:83–88
Rosario K, Breitbart M, Harrach B, Segalés J, Delwart E, Biagini P, Varsani A (2017) Revisiting the taxonomy of the family Circoviridae: establishment of the genus Cyclovirus and removal of the genus Gyrovirus. Arch Virol 162:1447–1463
Shivaprasad HL, Hill D, Todd D, Smyth JA (2004) Circovirus infection in a Gouldian finch (Chloebia gouldiae). Avian Pathol 33:525–529
Soike D, Albrecht K, Hattermann K, Schmitt C, Mankertz A (2004) Novel circovirus in mulard ducks with developmental and feathering disorders. Vet Rec 154:792–793
Stewart ME, Perry R, Raidal SR (2006) Identification of a novel circovirus in Australian ravens (Corvus coronoides) with feather disease. Avian Pathol 35:86–92
Woods LW, Latimer KS, Barr BC, Niagro FD, Campagnoli RP, Nordhausen RW, Castro AE (1993) Circovirus-like infection in a pigeon. J Vet Diagn Invest 5:609–612
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Our work was supported by the Momentum Program of the Hungarian Academy of Sciences. EF and SM were recipients of the Bolyai scholarship.
Conflict of interest
The authors declare no competing interests.
Acknowledgements
We thank Ádám Dán at Veterinary Diagnostic Directorate, National Food Chain Safety Office (Budapest, Hungary) for sharing the cloacal swab sample containing the novel duck cyclovirus.
Ethical approval
This article does not contain any studies with animals performed by any of the authors.
Additional information
Handling Editor: Sheela Ramamoorthy.
Rights and permissions
About this article

Cite this article
Fehér, E., Kaszab, E., Forró, B. et al. Genome sequence of a mallard duck origin cyclovirus, DuACyV-1. Arch Virol 162, 3925–3929 (2017). https://doi.org/10.1007/s00705-017-3566-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-017-3566-z