
Abstract
The dog provides a large animal model of familial dilated cardiomyopathy for the study of important aspects of this common familial cardiovascular disease. We have previously demonstrated a form of canine dilated cardiomyopathy in the Doberman pinscher breed that is inherited as an autosomal dominant trait and is associated with a splice site variant in the pyruvate dehydrogenase kinase 4 (PDK4) gene, however, genetic heterogeneity exists in this species as well and not all affected dogs have the PDK4 variant. Whole genome sequencing of a family of Doberman pinchers with dilated cardiomyopathy and sudden cardiac death without the PDK4 variant was performed. A pathologic missense variant in the titin gene located in an immunoglobulin-like domain in the I-band spanning region of the molecule was identified and was highly associated with the disease (p < 0.0001). We demonstrate here the identification of a variant in the titin gene highly associated with the disease in this spontaneous canine model of dilated cardiomyopathy. This large animal model of familial dilated cardiomyopathy shares many similarities with the human disease including mode of inheritance, clinical presentation, genetic heterogeneity and a pathologic variant in the titin gene. The dog is an excellent model to improve our understanding of the genotypic phenotypic relationships, penetrance, expression and the pathophysiology of variants in the titin gene.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Dilated cardiomyopathy (DCM) is a primary heart muscle disease characterized by left ventricular dilation and systolic dysfunction. It is one of the most common forms of familial cardiomyopathy in human beings, characterized by both genetic and allelic heterogeneity (Morales and Hershberger 2015). Variants have been identified in over 60 genes, particularly those that encode for sarcomeric and cytoskeletal proteins. Variants in the titin gene (TTN) are reported most commonly (de Gonzalo-Calvo et al. 2017; Franaszczyk et al. 2017; Herman et al. 2012; Hershberger et al. 2013; Towbin 2014). Although causative variants are now well-known for this common cardiovascular disease, there remain many poorly understood aspects of this familial disease including the great degree of genetic heterogeneity, variable penetrance and expression, and possible indication for treatment for genotype positive, preclinical disease individuals.
The dog is a natural animal model of familial DCM that has been used to study many different aspects of the disease from its pathophysiology to stem cell therapy (Cheng et al. 2016; Hensley et al. 2017). The Doberman pinscher is one of the breeds of dogs most commonly affected by familial DCM (Martin et al. 2010). Dilated cardiomyopathy in the Doberman pinscher is characterized by left ventricular dilation and systolic dysfunction, with subsequent development of both atrial and ventricular tachyarrhythmias (Calvert et al. 1997). Familial aspects of this disease are well-described (Mausberg et al. 2011; Meurs et al. 2007). We have previously determined that DCM in Doberman pinschers is inherited as an autosomal dominant trait, at least in some families of dogs (Meurs et al. 2007). A splice site variant has been identified in the PDK4 gene, encoding a mitochondrial protein that is strongly associated with the development of DCM in some families (Meurs et al. 2012). However, the PDK4 variant does not explain all cases of this familial canine disease even within this one breed, suggesting that even within a single breed of dog genetic heterogeneity is likely (Owczarek-Lipska et al. 2013). In this study, we evaluate a family of Doberman pinschers with DCM but without the PDK4 variant. A deleterious variant in the titin (TTN) gene was identified in this family as well as other affected dogs and was strongly associated (p < 0.0001) with the development of DCM. Sudden death was commonly observed. The variant is predicted to change the structure of the protein and was demonstrated to decrease active tension in myofibers from affected dogs.
Methods
This study was conducted in accordance with the guidelines of the North Carolina State University Institutional Animal Care and Use Committee.
Two sibling (male, female) Doberman pinchers were presented for evaluation of clinical signs consistent with congestive heart failure with DCM at the North Carolina State University College of Veterinary Medicine at 4 years of age. Their mother had died unexpectedly of sudden cardiac death at 6 years of age. The cardiac status of the father was unknown and he was not available for evaluation. Physical examination, echocardiographic and electrocardiographic examination were performed by a board-certified veterinary cardiologist. The echocardiogram was performed using standard two-dimensional and M-mode echocardiographic methods (Thomas et al. 1993). Dogs were considered to be affected if they had a left ventricular end diastolic dimension of at least 4.8 cm and a fractional shortening less than 20%, without evidence of ongoing systemic disease or other congenital or acquired heart disease (Meurs et al. 2008).
Both sibling dogs were determined to be affected by 4 years of age and died of sudden cardiac death by 5 years of age. The female had been bred once and had seven existing offspring. Six offspring were examined by a board-certified veterinary cardiologist, and all six were diagnosed with DCM between the ages of 3 and 6 and were managed with medical therapy (Fig. 1). Four died of sudden cardiac death by 6 years of age.

M-mode echocardiogram of the left ventricle of an unaffected (control) Doberman pinscher (a) and an affected Doberman pinscher (b). Note the reduced interventricular septum (IVS) and left ventricular free wall (LVFW) motion and increased left ventricular lumen (LFFW) size in the affected dog
DNA samples were obtained from the two proband siblings and the six available offspring of the female proband. All affected dogs in this family lacked the previously reported PDK4 variant. Samples from the female proband, one of her male affected offspring, and three additional (two male, one female) unrelated affected Doberman pinschers lacking the PDK4 variant were selected for whole genome sequencing.
Approximately, 3 µg of DNA from each dog was submitted for library preparation and whole genome sequencing (WGS) at the University of North Carolina Chapel Hill High-Throughout Sequencing Facility (https://www.med.unc.edu/genomics). All sequencing experiments were designed as 125 bp paired-end reads and samples were run on either 1 or 2 lanes of an Illumina HiSeq 2500 high-throughput sequencing system.
Variant calling from WGS data was performed using a standardized bioinformatics pipeline for all samples as described previously (Friedenberg and Meurs 2016). Briefly, sequence reads were trimmed using Trimmomatic 0.32 to a minimum phred-scaled base quality score of 30 at the start and end of each read with a minimum read length of 70 bp, and aligned to the canFam3 reference sequence using BWA 0.7.13 (Bolger et al. 2014; Li and Durbin 2009; Lindblad-Toh et al. 2005). Aligned reads were prepared for analysis using Picard Tools 2.5 (http://broadinstitute.github.io/picard) and GATK 3.7 following best practices for base quality score recalibration and indel realignment (Broad Institute, Cambridge, MA) (DePristo et al. 2011; McKenna et al. 2010; Van der Auwera et al. 2013). Variant calls were made using GATK’s HaplotyeCaller walker, and variant quality score recalibration (VQSR) was performed using sites from dbSNP 146 and the Illumina 174K CanineHD BeadChip as training resources. We applied a VQSR tranche sensitivity cutoff of 99.9% to SNPs and 99% to indels for use in downstream analyses; genotype calls with a phred-scaled quality score < 20 were flagged but not removed from the variant callset.
Variants present in at least 4 of 5 affected dogs (to allow for possible areas of poor coverage) were selected and filtered against a database of variants from 250 non-Doberman pinscher dogs from 44 different dog breeds. Variants that were present in at least four of five affected dogs and in none of the non-Doberman pinschers were selected for further evaluation and were then categorized by Variant Effect Predictor 87 and prioritized by their functional impact (e.g., stop codon, change in amino acid, etc.) as well as potential cardiac involvement (evidence of cardiac expression, previous association with some form of cardiomyopathy) (McLaren et al. 2016). Variants of highest interest were evaluated for functional significance with Polyphen (http://genetics.bwh.harvard.edu/pph2/), SIFT (http://sift.jcvi.org/) and Provean (http://provean.jcvi.org/index.php) and were evaluated for previous identification in the canine population in the DogSD (http://bigd.big.ac.cn/dogsdv2/) SNP database.
Variants shown to have an important functional impact by altering a stop or start codon, creating a frameshift or altering or creating a splice site, or altering a conserved amino acid in a way in which was predicted to be deleterious by at least two of the three in silico programs (SIFT, Provean, Polyphen) and were believed to have cardiac importance through expression or function within the heart were selected for additional evaluation. Variants were then evaluated by Sanger Sequencing of 135 affected and 122 unaffected Doberman pinchers from a DNA database maintained at the NCSU College of Veterinary Medicine. Affected phenotype was defined as described above and the unaffected phenotype was based on the presence of echocardiographically determined normal left ventricular dimensions, a fractional shortening of at least 25% and an age of at least 10 years. The variants were tested for allelic association with DCM using a Fisher’s exact test. A p value of < 0.05 was considered significant. Variants significantly associated with disease were next genotyped in an additional 258 non-Doberman pinscher dogs for a total of 508 (including the 250 from the pipeline indicated above) non-Doberman pinschers genotyped from 87 different breeds (Supplemental Table 2). Penetrance was determined for the most promising variants that were evaluated in affected Doberman Pinschers lacking the PDK4 variant by Sanger sequencing.
Titin isoform (N2BA and N2B) expression analysis of LV tissues was performed as previously described (Lahmers et al. 2004; Warren et al. 2003) using samples from four control dogs with the wildtype genotype and seven affected dogs (four heterozygous and three homozygous for the variant). The solubilized samples were electrophoresed on 1% Seakem Gold Agarose gels (Lonza, Allendale, NJ) using a Vertical Agarose Gel System as previously described (Warren et al. 2003). Gels were run for 3 h 20 min (@15 mA per gel), stained overnight using Coomassie brilliant blue (Acros Organics, Pittsburgh, PA), scanned using a commercial scanner (Epson Corporation, Long Beach CA) and analyzed using One-D scan (Scanalytics Inc, Rockville MD). Each sample was loaded in a range of six volumes (microliters) and the integrated optical density (OD) of titin and myosin heavy chain (MHC) was determined as a function of volume. Further, the slope of the linear relationship was obtained for each protein to quantify expression ratios. For titin Western blots, solubilized samples were run on a 0.8% Seakem Gold Agarose gel as described. On day 1, gels were run for 2 h 40 min, transferred onto a PVDF membrane (Immobilon-FL®, Millipore, Burlington, MA) using a semi-dry transfer unit (Trans-Blot Cell, Bio-Rad, Hercules CA) for 2 h 30 min, stained using Ponceau-S (Sigma, Burlington, MA) to visualize the total protein transferred and let dry overnight. On day 2, membranes were scanned and probed overnight with anti-titin N-terminus (Z1Z2, Abnova, Cat.No. H00007273-M06) and anti-titin C-terminus (M8M9, http://www.myomedix.com) antibodies. On day 3, membranes were labeled with secondary antibodies conjugated to fluorescent dyes with infrared (IR) excitation spectra (CF680, Goat anti-Rabbit, 20067-1 and CF790, Goat anti-Mouse 20342, Biotium Hayward, CA). Blots were then scanned using Odyssey Infrared Imaging System (LI-COR Biosciences, Lincoln, NE) and the images analyzed using LI-COR software (Image Studio Lite 5.2). Expression data was analyzed between the two groups with a Student’s t test.
Finally, ultrastructural and mechanical analysis on LV tissue of two control (CC) and two homozygous (TT) animals was performed. Frozen LV cardiac tissues from two affected Doberman pinchers homozygous positive for the variant and PDK4 variant negative and from two unaffected dogs with the wildtype genotype (controls), were warmed up from − 80 to − 20 °C in a 50% glycerol/relaxing solution [(in mM): 40 BES, 10 EGTA, 6.56 MgCl2, 5.88 Na-ATP, 1 dithiothreitol (DTT), 46.35 K-propionate, 15 creatine phosphate, pH 7.0] containing protease inhibitors [(in mM): 0.1 E64, 0.47 leupeptin and 0.25 phenylmethylsulfonyl fluoride (PMSF)] for 24 h. The samples were placed on ice and fiber bundles were dissected and skinned in relaxing solution containing 1% Triton X-100 and protease inhibitors for 3 h at 4 °C in a rotator. Skinned fiber bundles were washed thoroughly with relaxing solution, stretched from the slack length at ~ 15% in a sylgard coated petri dish and processed by conventional TEM. Briefly, fiber bundles were fixed in a mix solution of 3.7% paraformaldehyde, 3% glutaraldehyde and 0.2% tannic acid in PBS (10 mM, pH 7.2), and post-fixed in 1% OsO4 in PBS for 30 min at 4 °C. Subsequently, samples were dehydrated in an ethanol graded series, infiltrated with propylene oxide, and transferred to a mix of 1:1 propylene oxide:Araldite 502/Embed 812 (Epon-812, EMS), then to a pure Araldite 502/Embed 812 resin and polymerized for 48 h at 60 °C. Longitudinal sections of 100 nm were obtained with a Reichert-Jung ultramicrotome and contrasted with 1% potassium permanganate and lead citrate. Samples were observed in a TECNAI Spirit G2 transmission electron microscope (FEI) operated at 100 kV, and images acquired with a side-mounted AMT Image Capture Engine V6.02 (4Mpix) digital camera.
Mechanical studies were carried out on LV wall muscle strips isolated from four control (CC) and two homozygous (TT) animals. Mid-myocardial fibers from tissue stored at − 20 °C in 50% glycerol/relaxing solution were identified and dissected out in 100–200 µm diameter strips, approximately 1500 µm long (Ottenheijm et al. 2010). The strips were briefly washed with relaxing solution then skinned for 1 h at 4 °C on a 2D rocker. The strips were washed with a relaxing solution, then aluminum T-clips were attached to the ends for mounting. One end was attached to a force transducer (AE801; SensorOne) and the other to a servo-motor (308B; Aurora) in a bath of relaxing solution (Granzier and Labeit 2006). Sarcomere length (SL) was measured using a laser diffraction system. Active force was measured by stretching the preparation to SL 2.0 µm and activating it using a pCa 4.0 solution (in mM: 40 BES, 10 CaCO3 EGTA, 6.29 MgCl2, 6.12 Na-ATP, 1 DTT, 45.3 potassium propionate, 15 creatine phosphate) with protease inhibitors. Tissue producing zero active force was immediately discarded. Slack length and cross-sectional area (CSA) of the preparation was measured to calibrate percent stretch and normalize measured forces (tension = mg/mm2). To measure total passive tension, a stretch–hold–release protocol was used in which the tissue was stretched to a given SL within the physiological range (2.0–2.3 µm) at 10%/s, held for 90 s, and released back to slack length. When SL could not be reliably measured, it was assumed to track with muscle length as it does in other samples. In cases like this, the stretch calibration from an average control sample was applied. The preparation was then extracted using sequential treatments (40 min) with 0.6 M KCl in relaxing solution followed by 1.0 M KI in relaxing solution to depolymerize the thick filaments and remove the anchoring points of titin, keeping ECM intact. The stretch–hold–release protocol was then repeated to measure the ECM-based passive tension. Titin-based passive tension was calculated by subtracting the ECM-based passive tension from the total passive tension. Data was analyzed with a non-parametric Mann–Whitney test.
A protein sequence analysis was used to access information on the predicted consequences of the amino acid change (McLaren et al. 2016; Laddach et al. 2017; Pires et al. 2014a, b). The structural model was produced using the JSmol applet (http://www.jmol.org) embedded in the TITINdb webserver (http://fraternalilab.kcl.ac.uk/TITINdb/). The protein sequence alignment is a subset of the titin orthologue multiple sequence alignment downloaded from Ensembl (Vilella et al. 2009).
Results
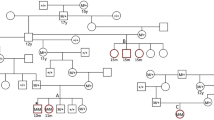
After filtering, 31,808 variants were identified in at least 4 of 5 affected Doberman pinscher dogs (cases) and absent in the 250 non-Doberman pinscher dogs from 44 different dog breeds (controls). Fourteen variants were predicted to create a gain of stop, 4 caused a loss of stop, 90 created a frameshift, 16 created an inframe deletion, 20 an inframe insertion, 60 altered a splice site and 1563 altered a missense mutation. The remaining variants were synonymous, located in a 3′ or 5′ untranslated region, upstream or downstream or intronic. One hundred and forty-three of the variants believed to be of the greatest importance as defined above were evaluated for Sanger sequencing (Supplemental Table 1). Five variants had a p < 0.1 (Table 1). However, only one, a missense C/T variant in the titin gene (ENSCAFT00000022319) at canine chromosome 36:22,321,955, was significantly associated (p < 0.0001) with DCM in the initial Sanger sequencing and identified in all of the affected dogs in the family of Doberman pinschers with DCM (Figs. 2, 3). The variant was not observed in any of the 508 unaffected non-Doberman pinscher dogs of 87 breeds from the canine whole genome database and was not identified in DogSD (http://bigd.big.ac.cn/dogsdv2/) as a known canine SNP (Supplemental Table 2). Within the family of affected Dobermans pinschers, the two probands dogs were heterozygous, four of six affected offspring were homozygous and two were heterozygous for the variant, consistent with an autosomal dominant mode of inheritance (Fig. 3). There was no apparent difference in clinical severity with all dogs developing the disease by 6 years of age, and six (three heterozygous and three homozygous) of the eight dogs dying of sudden death by 6 years of age.

Chromatogram of DNA sequence from unaffected (control) dog (a) and affected dog (b). Arrows indicate location of DNA variant (C in unaffected dog, T in affected dog)

Pedigree from a family of Doberman pinschers with dilated cardiomyopathy. Circles indicate females, squares indicate males. Black symbols indicate affected dogs; patterned symbols indicate unknown phenotype. Symbols with a line indicate deceased animals. The dog’s genotype is indicated next to each symbol. TT homozygous variant, CT heterozygous variant, CC wildtype homozygous
The variant had an association of p < 0.0001 to disease in a population of 125 unrelated affected Doberman pinschers. The CT and TT genotypes were both significantly associated with the DCM population in comparison to the control population by p < 0.003 and p < 0.0001, respectively. The variant had a calculated disease penetrance of 47% in DCM PDK4 negative dogs. Both variants (PDK4 and titin) were genotyped in 96 affected dogs and 124 unaffected dogs (Table 2). One, or both variants were identified in 99% of the affected dogs and in 73% of unaffected dogs, consistent with the incomplete penetrance of both variants for this disease.
The TTN variant is predicted to change the amino acid from glycine to arginine; glycine at this location is highly conserved in a wide range of species and corresponds in humans to residue p.8898G > R (ENST00000589042.5) in the immunoglobulin-like domain I71 (Fig. 4). I71 is located in the middle Tandem Ig segment of titin’s molecular spring region of the cardiac N2BA isoform. The variant was predicted to be a deleterious change by variant prediction algorithms. Polyphen (http://genetics.bwh.harvard.edu/pph2/), predicted the variant to be likely damaging (0.999; scores of 0.85–1.0 are predicted to be deleterious); SIFT (http://sift.jcvi.org/) predicted it to be a deleterious change (0.03; scores of 0–0.05 are predicted to be deleterious) and Provean (http://provean.jcvi.org/index.php) predicted it to be a deleterious change (− 5.113; scored of − 2.5 or less are predicted to be deleterious).

a The DNA variant is predicted to change an amino acid in an immunoglobulin-like domain from glycine to arginine (location indicated by circle), which would correspond in human to p.8898G>R in Ig domain I71. b Glycine is a highly conserved amino acid at this location. c Schematic of sarcomere with the indicated location of titin and (at bottom) the domain composition of titin’s molecular spring region (N2BA isoform). I71 is found in titin’s middle tandem Ig segment
A titin protein expression analysis was performed using left ventricular (LV) wall tissue that had been flash frozen. No major differences were found in titin expression when comparing control animals with animals that were either heterozygous for the variant (CT) or homozygous (TT) (Fig. 5a). The total titin–myosin heavy chain ratio was unaltered [0.20 ± 0.04 (CC), 0.17 ± 0.03 (CT), 0.29 ± 0.06 (TT)]. A Western blot analysis using the Z1Z2 antibody that detects titin’s N-terminus and the M8M9 antibody that detect titin’s C-terminus revealed that both N2BA and N2B titin in mutant animals were full-length titin (Fig. 5b). There was no apparent difference in the expression ratio of N2BA:N2B titin (Fig. 5a, top). The ratio of T2 (large degradation product), to T1 (full-length titin) revealed a trend towards being higher in animals carrying the variant (Fig. 5c, bottom), suggesting that there might be more protein degradation in animals carrying the variant.

a Protein gel revealing titin in unaffected (control) Doberman pinscher dog (CC), a heterozygous dog (CT) and a homozygous dog (TT). Titin appears normal in all genotypes. b Western blot analysis using the Z1Z2 antibody raised to titin’s N-terminus (top) and M8M9 raised to titin’s C-terminus (bottom) show that the bands identified as N2BA and N2B titin on protein gels are full-length molecules in all genotypes. c Top: quantitative analysis of expression ratio of N2BA titin (top band on gel) vs. N2B titin (bottom band) reveals no difference. Bottom: the ratio between T2 (large degradation product of titin) and T1 (full-length titin) shows a trend towards an increase in heterozygous and homozygous dogs. Mean and standard error of the mean are shown
Mechanical studies performed on LV wall muscle strips isolated from two control (CC) and two homozygous (TT) animals indicate a marginal (p = 0.057) reduction in maximal active tension in the homozygous animals (Fig. 6a) but no difference in passive tension (Fig. 6b).

Active tension (a) and passive tension (b) in cardiac tissue from unaffected (control) Doberman pincher (CC) and DCM dogs (TT). a Maximal active tension, at pCa 4.0 and measured at SL 2.0 mm, was higher in the unaffected dogs (p = 0.057). b No significant differences in titin-based passive tension were observed
Electron microscopy revealed focal and myopathic abnormalities in homozygous animals that include myofibrillar disarray and less dense packing of myofibrils, with sarcomeres that vary greatly in sarcomere length. In addition, Z-disk streaming was regularly observed and the Z-disks varied in thickness (Fig. 7). Similar changes including Z-disk streaming and myofibrillar disarray were observed in sections of the biceps femoris muscle of a homozygous dog (TT), results not shown.

Representative electron micrographs of cardiac tissue from two unaffected (control) Doberman pinchers dogs (CC) and two DCM dogs homozygous for the TTN mutation (TT). In sections from unaffected dogs (left two panels obtained from two different animals), the sarcomeres show a regular structure, while in the sections from the affected dogs (middle two panels and right two panels obtained from two different animals) sarcomere misalignment is observed, as well as disarrangement of myofilaments, wide spaces between myofibrils, Z-disk streaming with increased Z-disk thickness, and variation in I-band width. (Scale bar 1 µm.)
Discussion
The canine variant reported here is in titin, the gene most commonly associated with DCM in human beings (de Gonzalo-Calvo et al. 2017; Franaszczyk et al. 2017; Herman et al. 2012; Kimura 2016; LeWinter and Granzier 2014). Titin is the largest known protein and is expressed in both cardiac and skeletal muscle where it acts as a molecular spring contributing to both passive stiffness of muscle and active contraction, as well as biomechanical sensing and signaling (de Gonzalo-Calvo et al. 2017; Herman et al. 2012; Kimura 2016; Linke 2018; Granzier et al. 2009). In the myocyte, titin filaments span one-half of each sarcomere by anchoring the amino-terminus to the Z-disk and the carboxy-terminus to the M-line (Linke 2018; Neiva-Sousa et al. 2015; Gigli et al. 2016). Titin’s functional regions are named by their locations within the sarcomere including the Z-disk, I-band, A-band, and M-band regions (Granzier and Labeit 2002; Gerull et al. 2006). Titin gene variants that cause DCM in people have been located throughout the molecule, consistent with the allelic heterogeneity of this disease, although most have been found in the A band region (Herman et al. 2012; LeWinter and Granzier 2014; Neiva-Sousa et al. 2015; Gigli et al. 2016; Gerull et al. 2002, 2006; Itoh-Satoh et al. 2002; Schafer et al. 2017).
The missense variant reported here was identified within an immunoglobulin-like domain, in humans referred to as I71 (Bang et al. 2001). I71 is part of the middle tandem Ig segment that compromises one of the three spring elements within cardiac titin. I71 is included in both skeletal muscle titin and in the N2BA isoform of cardiac titin but not in the N2B cardiac isoform (Herman et al. 2012; Schafer et al. 2017; Bang et al. 2001; Labeit et al. 2006). Thus, the effect of the variant is likely to be isoform specific. Although a human variant causing DCM has not been identified in this exact location, a missense variant has been observed within the orthologous exon in humans with an arrhythmogenic right ventricular cardiomyopathy phenotype (Taylor et al. 2011).
It is unclear exactly how the variant identified here leads to the development of DCM. In silico programs predicted that the protein is not truncated but that rather its structure is altered. Thus, it is possible that I71 is either permanently unfolded or easily unfolds when force is exerted on the domain during diastole when sarcomeres are stretched. An unfolded domain is likely to have a heightened sensitivity to degradation, a notion that is supported by the increased T2 (degradation product) to T1 (full-length molecule) ratio (Fig. 5c, bottom) (Anderson et al. 2013). Such proteolysis would be expected to reduce passive tension, but the effect is likely to be modest because there is less N2BA titin (I71 is included) than N2B titin (I71 is absent). Indeed, there was no difference in the measured titin-based passive tension (Fig. 6b). This is in contrast to a recent study by Vikhorev et al. on human cardiac myofibrils from patients with DCM due to truncation variants in TTN (TTNtv); the authors speculated that the reduced passive tension is important in the pathology of TTNtv DCM (Vikhorev et al. 2017). Our study reveals that DCM can develop when passive tension is normal.
The active tension that we obtained also contrasts with the study of Vikhorev et al. in that we found that maximal active tension is reduced (p = 0.057), whereas Vikhorev et al. found no difference (Vikhorev et al. 2017). The reduced active tension of our study is consistent with that of myofibers engineered from human iPSC-CMs heterozygous for TTNtv, which also showed a deficit in active tension generation (Hinson et al. 2015). It is not clear how a missense variant in I71 directly leads to a reduced level of maximal active tension and it seems likely therefore that the active tension reduction is a secondary effect that arises from the myofibrillar disarray and less dense packing of myofibrils observed in our electron microscopy study. Less dense packing of myofibrils and sarcomere disorganization is also observed in engineered heart tissues consisting of iPSC-derived cardiac myocytes obtained from patients with TTNtv (Vikhorev et al. 2017). We speculate therefore that the missense variant in I71 leads to less dense packing of myofibrils and that the ensuing overall tension reduction at the level of the myocyte results in pathological remodeling and DCM. Clearly, more work is required to determine the mechanism by which a missense variant in I71 leads to DCM. Additionally, the number of animals in which good quality tissue was available post mortem was small and we were only able to evaluate samples from two homozygous variant dogs and two homozygous wild type dogs. A larger number of samples, including some from heterozygous dogs would improve the strength of these findings.
Dilated cardiomyopathy is a heterogeneous condition. The penetrance varies depending on the causative mutation and other factors (environment, lifestyle, etc) that may impact development of disease (Hershberger et al. 2013). This is a phenomenon common to familial cardiomyopathies, and in some forms of cardiomyopathy, the penetrance may be as low as 20–30%, meaning that only 20–30% of those with the disease-causing mutation will demonstrate the disease (Sen-Chowdhry et al. 2005). The factors associated with whether a mutation is fully penetrant or has incomplete penetrance are poorly understood but can include age, sex, environment, lifestyle, and modifying genetic factors (Hershberger et al. 2013). In the family described here, all sibling dogs with the variant eventually developed the disease. However, in the overall population of Doberman pinschers evaluated, many dogs had the variant but have not developed the disease. This could indicate that the dogs in the family share genetic or environmental factors which influence onset of the disease, that age of onset is critical to developing the condition or simply an ascertainment bias.
The identification of a variant in the titin gene in this spontaneous canine model of DCM completes the characterization of a large animal model of familial DCM associated with titin. As such, it is an excellent model to improve our understanding of genotypic phenotypic relationships and the value of early medical and/or behavioral intervention in gene positive individuals. A key role for this model may be the elucidation of factors that influence penetrance and expression of disease, since the Doberman pinscher is a purebred dog from a closed gene pool with a restricted amount of genetic variation. Additionally, monitoring diet, exercise and hormonal balance are all easily achievable in this large animal model and tend to relate closely to that of human beings, yet the affected dog’s life time is short enough that it is reasonable to follow the development of the phenotype over a 4–6 year period.
References
Anderson BR, Bogomolovas J, Labeit S, Granzier H (2013) Single molecule force spectroscopy on titin implicates immunoglobulin domain stability as a cardiac disease mechanism. J Biol Chem 288:5303–5315
Bang ML, Centner T, Fornoff F, Geach AJ, Gotthardt M, McNabb M et al (2001) The complete gene sequence of titin, expression of an unusual approximately 700-kDa titin isoform, and its interaction with obscurin identify a novel Z-line to I-band linking system. Circ Res 89:1065–1072
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120
Calvert CA, Pickus CW, Jacobs GJ, Brown J (1997) Signalment, survival, and prognostic factors in Doberman pinschers with end-stage cardiomyopathy. J Vet Intern Med 11:323–326
Cheng Y, Hogarth KA, O’Sullivan ML, Regnier M, Pyle WG (2016) 2-Deoxyadenosine triphosphate restores the contractile function of cardiac myofibril from adult dogs with naturally occurring dilated cardiomyopathy. Am J Physiol Heart Circ Physiol 310:H80–H91
de Gonzalo-Calvo D, Quezada M, Campuzano O, Perez-Serra A, Broncano J, Ayala R et al (2017) Familial dilated cardiomyopathy: a multidisciplinary entity, from basic screening to novel circulating biomarkers. Int J Cardiol 228:870–880
DePristo MA, Banks E, Poplin RE, Garimella KV, Maguire JR, Hartl C et al (2011) A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43:491–498
Franaszczyk M, Chmielweski P, Truszkowska G, Stawinski P, Michalak E, Rydzanicz M et al (2017) Titin truncating variants in dilated cardiomyopathy—prevalence and genotype-phenotype correlations. PLoS One 12:e0169007
Friedenberg SG, Meurs KM (2016) Genotype imputation in the domestic dog. Mamm Genome 27:485–494
Gerull B, Gramlich M, Atherton J, McNabb M, Trombitas K, Sasse-Klaassen S et al (2002) Mutations of TTN, encoding the giant muscle filament titin, cause familial dilated cardiomyopathy. Nat Genet 30:201–204
Gerull B, Atherton J, Geupel A, Sasse-Klaasen S, Heuser A, Frenneaux M et al (2006) Identification of a novel frameshift mutation in the giant muscle filament titin in a large Australian family with dilated cardiomyopathy. J Mol Med 84:478–483
Gigli M, Begay RL, Morea G, Graw SL, Sinagra G, Taylor MRG et al (2016) A review of the giant protein titin in clinical molecular diagnostics of cardiomyopathies. Front Cardiovasc Med 3:21
Granzier H, Labeit S (2002) Cardiac titin: an adjustable multi-functional spring. J Physiol 541:335–342
Granzier HL, Labeit S (2006) The giant muscle protein titin is an adjustable molecular spring. Exerc Sport Sci Rev 34:50–53
Granzier HL, Radke MH, Peng J, Westermann D, Nelson OL, Rost K, King NMP et al (2009) Truncation of titin’s elastic PEVK region leads to cardiomyopathy with diastolic dysfunction. Circ Res 105:557–564
Hensley MT, Tang J, Woodruff K, Defrancesco T, Tou S, Williams CM et al (2017) Intracoronary allogeneic cardiosphere-derived stem cells are safe for use in dogs with dilated cardiomyopathy. J Cell Mol Med 21:1503–1512
Herman DS, Lam L, Taylor MRG, Wang L, Teekakirikul P, Christodoulou D et al (2012) Truncations of titin causing dilated cardiomyopathy. N Engl J Med 366:619–628
Hershberger RE, Hedges DJ, Morales A (2013) Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol 10:531–547
Hinson JT, Chopra A, Nafissi N, Polacheck WJ, Benson CC, Swist S et al (2015) Titin mutations in iPS cells define sarcomere insufficiency as a cause of dilated cardiomyopathy. Science 349:982–986
Itoh-Satoh M, Hayashi T, Nishi H, Koga Y, Arimura T, Koyanagi T et al (2002) Titin mutations as the molecular basis for dilated cardiomyopathy. Biochem Biophys Res Commun 291:385–393
Kimura A (2016) Molecular genetics and pathogenesis of cardiomyopathy. J Hum Genet 61:41–50
Labeit S, Lahmers S, Burkart C, Fong C, McNabb M, Witt S et al (2006) Expression of distinct classes of titin isoforms in striated and smooth muscles by alternative splicing, and their conserved interaction with filamins. J Mol Biol 362:664–681
Laddach A, Gautel M, Fraternali F (2017) TITINdb-a computational tool to assess titin’s role as a disease gene. Bioinformatics 33:3482–3485
Lahmers S, Wu Y, Call DR, Labeit S, Granzier H (2004) Developmental control of titin isoform expression and passive stiffness in fetal and neonatal myocardium. Circ Res 94:505–513
LeWinter MM, Granzier HL (2014) Cardiac titin and heart disease. J Cardiovasc Pharmacol 63:207–212
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25:1754–1760
Lindblad-Toh K, Wade CM, Mikkelson TS, Karlsson EK, Jaffe DB, Kamal M et al (2005) Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature 438:803–819
Linke WA (2018) Titin gene and protein functions in passive and active muscle. Annu Rev Physiol 80:389–411
Martin MW, Stafford Johnson MJ, Strehlau G, King JN (2010) Canine dilated cardiomyopathy: a retrospective study of prognostic findings in 367 clinical cases. J Small Anim Pract 51:428–436
Mausberg TB, Wess G, Simak J, Keller L, Drogemuller M, Drogemuller C et al (2011) A locus on chromosome 5 is associated with dilated cardiomyopathy in Doberman pinschers. PLoS One 6:e20042
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A et al (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20:1297–1303
McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GRS, Thormann A et al (2016) The ensembl variant effect predictor. Genome Biol 17:122
Meurs KM, Fox PR, Norgard M, Spier AW, Lamb A, Koplitz SL et al (2007) A prospective genetic evaluation of familial dilated cardiomyopathy in the Doberman pinscher. J Vet Intern Med 21:1016–1020
Meurs KM, Hendrix KP, Norgard MM (2008) Molecular evaluation of five cardiac genes in Doberman pinschers with dilated cardiomyopathy. Am J Vet Res 69:1050–1053
Meurs KM, Lahmers S, Keene BW, White SN, Oyama MA, Maucell E et al (2012) A splice site mutation in a gene encoding for PDK4, a mitochondrial protein, is associated with the development of dilated cardiomyopathy in the Doberman pinscher. Hum Genet 131:1319–1325
Morales A, Hershberger RE (2015) The rationale and timing of molecular genetic testing for dilated cardiomyopathy. Can J Cardiol 31:1309–1312
Neiva-Sousa M, Almeida-Coelho J, Falcao-Pires I, Leite-Moreira AF (2015) Titin mutations: the fall of Goliath. Heart Fail Rev 20:579–588
Ottenheijm CA, Hooljiman P, DeChene ET, Stienen GJ, Beggs AH, Granzier H (2010) Altered myofilament function depresses force generation in patients with nebulin-based nemaline myopathy (NEM2). J Struct Biol 170:334–343
Owczarek-Lipska M, Mausberg TB, Stephenson H, Dukes-McEwan H, Wess G, Leeb T (2013) A 16-bp deletion in the canine PDK4 gene is not associated with dilated cardiomyopathy in a European cohort of Doberman pinschers. Anim Genet 44:239
Pires DE, Ascher DB, Blundell TL (2014a) DUET: a server for predicting effects of mutations on protein stability using an integrated computational approach. Nucleic Acids Res 42:W314–W319
Pires DE, Ascher DB, Blundell TL (2014b) mCSM: predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics 30:335–342
Schafer S, de Marvao A, Adami E, Fiedler LR, Ng B, Khin E et al (2017) Titin-truncating variants affect heart function in disease cohorts and the general population. Nat Genet 49:46–53
Sen-Chowdhry S, Syrris P, McKenna WJ (2005) Genetics of right ventricular cardiomyopathy. J Cardiovasc Electrophysiol 16:927–935
Taylor M, Graw S, Sinagra G, Barnes C, Slavov D, Brun F et al (2011) Genetic variation in titin in ARVC-overlap syndromes. Circulation 124:876–885
Thomas WP, Gaber CE, Jacobs GJ, Kaplan PM, Lombard CW, Moise NS et al (1993) Recommendations for standards in transthoracic two-dimensional echocardiography in the dog and cat. J Vet Intern Med 7:247–252
Towbin JA (2014) Inherited cardiomyopathies. Circ J 78:2347–2356
Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, del Angel G, Levy-Moonshine A et al (2013) From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics 43:1110–1133
Vikhorev PG, Smoktunowicz N, Munster AB, Copeland O, Kostin S, Montgiraud C et al (2017) Abnormal contractility in human heart myofibrils from patients with dilated cardiomyopathy due to mutations in TTN and contractile protein genes. Sci Rep 7:14829
Vilella AJ, Severin J, Ureta-Vidal A, Heng L, Durbin R, Birney E (2009) EnsemblCompara GeneTrees: complete, duplication-aware phylogenetic trees in vertebrates. Genome Res 19:327–335
Warren CM, Krzesinski PR, Greaser ML (2003) Vertical agarose gel electrophoresis and electroblotting of high-molecular-weight proteins. Electrophoresis 24:1695–1702
Funding
Research reported in this publication was supported by the Heart Lung and Blood Institute of the National Institutes of Health under award number R35HL144998.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest. North Carolina State University offers the canine titin variant test as a risk assement tool for dogs.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Meurs, K.M., Friedenberg, S.G., Kolb, J. et al. A missense variant in the titin gene in Doberman pinscher dogs with familial dilated cardiomyopathy and sudden cardiac death. Hum Genet 138, 515–524 (2019). https://doi.org/10.1007/s00439-019-01973-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-019-01973-2