
Abstract
Three cycloviruses (genus Cyclovirus, family Circoviridae) were recovered from a dragonfly (Odonata: Anisoptera) captured in Fuzhou, China. The three cycloviruses, named dragonfly associated cyclovirus 9, 10 and 11 (DfCyV-9, -10, -11), respectively, show 56.1-79.6% genome-wide identity to known cycloviruses and 61.6-65.1% among themselves. Thus, according to the current species demarcation criteria, they represent three novel cycloviruses. Notably, DfCyV-10 has a predicted replication-associated protein (Rep) that is most similar to that of bat associated cyclovirus 2 (BatACyV-2), a cyclovirus discovered in China, with 79.4% amino acid sequence identity, but a putative capsid protein (Cp) most similar to that of BatACyV-10, a cyclovirus discovered in Brazil, with 71.7% amino acid sequence identity. These data are useful for understanding the diversity and evolution of cycloviruses, especially those found in insects.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Circular single-stranded DNA (ssDNA) viruses of the family Circoviridae are grouped into two genera, named Circovirus (29 species) and Cyclovirus (45 species), that are distinguished based on their genome arrangement [16]. While several members of the genus Circovirus, including porcine circovirus type 2 and beak and feather disease virus, cause diseases in their host [1, 19], no pathogenic effects have been described for cycloviruses, although several cycloviruses have been recovered from cerebrospinal fluid (CSF), blood serum, and respiratory secretions of patients suffering from various diseases [13, 17, 18]. The ssDNA genome of either a circovirus or a cyclovirus is about 2000 nt in size, containing at least two inversely arranged open reading frames (ORFs) encoding a replication-associated protein (Rep), which is involved in the initiation of rolling-circle replication (RCR) of the virus, and a capsid protein (Cp). The intergenic region between the 5′ ends of Rep and Cp contains a putative stem-loop structure with a conserved nonanucleotide motif (TAGTATTAC or its variants) at its apex, which serves as the site for the origin of RCR (ori) [2]. For circoviruses, the conserved nonanucleotide motif is located in the Rep-encoding strand of the genome, whereas for cycloviruses, this motif is found in the Cp-encoding strand [4, 16]. Circoviruses have a somewhat larger genome and a longer 3′ intergenic region than cycloviruses [8].
In addition to a wide range of vertebrate animal species, cycloviruses may also have invertebrates as their hosts. To date, several cycloviruses have been discovered in dragonflies and in a cockroach of the species Eurycotis floridana (Walker) [3, 12, 14,15,16]. In addition, many cycloviruses recovered from the faeces of bats are thought to originate from their insect prey [6, 9, 20].
Dragonflies (Odonata: Anisoptera) are thought to be a valuable sampling tool for identifying insect ssDNA viruses, including cycloviruses [3, 14, 15]. Several papers have documented a great diversity of cycloviruses in dragonflies collected from various locations around the world, including the USA and New Zealand [3, 14, 15]. However, similar studies have not been reported for dragonflies collected from Asian countries. In this paper, we report three novel dragonfly-associated cycloviruses from China.
Sequencing methods
A dragonfly (Odonata: Anisoptera) was captured in Fuzhou, China, on September 10, 2016. The abdomen of the dragonfly was dissected and homogenized in 1.5 ml of SM buffer (0.1 M NaCl, 50 mM Tris/HCl [pH 7.4], 10 mM MgSO4). Virus particles were separated from the homogenates by filtration, and nucleic acids were extracted from these particles as described by Rosario et al. [14, 15]. Nucleic acids were extracted from the virus particles using a QIAamp MinElute Virus Spin Kit (QIAGEN). Rolling-circle amplification (RCA) was used to enrich for circular DNA molecules. The RCA product was digested with EcoRI, BamHI HindIII and PstI, respectively, and the products of each enzyme digestion were electrophoresed through a 1% (w/v) agarose gel. Bands with a size ranging from 1500 to 2000 bp were recovered, purified, ligated to the corresponding enzyme-digested pGEM-T (Promega Corporation) vector. The recombinant plasmids obtained were used to transform Escherichia coli and cloned. Positive colonies were selected to sequence the inserts of interest with flanking primers. After assembling the sequence with the aid of DNASTAR Lasergene 10 software (DNASTAR Inc.) and manual inspection, three circular ssDNAs, each representing the genome of a distinct cyclovirus, were obtained. The three cycloviruses were named “dragonfly associated cyclovirus 9”, “10” and “11” (DfCyV-9, -10, -11), respectively. The genome sequences of the three cycloviruses were deposited in the GenBank database, and the accession numbers MG779477, MG779478 and MG779479 have been assigned to DfCyV-9, -10 and -11, respectively.
Sequence properties
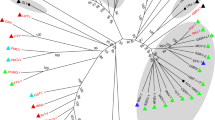
DfCyV-9 to -11 show 61.6% to 65.1% genome-wide nucleotide sequence identity to each other, as determined using the Sequence Demarcation Tool (SDT) v1.2, after an alignment the three cycloviral genomes with the MUSCLE program [5, 10]. When compared with members of the 43 approved species of cycloviruses, DfCyV-9-11 was found to be most similar to bat associated cyclovirus 3 (BatACyV3; JF938081), bat associated cyclovirus 2 (BatACyV-2; JF938079) and human associated cyclovirus-3 (HuACyV-3; GQ404846) with 66.7%, 79.6% and 66.8% genome-wide nucleotide identity, respectively (Fig. 1A).

a Genome-wide pairwise nucleotide sequence identities among dragonfly associated cyclovirus 9, 10 and 11 (DfCyV-9, -10, -11) and members of the genus Cyclovirus. DfCyV-9, -10, and -11 are indicated by red arrows. b Schematic genome organization of DfCyV-9, -10, and -11. The putative stem-loop structure of each cyclovirus is shown to the right of the circular genome
The genome of DfCyV-9-11 contains the conserved nonamer sequence TAGTATTAC located on the apex of a putative stem-loop structure (Fig. 1B). As with other cycloviruses, two major ORFs, one encoding a replication-associated protein (Rep) and the other encoding a putative capsid protein (Cp), are found in the genome of DfCyV-9-11 [8]. The two ORFs are inversely arranged, with the Cp-encoding ORF in the strand containing the nonamer sequence TAGTATTAC and the Rep-encoding ORF in the opposite strand (Fig. 1B), consistent with known cycloviruses [15, 16]. The intergenic region between the 5′ ends of the Rep- and Cp-encoding ORFs is 241, 211, and 224 nt long for DfCyV-9, -10, and -11, respectively. Similar to other cycloviruses, DfCyV-9, -10, and -11 lack (DfCyV-FZ-1 and -3) or have a very short (DfCyV-FZ-2) intergenic region between the 3′ ends of the two major ORFs (Fig. 1B).
The predicted Rep proteins of DfCyV-FZ-1, -2, and -3, which are 278, 243 and 280 amino acids in length, respectively, share between 38% and 79% pairwise amino acid identity with those of known cycloviruses (Supplemental Fig. 1A). The predicted Cp proteins of DfCyCV 1, 2, and 3 are 277, 215, and 216 amino acids in length, respectively, and show less than 47% pairwise amino acid sequence identity to the Cp of previously described cycloviruses. Interestingly, the predicted Cp of DfCyV-10 shows an unexpectedly high level of sequence identity to those of two other cycloviruses, bat associated cyclovirus 10 (BatACyV-10; KM382270), a cyclovirus discovered in Brazil, and human associated cyclovirus 6 (HuACyV-6; GQ404854), a cyclovirus discovered in Nigeria, at 71.7% and 71.0%, respectively. In contrast, the closest match of the Rep protein of DfCyV-10 is the Rep from BatACyV2, a cyclovirus discovered in China, based on sequence analysis with SDT v1.2, with 79.4% amino acid sequence identity. The amino acid sequence identity between the Rep of DfCyV-10 and that of BatACyV10 and HuACyV6 is much lower than this value, at 56.7% and 59.3%, respectively (Supplemental Fig. 1B).
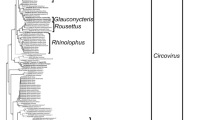
A phylogenetic tree was built using the aligned full-genome sequences of DfCyV-9, 10, and 11 and those of members of approved cycloviral species [16]. As shown in Fig. 2, DfCyV-DfCyV-9, 10, and 11 fell within distinct clusters within the tree. DfCyV-9 was separated from DfACyV-2 and HuACyV-10 but was more closely clustered with HuACyV-10. DfCyV-10 was separated from BatACyV-1 and BatACyV-2 but was more closely related to BatACyV-2. DfCyV-11 formed a cluster with three cycloviruses, BatACyV-4, BatACyV-7 and HuACyV-7, and was placed at a basal position relative to the latter three cycloviruses. Overall, the phylogenetic analysis supports the notion that DfCyV-9, 10, and 11 represent three new species of cycloviruses.

Phylogenetic tree showing the taxonomic position of dragonfly associated cyclovirus 9, 10 and 11 (DfCyV-9, -10, and -11, in red). The phylogenetic tree was constructed by the maximum-likelihood (ML) method implemented in IQ-TREE [11], using the TN+R4 substitution model, which was selected using the Bayesian information criterion by ModelFinder [7]. The tree was rooted using the genome sequence of PCV-2 as an outgroup
In summary, this paper is the first to report dragonfly-associated cycloviruses from China. Our study further demonstrates the high diversity of cycloviruses associated with insects.
References
Allan GM, Ellis JA (2000) Porcine circoviruses: a review. J Vet Diag Invest 12:3–14
Biagini P, Bendinelli M, Hino S, Kakkola L, Mankertz A, Niel C, Okamoto H, Raidal S, Teo CG, Todd D (2012) Family Circoviridae. In: King AMQ, Lindberg AM, Pallansch MA, Palmenberg AC, Simmonds P (eds) Virus taxonomy: classification and nomenclature of viruses: ninth report of the International Committee on Taxonomy of Viruses. Academic Press, London, pp 343–349
Dayaram A, Potter KA, Moline AB, Rosenstein DD, Marinov M, Thomas JE, Breitbart M, Rosario K, Argüello-Astorga GR, Varsani A (2013) High global diversity of cycloviruses amongst dragonflies. J Gen Virol 94:1827–1840
Delwart E, Li L (2012) Rapidly expanding genetic diversity and host range of the Circoviridae viral family and other Rep encoding small circular ssDNA genomes. Virus Res 164:114–121
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucl Acids Res 32:1792–1797
Ge X, Li J, Peng C, Wu L, Yang X, Wu Y, Zhang Y, Shi Z (2011) Genetic diversity of novel circular ssDNA viruses in bats in China. J Gen Virol 92:2646–2653
Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS (2017) ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods 14:587
Li L, Kapoor A, Slikas B, Bamidele OS, Wang C, Shaukat S, Masroor MA, Wilson ML, Ndjango J-BN, Peeters M (2010) Multiple diverse circoviruses infect farm animals and are commonly found in human and chimpanzee feces. J Virol 84:1674–1682
Male MF, Kraberger S, Stainton D, Kami V, Varsani A (2016) Cycloviruses, gemycircularviruses and other novel replication-associated protein encoding circular viruses in Pacific flying fox (Pteropus tonganus) faeces. Infec Gen Evo 39:279–292
Muhire BM, Varsani A, Martin DP (2014) SDT: a virus classification tool based on pairwise sequence alignment and identity calculation. PloS One 9:e108277
Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ (2014) IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evo 32:268–274
Padilla-Rodriguez M, Rosario K, Breitbart M (2013) Novel cyclovirus discovered in the Florida woods cockroach Eurycotis floridana (Walker). Arch Virol 158:1389–1392
Phan TG, Luchsinger V, Avendano LF, Deng X, Delwart E (2014) Cyclovirus in nasopharyngeal aspirates of Chilean children with respiratory infections. J Gen Virol 95:922–927
Rosario K, Marinov M, Stainton D, Kraberger S, Wiltshire EJ, Collings DA, Walters M, Martin DP, Breitbart M, Varsani A (2011) Dragonfly cyclovirus, a novel single-stranded DNA virus discovered in dragonflies (Odonata: Anisoptera). J Gen Virol 92:1302–1308
Rosario K, Dayaram A, Marinov M, Ware J, Kraberger S, Stainton D, Breitbart M, Varsani A (2012) Diverse circular ssDNA viruses discovered in dragonflies (Odonata: Epiprocta). J Gen Virol 93:2668–2681
Rosario K, Breitbart M, Harrach B, Segalés J, Delwart E, Biagini P, Varsani A (2017) Revisiting the taxonomy of the family Circoviridae: establishment of the genus Cyclovirus and removal of the genus Gyrovirus. Arch Virol 162:1447–1463
Smits SL, Zijlstra E, van Hellemond JJ, Schapendonk CME, Bodewes R, Schürch AC, Haagmans BL, Osterhaus ADME (2013) Novel cyclovirus in human cerebrospinal fluid, Malawi, 2010–2011. Emer Infect Dis 19:1511–1513
Tan LV, van Doorn HR, Nghia HD, Chau TT, Tu LTP, de Vries M, Canuti M, Deijs M, Jebbink MF, Baker S, Bryant JE, Tham NT, BKrong NTTC, Boni MF, Loi TQ, Phuong LT, Verhoeven JT, Crusat M, Jeeninga RE, Schultsz C, Chau NV, Hien TT, van der Hoek L, Farrar J, de Jong MD (2013) Identification of a new cyclovirus in cerebrospinal fluid of patients with acute central nervous system infections. MBio 4:e00213–e00231
Todd D (2000) Circoviruses: immunosuppressive threats to avian species: a review. Avian Pathol 29:373–394
Wu Z, Yang L, Ren X, He G, Zhang J, Yang J, Qian Z, Dong J, Sun L, Zhu Y (2016) Deciphering the bat virome catalog to better understand the ecological diversity of bat viruses and the bat origin of emerging infectious diseases. ISME J 10:609–620
Acknowledgments
This work was supported by a fund from the State Tobacco Monopoly Administration (110201601024(LS-04)) and an FAFU fund for excellent young scholars (xjq201622). We are grateful to Dr. Fangluan Gao at the College of Plant Protection, Fujian Agriculture and Forestry University, for his help in constructing the phylogenetic tree.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The author declares no competing interests.
Ethical approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. This article does not contain any studies with human participants performed by any of the authors.
Additional information
Handling Editor: T. K. Frey.
Electronic supplementary material
Below is the link to the electronic supplementary material.
705_2018_3876_MOESM1_ESM.png
Supplementary Fig. 1 Pairwise comparisons among dragonfly associated cyclovirus 9, 10 and 11 (DfCyV-9, -10, and -11) and members of the genus Cyclovirus, including identity values based on the replication-associated (Rep) (A) and capsid (Cp) (B) protein coding sequences
Rights and permissions
About this article

{kind=link}
Cite this article
Islam, S.U., Lin, W., Wu, R. et al. Complete genome sequences of three novel cycloviruses identified in a dragonfly (Odonata: Anisoptera) from China. Arch Virol 163, 2569–2573 (2018). https://doi.org/10.1007/s00705-018-3876-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-018-3876-9