
Abstract
Titin (TTN), the largest protein in the human body, forms powerful elastic filaments along the sarcomere of cardiomyocytes. This multifunctional protein is involved in numerous cellular processes, including sarcomeric assembly, stabilization and mechanosensing. Along physiological sarcomere lengths, TTN is also the most important determinant of the passive tension of cardiac muscle. However, as the giant Goliath was brought down by David’s slingshot, so single-base-pair mutations in the gene encoding TTN (TTN) can ultimately impair to some degree a normal cardiac function. Since the first report on the involvement of TTN mutations in the development of hypertrophic cardiomyopathy, in 1999, dozens of other mutations have been described and associated with the onset of cardiac disease. In this review, we aim to explore some of the mechanisms underlying the functions of TTN, as well as the pathophysiological consequences arising from the expression of abnormal TTN isoforms resulting from mutations located along TTN.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Titin as a sarcomeric multifunctional protein
The sarcomere is classically defined as the basic motor unit of the cardiomyocyte. Its intrinsic contractile properties arise from the interdigitation of thin and thick myofilaments and their ability to slide along each other in response to changes in intracellular levels of calcium, leading to sarcomere shortening and/or to the production of contractile force. Thin filaments are mainly composed of a double-stranded α-helical polymer of actin (F-actin), as well as tropomyosin and troponin, while thick filaments consist of a bipolar assembly of multiple myosin II molecules. Studies in vitro have shown that, although monomers of both actin and myosin can spontaneously polymerize into filamentous structures, the combination of these self-assembled proteins does not automatically generate organized sarcomere-resembling structures [1]. Therefore, additional proteins including nebulin and obscurin are required for sarcomere assembly, with the leading role being attributed to titin [2].
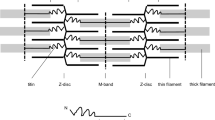
Titin (TTN), also known as connectin, is the largest human protein—with isoforms ranging from 2970 to ∼3800 kDa, and the only single molecular element spanning from the Z-disk to the M-line [3]. TTN amino-terminus is strongly attached to the Z-disk through at least four binding sites for α-actinin and overlaps with another TTN amino-terminus from the adjacent sarcomere [4]. The carboxy-terminus is anchored to the M-line through multiple interactions with myosin and C-protein, and with two TTN regions from the adjacent half-sarcomere [5]. Along its length of over 1 µm, TTN helps to define the axial position of several sarcomeric elements, including myosin, actin, α-actinin, T-cap/telethonin, myomesin, myosin binding protein-C and obscurin [6], a role that has granted TTN the epithet of molecular ruler and blueprint of the sarcomere [1].
Additionally, as active force is developed during contraction, several sarcomeric structures become susceptible to mechanical stress. Consequently, the cardiomyocyte must somehow sense this increasing tension and counteract it with additional cross-links between actin and myosin filaments. The presence of a constitutively expressed Ser/Thr kinase (TK) domain near TTN carboxy-terminus suggests that this molecule also plays a pivotal role in the cardiomyocyte mechanical stress sensing mechanism.
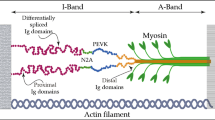
Besides its importance in sarcomeric assembly, stabilization and mechanosensing, TTN, along with collagen, is the major determinant of the myocardial passive tension [3]. In fact, it has been demonstrated that while at high sarcomere lengths the contribution to myocardial stiffness is mostly due to collagen, at low lengths most of the passive tension is determined by TTN [7]. This phenomenon originates from the ability of TTN to stretch under applied force and to recoil to the original length upon force removal. Since the portion of TTN laying in the A-band of the sarcomere, composed of regular patterns of immunoglobulin (Ig)-like and fibronectin type 3 repeats, is virtually inextensible, the elasticity of TTN derives from the I-band segment. This portion contains tandem Ig-like repeats, a region rich in proline (P), glutamate (E), valine (V) and lysine (K) arranged in up to 60 motifs of ∼28 residue repeats, known as PEVK, and a region located in the central I-band portion comprising three Ig domains interspersed with a 572 amino acid unique sequence, named N2B (Fig. 1). Some long TTN isoforms additionally present I-band segments found between N2B and the PEVK, the N2A domain. In resting sarcomere, TTN is found in a highly folded state; upon stretching, different portions of the protein sequentially unfold. The links between the tandem Ig-like repeats are the first to extend, followed by the PEVK region and finally by the N2 domains [8]. The tandem Ig-like repeats seem to remain collapsed even when the upper end of the physiological sarcomere length range is reached, unfolding only in non-physiological conditions (Fig. 2).

Titin structure and its position within the sarcomere. Titin spans the sarcomere from the M-line to the Z-disk. The fibronectin/immunoglobulin repeats are virtually inextensible, whereas PEVK, N2 and immunoglobulin domains are responsible for the elasticity of TTN. Figure was produced using Servier Medical Art

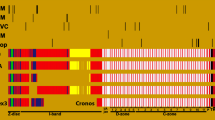
Mutations associated with cardiomyopathies distributed along the canonical TTN sequence (UniProtKB: Q8WZ42-1). The color of each mutation represents the associated pathology. The type of domain in TTN is represented by the color of each block in the sequence. Abbreviations DCM stands for dilated cardiomyopathy, HCM hypertrophic cardiomyopathy, ARVC arrhythmogenic right ventricular cardiomyopathy, RCM restrictive cardiomyopathy, Ig immunoglobulin, PEVK region rich in proline (P), glutamate (E), valine (V) and lysine (K), TK titin Ser/Thr kinase
Three isoform classes of TTN are expressed in cardiomyocytes, being the different properties of each defined by their unique I-band segment sequences, as the length and structure of the portion of TTN laying in the A-band are extremely conserved among vertebrates [3]. The first TTN class includes ∼3000-kDa isoforms containing the N2B region, therefore becoming collectively known as N2B TTN. A second TTN class named N2BA includes isoforms ranging from ∼3300 to 3500 kDa, and it contains, as its name implies, both the N2B and N2A domains [9]. Because N2BA TTN presents a longer extensible I-band region, isoforms from this class are much more compliant than N2B TTN. Both N2BA and N2B are expressed in the sarcomere, being the stiffness of the cardiomyocyte defined by their ratio. In the human left ventricle, the N2BA/N2B ratio is ∼0.55 [10], while right ventricles showed higher ratios than left ventricles [11], whereas atria seems to preferentially express N2BA isoforms [3]. Finally, a third TTN class is found only during fetal/neonatal life. These large, highly compliant N2BA isoforms (∼3600 to 3800 kDa) contain additional spring elements in both tandem Ig and PEVK regions and are typically associated with low cardiomyocyte passive tension [12]. During perinatal development, the fetal/neonatal N2BA class is gradually replaced by smaller N2B isoforms, which, as previously stated, become the predominant TTN isoforms in the adult left ventricle. Although the I-band segment sequence of each isoform primarily defines the stiffness of TTN, passive tension can also be modulated by posttranslational modifications or by calcium. The N2B domain has been identified as a substrate for protein kinase A (PKA), which, upon TTN phosphorylation, reduces passive tension [13]. Several protein kinases can modulate passive stiffness by phosphorylating TTN: PKG [14, 15], CaMKII [16], ERK2 [17] and PKC [18] are known to phosphorylate titin. Both titin isoforms N2BA (on N2A domain) and N2B (on N2B unique sequence) are phosphorylated by PKG. Although PKG phosphorylates more than one site in TTN, it reduces titin-based stiffness via phosphorylation of a serine residue (Ser-469) within the N2B unique sequence in N2B isoform, thereby acting to improve diastolic function in human hearts to reduce passive stiffness [14]. For instance, in heart failure with preserved ejection fraction, reduced cardiomyocyte PKG activity leads to myocardial diastolic dysfunction, causing a higher cardiomyocyte passive tension. This was reverted by in vitro administration of PKG [15]. The inhibition of the breakdown of cyclic guanosine monophosphate by phosphodiesterase 5A through sildenafil raises myocardial PKG activity, which has proven to be effective in decreasing diastolic tone. Also CaMKIIδ, the isoform that predominates in the heart, can phosphorylate specific sites within N2B unique sequence (Ser-4043) and PEVK (Ser-12884) in N2B and N2BA isoforms in mouse and human hearts’ titin, lowering cardiomyocyte passive tension [16]. Raskin et al. [17] showed an additional kinase that targets TTN and therefore regulates the muscle compliance. TTN undergoes in situ phosphorylation by the extracellular signal-regulated kinase 2 (ERK2) in N2B residues Ser-3873, Ser-3915 and Ser-3965. This process likely involves the negative regulator four-and-a-half LIM domains protein 1 (FHL1). For instance, in hypertrophic cardiomyopathy, the FHL1 gene is up-regulated [19], which impairs ERK2-mediated titin N2B phosphorylation and those mutant proteins lead to systolic and/or diastolic dysfunctions such as an increase in diastolic tension and ventricular stiffness and hypertrophy. Also PKC, particularly the α-isoform, phosphorylates both titin isoforms in two highly conserved sites of the PEVK region (Ser-11878 and Ser-12022), but, contrary to most kinases, it leads to an increase in titin-based passive tension [18]. Another difference between PKA and PKG to PKC is that while the first two phosphorylate the N2B element and thus are circumscribed to myocardium, the latter phosphorylates the PEVK region and probably also affects skeletal muscle. Hence, PKA, PKG, CaMKIIδ and ERK2 protein kinases alter passive tension in the same direction on phosphorylating the N2B fragment of the N2B isoform, while PKCα is the only that induces an increase in titin stiffening. Ca2+, on the other hand, modifies the cardiomyocyte passive tension by two mechanisms: directly, since it increases TTN stiffness when bound to E-rich motifs in the PEVK region [20], or indirectly via S100A1, by modulating the interaction between PEVK and actin [21]. S100A1, a soluble calcium-binding protein found at high physiological concentrations in the myocardium, inhibits PEVK–actin interactions in the sarcomere when activated by calcium physiological levels; thus, viscosity can be turned off during systole when high calcium levels are present in the cytosol and turned on again during diastole when calcium levels fall [21]. Recent studies have also related oxidative stress (OS) with changes in the cardiomyocyte passive tension [22]. Apparently, OS induces the formation of disulfide bridges within the N2B domain, which reduce the length of N2B and stiffens TTN. Recently, Alegre-Cebollada et al. [23] showed that, upon unfolding of Ig domains in the I-band, titin is S-glutathionylated in cryptic cysteines, leading to a mechanical weakening and inhibition of folding which ultimately causes a decrease in stiffness of the cardiomyocyte and consequently increases its elasticity at physiological sarcomere lengths. Since the amino acid sequence of the Ig domains in the I-band is highly abundant in cysteine residues, it would be interesting to know whether titin is also subjected to other posttranslational modifications like sulfenylation, carbonylation and nitrosylation. In fact, given the important regulatory role of the nitric oxide in cardiac function and specifically of the S-nitrosylation in cell signaling, it is possible to think that S-nitrosylation can be another route to alter the elasticity of the titin. Also, although it was proven that the levels of oxidative and nitrosative species can alter the integrity of the titin in an infection state [24], it remains unclear whether titin is subjected to carbonylation.
Many TTN isoforms are found throughout the human heart, but interestingly, all of them are encoded by the same gene (TTN). Located on a 294-kb region of the long arm of chromosome 2 (2q31), TTN contains 363 exons, the first 251 encoding for the regions laying in the I-band and the remaining 112 encoding for the A-band segment [8, 9]. Although the entire gene could in theory encode for a 4200-kDa protein with 38,138 amino acid residues [9], extensive alternative splicing over a third of TTN exons is responsible for the generation of a multitude of isoforms ranging from 2970 to ∼3800 kDa [3]. Due to TTN’s multifunctional role in the cardiomyocyte, mutations in its encoding gene can potentially impair a normal cardiac function. The pathophysiological consequences resulting from mutations in TTN will be dealt within the next sections.
Abnormal sarcomerogenesis induced by mutations in TTN
Due to the relevance of TTN in sarcomeric assembly, it would be expected for mutations in TTN to cause significant disturbances to cardiac sarcomerogenesis. Ethical concerns, however, virtually impair any study of this process in human beings (one would hardly justify the screening of human embryos aiming for cardiac anomalies). Therefore, current knowledge on abnormal sarcomerogenesis has been growing from experiments with transfected cell lines and transgenic animals. In 2003, Gotthardt et al. [25] investigated the role of TTN Ser/Thr kinase (TK) domain in sarcomeric assembly, by deleting the two exons (358/359) encoding TK plus flanking regions. Deletion of the TK domain from cardiac TTN during early embryonic development resulted in the death of all homozygous specimens. Three years later, the same group also published results with mice heterozygous for the 358/359 TTN mutation [26]. Contrary to homozygous individuals, the embryonic development at organ, cellular and ultrastructural levels of heterozygous mice did not differ from wild-type (WT) animals. In the same year, Musa et al. [27] assessed the repercussion in sarcomerogenesis of deletions in the TK domain in one or both alleles of mouse embryonic stem cells that would be subsequently differentiated into beating cardiomyocytes. The authors reported that following 10 days of differentiation, cultures of WT and heterozygous cells for the mutant TTN allele presented a strong beating phenotype, relatively similar sarcomeric appearance, molecular content and length. In contrast, cells homozygous for the TTN mutant allele failed to beat. Moreover, although a wide variety of sarcomeric proteins could be detected in the homozygous mutant cells, myofibrillar organization seemed completely absent. With this study, the authors demonstrated not only that the integrity of TTN carboxy-terminus is critical for myosin filament assembly, M-line formation and maturation of the Z-disk, but also that sarcomeric assembly can still occur in the presence of a TTN TK domain mutation, as long as a compensatory WT allele is still present. In the past decade, important contributions came also from studies performed with an improbable exotic creature, the zebrafish (Danio rerio). Zebrafish is a small freshwater fish used as an in vivo vertebrate model in cardiovascular research due to some peculiar characteristics: The genome is fully sequenced and easy to manipulate, and embryos are transparent, develop rapidly ex utero and are able to survive in the absence of blood flow up to 5 days, since oxygen is delivered to cells via a simple diffusion mechanism [28]. These features allow the study of mutant embryos with aberrant sarcomerogenesis, opposing to mammalian embryos, which require a fully functional cardiovascular system to survive. One zebrafish mutant, pickwick, found during a large-scale mutagenesis screening, presents a recessive lethal mutation in TTN. Xu et al. [28] identified a transversion of a single base pair (TTA → TGA) in the N2B region of pickwick that caused a change of a leucine to a stop codon. The major physiological consequence of this mutation was the generation of little systolic force. The molecular mechanism behind this observation was attributed to the inability of pickwick to produce organized sarcomeres. When transplanted to WT hearts, mutated cardiomyocytes remained poorly contractile and bulged discoordinately outward at each systolic contraction, which could also be explained by the lack of recoil tension normally offered by WT TTN. Interestingly, the authors discussed the resemblance between the pickwick phenotype (dilated heart with thin walls and poor contractions) and human dilated cardiomyopathy, which could suggest a similar genetic etiology.
Studies with transfected cell lines and transgenic animals presenting TTN mutations reinforced the importance of TTN in sarcomerogenesis. Results showed that TTN homozygous mutations are, as expected, much more severe, leading to a complete absence of sarcomeric assembly, while their heterozygous counterparts can still encode functional sarcomeres, although with altered organizational characteristics that may impair a normal cardiac function in adulthood. These findings go in accordance with Carmignac et al. [29], who described several patients with homozygous TTN mutations who died of heart disease before reaching 20 years old, and Satoh et al. [30] and Gerull et al. [31, 32], who described much older patients with heterozygous TTN defects, but never homozygous.
Cardiomyopathies induced by mutations in TTN
In 2006, the American Heart Association (AHA) defined cardiomyopathies as “a heterogeneous group of diseases of the myocardium associated with mechanical and/or electrical dysfunction that usually (but not invariably) exhibit inappropriate ventricular hypertrophy or dilatation and are due to a variety of causes that frequently are genetic. Cardiomyopathies either are confined to the heart or are part of generalized systemic disorders, often leading to cardiovascular death or progressive heart failure-related disability” [33]. In the same document, the AHA also proposed that primary cardiomyopathies, those predominantly involving the heart, should be divided into three categories according to the etiology of the disease: (1) genetic, which would include hypertrophic cardiomyopathy (HCM) and arrhythmogenic right ventricular cardiomyopathy (ARVC), (2) mixed, where dilated cardiomyopathy (DCM) and restrictive cardiomyopathy (RCM) would fit, and (3) acquired. In 2008, the European Society of Cardiology also proposed a new classification model for cardiomyopathies, grouping diseases according to their morphological and functional phenotypes [34]. Five groups were then created and named: (1) ARVC, (2) DCM, (3) HCM, (4) RCM and (5) unclassified.
The etiology of cardiomyopathies was practically unknown until the first genome-wide linkage studies performed during the decade of 1980. We now recognize the impact of mutations in genes encoding for sarcomere-related proteins in the onset and establishment of cardiomyopathy. The gene most times associated with this group of diseases encodes for the β-myosin heavy chain. Surprisingly, although TTN is by far the largest human protein, far fewer mutations in TTN have been related to cardiomyopathy, with the first description being published only in 1999 [30]. Recently, Greaser has addressed this issue and suggested several explanations for the lack of identified TTN mutations [35]. The first reason would be the exceptional size of TTN, which impairs full cDNA sequencing. Secondly, since TTN originates many different transcripts, it becomes hard to recognize whether a certain isoform results from a mutation or from physiological alternative splicing. Thirdly, since a large portion of TTN consists of several tandem Ig-like encoding sequences, mutations in these regions may have an almost irrelevant effect in TTN length and function. Last but not the least, as the physiological mechanism of TTN in several cellular processes remains poorly understood, so does its relation to cardiomyopathy. Nevertheless, TTN mutations have been strongly associated with four cardiomyopathies: DCM, HCM, ARVC and RCM. The following subsections will deal with each.
Dilated cardiomyopathy
Dilated cardiomyopathy (DCM) is a heart disease characterized by the enlargement of the cardiac chambers and systolic dysfunction. Although more than half of reported cases of DCM result from coronary artery disease or environmental factors, the familial form of the disease is found in ~35 % of patients [2]. The inheritance of DCM suggests genetic abnormalities typically associated with a dominant autosomal pattern of transmission, although X-linked, autosomal recessive and mitochondrial DNA mutations patterns may be observed as well [33]. The etiology of familial DCM is generally grouped into five distinct classes: (1) mutations affecting cytoskeletal proteins, (2) mutations in the genes encoding for sarcomeric proteins, (3) mutations in components of the nuclear envelope, (4) mutations that impair the supply and regulation of energy metabolism and (5) mutations in ion channels. Among the distinct classes of mutated genes, the cytoarchitectural abnormalities are by far the most common trigger in the development of DCM, as disrupted force transmission at the Z-disk impairs an efficient cardiomyocyte contractility. In the past 10 years, several TTN mutations have been identified in humans resulting in DCM [29, 31, 32, 36–39]. In 2002, two research groups published articles describing DCM patients carrying TTN mutation. Itoh-Satoh et al. [36] described four TTN mutations in the heterozygous state in DCM patients that were not found in healthy controls. Two of the mutations, Val54Met and Ala743Val, were located in the Z-disk region of TTN and were associated with familial DCM. In contrast, the other two mutations were located in the N2B region and patients did not report any history of cardiac disease in the family. Using yeast two-hybrid assays, it was demonstrated that both mutations in the Z-disk region induced functional alterations in TTN, as Ala743Val and Val54Met decreased the binding affinity of TTN to α-actinin and T-cap/telethonin, respectively. The authors inferred that the decreased binding between TTN and these proteins probably reduced the cardiomyocyte resistance to tension, making it easily extendible in the presence of mechanical stretch. One of the N2B mutations was found to be a nonsense mutation, leading to a shorter truncated TTN isoform. In the same year, Gerull et al. [32] described two families (A1 and MAO) with DCM, presenting a two-base-pair (bp) insertion mutation in TTN exon 326 and a TTN missense mutation (Trp930Arg) located in a highly conserved hydrophobic core sequence located in the Z-disk–I-band transition zone (Ig-Z4), respectively. The mutation in the A1 family resulted in the expression of a much smaller TTN isoform since a stop codon was found just twelve nucleotides ahead of the 2-bp insertion. Regarding the MAO family mutation, yeast two-hybrid assays failed to identify any binding protein for the Ig-Z4 domain. However, the authors suggested that Trp930Arg may have disrupted the β-sandwich structure of the Ig-Z4 domain, an element thought to play a key role in the organization and maintenance of the Z-disk. A few years later, the same research group described a novel TTN frameshift mutation in a large Australian family with DCM [31]. The mutation, a heterozygous 1-bp deletion in TTN exon 335, caused a frameshift, resulting in a premature stop codon following the addition of ten novel amino acid residues (Glu28386LysfsX10). In 2005, Matsumoto et al. described two TTN mutations in Japanese patients with DCM that could alter the cardiac function through an alternative mechanism. One of the mutations, Gln4053ter, located in the N2B region of TTN, which had already been described by this group [36], was now found to decrease the binding between TTN and four-and-a-half LIM domains protein 2 (FHL2) [39]. Since FHL2 is known to bind enzymes of the energetic metabolism (adenylate kinase, phosphofructokinase and creatinine kinase), the decreased binding between FHL2 and the N2B domain of TTN may have reduced the recruitment of these enzymes, consequently leading to dysfunction of metabolic pathways. In 2007, Carmignac et al. [29] described for the first time two homozygous deletions causing a truncation in the M-line region of TTN. Although compatible with life, homozygous deletions found among elements of the two families of Moroccan and Sudanese origin resulted in the premature death of five patients at ages ranging from 8 to 19.5 years. Moreover, contrary to all other DCM-related TTN mutations described to date, the subjects also presented generalized muscle weakness in the lower limbs (psoas, gluteus maximus, tibialis anterior and peroneus muscles), proximal upper limbs, neck and trunk flexors, and facial muscles. In 2012, several more TTN mutations were characterized. Yoskovitz et al. [37] described a new TTN mutation (Ser19628IlefsX1) in an Arab family residing in Galilee, Israel. This family included members previously diagnosed with symptomatic heart failure and DCM, members who died from heart failure and elements who had heart transplantation or were referred for this procedure. Sequence analysis identified a single nucleotide insertion in exon 326 causing protein truncation after 19,628 amino acids, resulting in a smaller TTN isoform. In another study involving DCM patients from the USA, UK and Italy, Herman et al. [38] found, among nonsense, frameshift and splicing subtypes, more than 70 new TTN mutations responsible for TTN truncation. The authors noted that many of the truncated isoforms lacked the M-line region, which is thought to be responsible for mechanosensing, and therefore could impair a normal regulation of sarcomeric force. More recently, using a whole-exome sequencing technique, Roncarati et al. [40] described an association between a newly described mutation in TTN (Leu4855Phe) and a previously known lamin A/C gene (LMNA) mutation. Several members of the Italian family where the study was conducted presented a wide array of cardiological symptoms, ranging from mild dyspnea to severe congestive heart failure requiring heart transplantation. Using the same technique, Chauveau et al. [41] also described seven novel homozygous or compound heterozygous TTN mutations, mostly located in the M-line region of TTN. Moreover, studies in vitro conducted by the authors documented the first-reported absence of a functional TTN TK domain in humans, resulting in severe antenatal phenotype.
The past few years have been prolific in the description of new DCM-related TTN mutations; therefore, several explanations on how abnormal TTN isoforms can ultimately cause DCM are being exploited. Observations suggest that these mutations may cause (1) short truncated TTN isoforms unable to span between the M-line and the Z-disk, frequently lacking the mechanosensing M-line region, (2) normal-sized TTN isoforms poorly attached to the Z-disk or (3) abnormal TTN isoforms involved in dysfunctional energetic metabolic pathways. The inclusion of these aberrant TTN isoforms in the cardiomyocyte results in “loose” and poorly contractile sarcomeres most likely involved in the onset of DCM.
Hypertrophic cardiomyopathy
Hypertrophic cardiomyopathy (HCM) is a disorder of the myocardium characterized by an unexplainable left ventricle hypertrophy, usually associated with a small cardiac cavity, cardiomyocyte enlargement, myocardial fibrosis and diastolic dysfunction. In approximately 50–70 % of all diagnosed cases of HCM, family history of the disease is concomitantly reported [42]. These familial forms of HCM typically follow an autosomal dominant pattern of transmission, consistent with the presence of some kind of genetic abnormality. The first mutation ever reported in a family with HCM history was published by Geisterfer-Lowrance et al. [43] in the beginning of the decade of 1990. At the time, the authors attributed the disease to a mutation in the β-myosin heavy chain gene, but since then, hundreds of other HCM-causing mutations have been identified [44]. Most forms of genetic-origin HCM have been grouped within two distinct classes: sarcomere-HCM or Z-disk-HCM [42]. This division is etiology-based only, as the clinical and histopathologic features of both classes are virtually the same. Sarcomere-HCM is caused by mutations in genes encoding for proteins belonging to the sarcomeric thick (α- and β-myosin heavy chains, essential and regulatory myosin light chains) and thin filaments (troponin T, I and C, α-tropomyosin and actin). An initial hypothesis stated that as abnormal sarcomeres resulted in weaker cardiomyocyte contractions, cardiac walls would become progressively thicker in an attempt to maintain a normal cardiac outflow [45]. This compensatory mechanism, most likely unsustainable, would soon become maladaptive, triggering the development of HCM. Kimura et al. [46], on the other hand, suggest that sarcomere-HCM may be more likely related to an increased sarcomeric sensitivity to Ca2+. The fact that cardiomyocytes fail to relax even at low cytoplasmic Ca2+ levels helps to explain the onset of diastolic dysfunction, followed by cardiac hypertrophy [42]. Z-disk-HCM arises from the presence of abnormal cytoskeletal Z-disk proteins (TTN, T-cap/telethonin, α-actinin and obscurin).
In 1999, Satoh et al. [30] reported for the first time the involvement of a TTN mutation in the establishment of HCM. A single-base-pair transversion (CGC → CTC), causing the replacement of an arginine for a leucine (Arg740Leu), was described, and subsequent yeast two-hybrid assays showed an increased binding affinity of TTN to α-actinin by ~40 %. Although the molecular mechanism underlying the establishment of HCM remained to be elucidated, the authors suggested that an impairment in force transmission during contraction could result in a compensatory cardiac hypertrophy. In 2005, Matsumoto et al. [39] investigated cardiac functional alterations due to mutations in the N2B region of TTN. Their study reported a HCM-associated Ser3799Tyr mutation that increased the binding affinity of TTN to FHL2. Although the association between the increased binding and cardiac hypertrophy remained to be solved, the energy depletion hypothesis of Ashrafian et al. [47] helps to explain this phenomenon. It is suggested that abnormal cardiomyocytes are unable to sustain the energy levels in subcellular compartments required for contraction and Ca2+ re-uptake into the sarcoplasmic reticulum. Consequently, the energetic dysfunction would trigger the development of cardiac hypertrophy. Arimura et al. [48] screened the presence of mutations in key sarcomeric proteins of 384 patients with HCM. Two new TTN mutations were found in the N2A domain, which increased the binding of TTN to a Z-disk protein, the cardiac ankyrin repeat protein (CARP). In the study conducted by Herman et al. [38] published in 2012, already characterized in the DCM section, a few TTN mutations associated with HCM were also described. Moreover, this study demonstrates that, despite its importance in the onset of HCM, the frequency of TTN mutations is actually much higher among patients with DCM [54 of 203 (27 %) vs. 3 of 231 (1 %) with HCM].
Arrhythmogenic right ventricular cardiomyopathy
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is an inheritable heart disease characterized by a dysfunctional dilated right ventricle with thin walls, where replacement of cardiomyocytes by fibrofatty tissue progressively occurs.
Most cases of ARVC are a consequence of mutations in the genes encoding proteins belonging to the desmosome complex. The resultant abnormal desmosomes are unable to maintain the adhesion between cardiomyocytes at the intercalated disks, triggering cell death and the installation of regional fibroadiposis. Desmosomal mutations, however, fail to be found in all familial inherited ARVC cases, suggesting that other genes must be linked to ARVC. In 2011, Taylor et al. [49] described for the first time that a set of TTN mutations could be the precursor of ARVC. It may seem odd for a sarcomeric protein to be involved in ARVC, but the fact that TTN plays a multitude of cellular roles and is functionally linked to the desmosome helps to explain how aberrant isoforms can influence electrical signaling across cardiomyocytes. One of the described mutations (Thr2896IIe), located in the tenth Ig-like domain of TTN, was discovered in individuals of one family with well-documented history of sudden death, heart transplantation or death resulting from progressive myocardial dysfunction and conduction disease. The Ig10 domain, located in the Ig-like region near the Z-disk, is involved in the development of passive tension, lacking any binding sites for regulatory proteins. Therefore, it would seem highly improbable for a single mutation in one of the so many Ig-like domains to alter the sarcomere ability to generate tension, to the point of causing disease. Accordingly, the authors found that it was not the inability to generate tension the cause for the establishment of ARVC. While passive tension was reduced by only 3 %, a much more relevant phenomenon was described; the Ig10 domain became extremely prone to proteolysis, making this mutated isoform of TTN much more unstable in physiological conditions. Despite these evidences, the exact pathophysiological role of mutated isoforms of TTN in the onset of ARVC remains to clarify.
Restrictive cardiomyopathy
Restrictive cardiomyopathy (RCM), the rarest of all cardiomyopathies, is a disease characterized by restrictive ventricular filling due to an increased myocardial stiffness, associated with atrial enlargement, but normal ventricular wall thickness and systolic function. Both sporadic and familial forms have been described, the latter mostly inherited by autosomal dominant transmission, but also by autosomal recessive and X-linked inheritance [50]. Mutations in several sarcomeric proteins including troponin I, β-myosin heavy chain, troponin T and α-cardiac actin had already been linked to familial forms of RCM, but only recently, a TTN mutation was implicated in the onset of RCM. Peled et al. [50] described a family comprising six affected members whose echocardiographic findings were compatible with RCM—mild-to-severe diastolic dysfunction along with preserved systolic function and normal left ventricular wall thickness. The authors characterized the replacement of adenine with guanine at position 109 of the TTN exon 266 resulting in the substitution of tyrosine by cysteine at position 7621 of TTN protein (Tyr7621Cys). The mutation is located at the A-/I-band junction region—the intersection between the compliant and the rigid components of TTN. It is suggested that the absence of the aromatic ring of the tyrosine likely disrupts the beta-sandwich structure which is integrated, leading, by unknown mechanisms, to RCM.
Conclusion
During the past decade, much has been learned about TTN mutations and their role in the establishment of cardiomyopathies. State-of-the-art molecular biology techniques have granted us the ability to find dozens of new TTN mutations worldwide, while novel transgenic animal models have been used to mimic human cardiac dysfunctions. Despite all these advances, many of the molecular and pathophysiological mechanisms underlying these cardiac disorders remain to be elucidated. There is also no apparent pattern in the distribution of mutations along the different domains along TTN sequence and the respective cardiomyopathies arising from them. There is still much to be unfolded, and therefore, one should keep in mind the necessity to continue investing effort, time and resources in this field of research, as a detailed knowledge on TTN-mutation-based cardiomyopathies may result in more accurate diagnosis and innovative therapeutics aiming to ameliorate or even prevent disease in patients with TTN mutations.
References
Ehler E, Gautel M (2008) The sarcomere and sarcomerogenesis. Adv Exp Med Biol 642:1–14
Kontrogianni-Konstantopoulos A, Ackermann MA, Bowman AL, Yap SV, Bloch RJ (2009) Muscle giants: molecular scaffolds in sarcomerogenesis. Physiol Rev 89(4):1217–1267. doi:10.1152/physrev.00017.2009
LeWinter MM, Granzier H (2010) Cardiac titin: a multifunctional giant. Circulation 121(19):2137–2145. doi:10.1161/CIRCULATIONAHA.109.860171
Gregorio CC, Trombitas K, Centner T, Kolmerer B, Stier G, Kunke K, Suzuki K, Obermayr F, Herrmann B, Granzier H, Sorimachi H, Labeit S (1998) The NH2 terminus of titin spans the Z-disc: its interaction with a novel 19-kD ligand (T-cap) is required for sarcomeric integrity. J Cell Biol 143(4):1013–1027
Obermann WM, Gautel M, Steiner F, van der Ven PF, Weber K, Furst DO (1996) The structure of the sarcomeric M band: localization of defined domains of myomesin, M-protein, and the 250-kD carboxy-terminal region of titin by immunoelectron microscopy. J Cell Biol 134(6):1441–1453
Sanger JW, Sanger JM (2001) Fishing out proteins that bind to titin. J Cell Biol 154(1):21–24
Hamdani N, Paulus WJ (2013) Myocardial titin and collagen in cardiac diastolic dysfunction: partners in crime. Circulation 128(1):5–8. doi:10.1161/CIRCULATIONAHA.113.003437
Walker JS, de Tombe PP (2004) Titin and the developing heart. Circ Res 94(7):860–862. doi:10.1161/01.RES.0000126698.37440.B0
Bang ML, Centner T, Fornoff F, Geach AJ, Gotthardt M, McNabb M, Witt CC, Labeit D, Gregorio CC, Granzier H, Labeit S (2001) The complete gene sequence of titin, expression of an unusual approximately 700-kDa titin isoform, and its interaction with obscurin identify a novel Z-line to I-band linking system. Circ Res 89(11):1065–1072
Neagoe C, Kulke M, del Monte F, Gwathmey JK, de Tombe PP, Hajjar RJ, Linke WA (2002) Titin isoform switch in ischemic human heart disease. Circulation 106(11):1333–1341
Neagoe C, Opitz CA, Makarenko I, Linke WA (2003) Gigantic variety: expression patterns of titin isoforms in striated muscles and consequences for myofibrillar passive stiffness. J Muscle Res Cell Motil 24(2–3):175–189
Lahmers S, Wu Y, Call DR, Labeit S, Granzier H (2004) Developmental control of titin isoform expression and passive stiffness in fetal and neonatal myocardium. Circ Res 94(4):505–513. doi:10.1161/01.RES.0000115522.52554.86
Yamasaki R, Wu Y, McNabb M, Greaser M, Labeit S, Granzier H (2002) Protein kinase A phosphorylates titin’s cardiac-specific N2B domain and reduces passive tension in rat cardiac myocytes. Circ Res 90(11):1181–1188
Kruger M, Kotter S, Grutzner A, Lang P, Andresen C, Redfield MM, Butt E, dos Remedios CG, Linke WA (2009) Protein kinase G modulates human myocardial passive stiffness by phosphorylation of the titin springs. Circ Res 104(1):87–94. doi:10.1161/CIRCRESAHA.108.184408
van Heerebeek L, Hamdani N, Falcao-Pires I, Leite-Moreira AF, Begieneman MP, Bronzwaer JG, van der Velden J, Stienen GJ, Laarman GJ, Somsen A, Verheugt FW, Niessen HW, Paulus WJ (2012) Low myocardial protein kinase G activity in heart failure with preserved ejection fraction. Circulation 126(7):830–839. doi:10.1161/CIRCULATIONAHA.111.076075
Hamdani N, Krysiak J, Kreusser MM, Neef S, Dos Remedios CG, Maier LS, Kruger M, Backs J, Linke WA (2013) Crucial role for Ca2(+)/calmodulin-dependent protein kinase-II in regulating diastolic stress of normal and failing hearts via titin phosphorylation. Circ Res 112(4):664–674. doi:10.1161/CIRCRESAHA.111.300105
Raskin A, Lange S, Banares K, Lyon RC, Zieseniss A, Lee LK, Yamazaki KG, Granzier HL, Gregorio CC, McCulloch AD, Omens JH, Sheikh F (2012) A novel mechanism involving four-and-a-half LIM domain protein-1 and extracellular signal-regulated kinase-2 regulates titin phosphorylation and mechanics. J Biol Chem 287(35):29273–29284. doi:10.1074/jbc.M112.372839
Hidalgo C, Hudson B, Bogomolovas J, Zhu Y, Anderson B, Greaser M, Labeit S, Granzier H (2009) PKC phosphorylation of titin’s PEVK element: a novel and conserved pathway for modulating myocardial stiffness. Circ Res 105(7):631–638, 617 p following 638. doi:10.1161/CIRCRESAHA.109.198465
Lim DS, Roberts R, Marian AJ (2001) Expression profiling of cardiac genes in human hypertrophic cardiomyopathy: insight into the pathogenesis of phenotypes. J Am Coll Cardiol 38(4):1175–1180
Labeit D, Watanabe K, Witt C, Fujita H, Wu Y, Lahmers S, Funck T, Labeit S, Granzier H (2003) Calcium-dependent molecular spring elements in the giant protein titin. Proc Natl Acad Sci USA 100(23):13716–13721. doi:10.1073/pnas.2235652100
Yamasaki R, Berri M, Wu Y, Trombitas K, McNabb M, Kellermayer MS, Witt C, Labeit D, Labeit S, Greaser M, Granzier H (2001) Titin-actin interaction in mouse myocardium: passive tension modulation and its regulation by calcium/S100A1. Biophys J 81(4):2297–2313. doi:10.1016/S0006-3495(01)75876-6
Grutzner A, Garcia-Manyes S, Kotter S, Badilla CL, Fernandez JM, Linke WA (2009) Modulation of titin-based stiffness by disulfide bonding in the cardiac titin N2-B unique sequence. Biophys J 97(3):825–834. doi:10.1016/j.bpj.2009.05.037
Alegre-Cebollada J, Kosuri P, Giganti D, Eckels E, Rivas-Pardo JA, Hamdani N, Warren CM, Solaro RJ, Linke WA, Fernandez JM (2014) S-glutathionylation of cryptic cysteines enhances titin elasticity by blocking protein folding. Cell 156(6):1235–1246. doi:10.1016/j.cell.2014.01.056
Dhiman M, Nakayasu ES, Madaiah YH, Reynolds BK, Wen JJ, Almeida IC, Garg NJ (2008) Enhanced nitrosative stress during Trypanosoma cruzi infection causes nitrotyrosine modification of host proteins: implications in Chagas’ disease. Am J Pathol 173(3):728–740. doi:10.2353/ajpath.2008.080047
Gotthardt M, Hammer RE, Hubner N, Monti J, Witt CC, McNabb M, Richardson JA, Granzier H, Labeit S, Herz J (2003) Conditional expression of mutant M-line titins results in cardiomyopathy with altered sarcomere structure. J Biol Chem 278(8):6059–6065. doi:10.1074/jbc.M211723200
Weinert S, Bergmann N, Luo X, Erdmann B, Gotthardt M (2006) M line-deficient titin causes cardiac lethality through impaired maturation of the sarcomere. J Cell Biol 173(4):559–570. doi:10.1083/jcb.200601014
Musa H, Meek S, Gautel M, Peddie D, Smith AJ, Peckham M (2006) Targeted homozygous deletion of M-band titin in cardiomyocytes prevents sarcomere formation. J Cell Sci 119(Pt 20):4322–4331. doi:10.1242/jcs.03198
Xu X, Meiler SE, Zhong TP, Mohideen M, Crossley DA, Burggren WW, Fishman MC (2002) Cardiomyopathy in zebrafish due to mutation in an alternatively spliced exon of titin. Nat Genet 30(2):205–209. doi:10.1038/ng816
Carmignac V, Salih MA, Quijano-Roy S, Marchand S, Al Rayess MM, Mukhtar MM, Urtizberea JA, Labeit S, Guicheney P, Leturcq F, Gautel M, Fardeau M, Campbell KP, Richard I, Estournet B, Ferreiro A (2007) C-terminal titin deletions cause a novel early-onset myopathy with fatal cardiomyopathy. Ann Neurol 61(4):340–351. doi:10.1002/ana.21089
Satoh M, Takahashi M, Sakamoto T, Hiroe M, Marumo F, Kimura A (1999) Structural analysis of the titin gene in hypertrophic cardiomyopathy: identification of a novel disease gene. Biochem Biophys Res Commun 262(2):411–417. doi:10.1006/bbrc.1999.1221
Gerull B, Atherton J, Geupel A, Sasse-Klaassen S, Heuser A, Frenneaux M, McNabb M, Granzier H, Labeit S, Thierfelder L (2006) Identification of a novel frameshift mutation in the giant muscle filament titin in a large Australian family with dilated cardiomyopathy. J Mol Med 84(6):478–483. doi:10.1007/s00109-006-0060-6
Gerull B, Gramlich M, Atherton J, McNabb M, Trombitas K, Sasse-Klaassen S, Seidman JG, Seidman C, Granzier H, Labeit S, Frenneaux M, Thierfelder L (2002) Mutations of TTN, encoding the giant muscle filament titin, cause familial dilated cardiomyopathy. Nat Genet 30(2):201–204. doi:10.1038/ng815
Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, Moss AJ, Seidman CE, Young JB, American Heart A, Council on Clinical Cardiology HF, Transplantation C, Quality of C, Outcomes R, Functional G, Translational Biology Interdisciplinary Working G, Council on E, Prevention (2006) Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation 113(14):1807–1816. doi:10.1161/circulationaha.106.174287
Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, Dubourg O, Kuhl U, Maisch B, McKenna WJ, Monserrat L, Pankuweit S, Rapezzi C, Seferovic P, Tavazzi L, Keren A (2008) Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 29(2):270–276. doi:10.1093/eurheartj/ehm342
Greaser ML (2009) Stressing the giant: a new approach to understanding dilated cardiomyopathy. J Mol Cell Cardiol 47(3):347–349. doi:10.1016/j.yjmcc.2009.06.011
Itoh-Satoh M, Hayashi T, Nishi H, Koga Y, Arimura T, Koyanagi T, Takahashi M, Hohda S, Ueda K, Nouchi T, Hiroe M, Marumo F, Imaizumi T, Yasunami M, Kimura A (2002) Titin mutations as the molecular basis for dilated cardiomyopathy. Biochem Biophys Res Commun 291(2):385–393. doi:10.1006/bbrc.2002.6448
Yoskovitz G, Peled Y, Gramlich M, Lahat H, Resnik-Wolf H, Feinberg MS, Afek A, Pras E, Arad M, Gerull B, Freimark D (2012) A novel titin mutation in adult-onset familial dilated cardiomyopathy. Am J Cardiol 109(11):1644–1650. doi:10.1016/j.amjcard.2012.01.392
Herman DS, Lam L, Taylor MR, Wang L, Teekakirikul P, Christodoulou D, Conner L, DePalma SR, McDonough B, Sparks E, Teodorescu DL, Cirino AL, Banner NR, Pennell DJ, Graw S, Merlo M, Di Lenarda A, Sinagra G, Bos JM, Ackerman MJ, Mitchell RN, Murry CE, Lakdawala NK, Ho CY, Barton PJ, Cook SA, Mestroni L, Seidman JG, Seidman CE (2012) Truncations of titin causing dilated cardiomyopathy. N Engl J Med 366(7):619–628. doi:10.1056/NEJMoa1110186
Matsumoto Y, Hayashi T, Inagaki N, Takahashi M, Hiroi S, Nakamura T, Arimura T, Nakamura K, Ashizawa N, Yasunami M, Ohe T, Yano K, Kimura A (2005) Functional analysis of titin/connectin N2-B mutations found in cardiomyopathy. J Muscle Res Cell Motil 26(6–8):367–374. doi:10.1007/s10974-005-9018-5
Roncarati R, Viviani Anselmi C, Krawitz P, Lattanzi G, von Kodolitsch Y, Perrot A, di Pasquale E, Papa L, Portararo P, Columbaro M, Forni A, Faggian G, Condorelli G, Robinson PN (2013) Doubly heterozygous LMNA and TTN mutations revealed by exome sequencing in a severe form of dilated cardiomyopathy. Eur J Hum Genet 21(10):1105–1111. doi:10.1038/ejhg.2013.16
Chauveau C, Bonnemann CG, Julien C, Kho AL, Marks H, Talim B, Maury P, Arne-Bes MC, Uro-Coste E, Alexandrovich A, Vihola A, Schafer S, Kaufmann B, Medne L, Hubner N, Foley AR, Santi M, Udd B, Topaloglu H, Moore SA, Gotthardt M, Samuels ME, Gautel M, Ferreiro A (2014) Recessive TTN truncating mutations define novel forms of core myopathy with heart disease. Hum Mol Genet 23(4):980–991. doi:10.1093/hmg/ddt494
Arimura T, Hayashi T, Kimura A (2007) Molecular etiology of idiopathic cardiomyopathy. Acta Myol 26(3):153–158
Geisterfer-Lowrance AA, Kass S, Tanigawa G, Vosberg HP, McKenna W, Seidman CE, Seidman JG (1990) A molecular basis for familial hypertrophic cardiomyopathy: a beta cardiac myosin heavy chain gene missense mutation. Cell 62(5):999–1006
Alcalai R, Seidman JG, Seidman CE (2008) Genetic basis of hypertrophic cardiomyopathy: from bench to the clinics. J Cardiovasc Electrophysiol 19(1):104–110. doi:10.1111/j.1540-8167.2007.00965.x
Lankford EB, Epstein ND, Fananapazir L, Sweeney HL (1995) Abnormal contractile properties of muscle fibers expressing beta-myosin heavy chain gene mutations in patients with hypertrophic cardiomyopathy. J Clin Investig 95(3):1409–1414. doi:10.1172/JCI117795
Kimura A, Ito-Satoh M, Hayashi T, Takahashi M, Arimura T (2001) Molecular etiology of idiopathic cardiomyopathy in Asian populations. J Cardiol 37(Suppl 1):139–146
Ashrafian H, Redwood C, Blair E, Watkins H (2003) Hypertrophic cardiomyopathy: a paradigm for myocardial energy depletion. Trends Genet 19(5):263–268. doi:10.1016/S0168-9525(03)00081-7
Arimura T, Bos JM, Sato A, Kubo T, Okamoto H, Nishi H, Harada H, Koga Y, Moulik M, Doi YL, Towbin JA, Ackerman MJ, Kimura A (2009) Cardiac ankyrin repeat protein gene (ANKRD1) mutations in hypertrophic cardiomyopathy. J Am Coll Cardiol 54(4):334–342. doi:10.1016/j.jacc.2008.12.082
Taylor M, Graw S, Sinagra G, Barnes C, Slavov D, Brun F, Pinamonti B, Salcedo EE, Sauer W, Pyxaras S, Anderson B, Simon B, Bogomolovas J, Labeit S, Granzier H, Mestroni L (2011) Genetic variation in titin in arrhythmogenic right ventricular cardiomyopathy-overlap syndromes. Circulation 124(8):876–885. doi:10.1161/CIRCULATIONAHA.110.005405
Peled Y, Gramlich M, Yoskovitz G, Feinberg MS, Afek A, Polak-Charcon S, Pras E, Sela BA, Konen E, Weissbrod O, Geiger D, Gordon PM, Thierfelder L, Freimark D, Gerull B, Arad M (2014) Titin mutation in familial restrictive cardiomyopathy. Int J Cardiol 171(1):24–30. doi:10.1016/j.ijcard.2013.11.037
Acknowledgments
This work was supported by the Portuguese Foundation for Science and Technology Grants PEst-C/SAU/UI0051/2014 and EXCL/BIM-MEC/0055/2012 through the Cardiovascular R&D Unit and by European Commission Grant FP7-Health-2010, MEDIA-261409.
Conflict of interest
Drs. Manuel Neiva-Sousa, João Almeida-Coelho, Inês Falcão-Pires and Adelino F. Leite-Moreira have no conflicts of interest or financial ties to disclose.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Neiva-Sousa, M., Almeida-Coelho, J., Falcão-Pires, I. et al. Titin mutations: the fall of Goliath. Heart Fail Rev 20, 579–588 (2015). https://doi.org/10.1007/s10741-015-9495-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10741-015-9495-6