
Abstract
Application of imputation methods to accurately predict a dense array of SNP genotypes in the dog could provide an important supplement to current analyses of array-based genotyping data. Here, we developed a reference panel of 4,885,283 SNPs in 83 dogs across 15 breeds using whole genome sequencing. We used this panel to predict the genotypes of 268 dogs across three breeds with 84,193 SNP array-derived genotypes as inputs. We then (1) performed breed clustering of the actual and imputed data; (2) evaluated several reference panel breed combinations to determine an optimal reference panel composition; and (3) compared the accuracy of two commonly used software algorithms (Beagle and IMPUTE2). Breed clustering was well preserved in the imputation process across eigenvalues representing 75 % of the variation in the imputed data. Using Beagle with a target panel from a single breed, genotype concordance was highest using a multi-breed reference panel (92.4 %) compared to a breed-specific reference panel (87.0 %) or a reference panel containing no breeds overlapping with the target panel (74.9 %). This finding was confirmed using target panels derived from two other breeds. Additionally, using the multi-breed reference panel, genotype concordance was slightly higher with IMPUTE2 (94.1 %) compared to Beagle; Pearson correlation coefficients were slightly higher for both software packages (0.946 for Beagle, 0.961 for IMPUTE2). Our findings demonstrate that genotype imputation from SNP array-derived data to whole genome-level genotypes is both feasible and accurate in the dog with appropriate breed overlap between the target and reference panels.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Identifying disease-associated genetic polymorphisms in the dog often relies on genotyping affected and unaffected animals using a SNP genotyping array such as the CanineHD BeadChip (Illumina, San Diego, CA) or the Canine SNP Genotyping Array (Affymetrix, Santa Clara, CA). These arrays contain 173,662 and 49,663 markers, respectively. Both of these arrays offer a relatively cost-effective method of genotyping hundreds of animals for tag SNPs dispersed throughout the genome. Use of these arrays has helped lead to the discovery of many disease-associated mutations and morphology-associated traits in the dog (Bannasch et al. 2010; Wilbe et al. 2010; Meurs et al. 2010; Ahonen et al. 2013). However, these arrays capture only a portion of the known canine genetic variation (Axelsson et al. 2013), and capturing additional variation could be helpful in both genome-wide association studies and genomic prediction models. In contrast, whole genome sequencing captures nearly all of the variation within a subject, but despite precipitous declines in costs over the past several years, it remains significantly more expensive than array-based genotyping (Check Hayden 2014).
Over the past decade, highly accurate methods of genotype imputation have been developed which allow for the prediction of large numbers of genetic variants from a much smaller subset of known genotypes (Li et al. 2009). Genotype imputation is a computational method for predicting these larger number of variants. Performing imputation on a whole genome-level requires a high-density, high-quality, species-specific reference panel, and has been performed successfully in humans, cows, and horses (Howie et al. 2012; Frischknecht et al. 2014; Brøndum et al. 2014; Daetwyler et al. 2014). Whole genome-level SNP imputation has also recently become more common in the analysis of both human and bovine GWAS data as a result of comprehensive reference panels provided by the 1000 Genomes and 1000 Bull Genomes consortia (Marchini and Howie 2010; Abecasis et al. 2010; Howie et al. 2012; Daetwyler et al. 2014).
SNP imputation has been reported in the dog as part of a genomic breeding value study in which SNPs from the Illumina array were imputed using genotypes derived from the smaller Affymetrix array (Guo et al. 2011); similar studies have been performed in pigs, sheep, and chickens (Huang et al. 2012; Heidaritabar et al. 2015; Bolormaa et al. 2015). However, to the best of our knowledge, whole genome-level SNP imputation has never before been reported in the dog, and no study has been performed examining the accuracy of SNP imputation in this species.
Here, we developed a reference panel of 83 canine whole genome sequences across 15 dog breeds and used these data to predict the genotypes of 268 dogs in three dog breeds at a whole genome-level. Our objectives were to demonstrate the feasibility of sequence-level genotype imputation in the dog and evaluate its accuracy using commonly referenced linkage disequilibrium-based software algorithms.
Materials and methods
Samples
We analyzed DNA samples from 180 Standard Poodles, 68 Boxers, 54 Great Danes, 6 Scottish Terriers, 5 Scottish Deerhounds, 4 Collies, 4 Doberman Pinschers, 4 West Highland White Terriers, 3 Irish Setters, 3 Rhodesian Ridgebacks, 3 Yorkshire Terriers, 2 Golden Retrievers, 1 Cavalier King Charles Spaniel, 1 German Shepherd, and 1 Shetland Sheepdog. Samples were collected as part of ongoing research in our laboratory. All DNA was collected from EDTA blood and was extracted using the standard protocol of the DNeasy Blood and Tissue Kit (Qiagen).
Whole genome sequencing
Whole genome sequencing was performed on 20 Standard Poodles, 20 Boxers, 6 Great Danes, 6 Scottish Terriers, 5 Scottish Deerhounds, 4 Collies, 4 Doberman Pinschers, 4 West Highland White Terriers, 3 Irish Setters, 3 Rhodesian Ridgebacks, 3 Yorkshire Terriers, 2 Golden Retrievers, 1 Cavalier King Charles Spaniel, 1 German Shepherd, and 1 Shetland Sheepdog for a total of 83 dogs. These samples included two Standard Poodle trios, one Rhodesian Ridgeback trio, and two Standard Poodle duos. Genotypes derived from this population of dogs comprised the reference panel for SNP imputation.
Approximately 3 μg of DNA was submitted for library preparation and whole genome sequencing at the University of North Carolina at Chapel Hill High-Throughput Sequencing Facility (51), the Medical University of South Carolina Proteogenomics Facility (13), the University of Washington High-Throughput Genomics Center (8), the University of Missouri DNA Core (6), or the Genomic Sciences Laboratory at North Carolina State University (5) (numbers in parenthesis represent the number of dogs sequenced at each institution). All sequencing experiments were designed as 100 or 125 bp paired-end reads and each sample was run on either 1 or 2 lanes of an Illumina HiSeq 2000 or 2500 high-throughput sequencing system.
Analysis of next-generation sequencing data was performed using a standardized bioinformatics pipeline. Sequence reads were trimmed using Trimmomatic 0.32 (Bolger et al. 2014) to a minimum Phred-scaled base quality score of 30 at the start and end of each read with a minimum read length of 70 bp, and aligned to the canFam3 reference sequence (Lindblad-Toh et al. 2005) using BWA 0.7.10 (Li and Durbin 2009). Aligned reads were prepared for analysis using GATK 3.4-46 (McKenna et al. 2010) following the best practices for base quality score recalibration and indel realignment specified by the Broad Institute, Cambridge, MA (DePristo et al. 2011; Van der Auwera et al. 2013). Variant calls were made using GATK’s HaplotypeCaller walker, and variant quality score recalibration (VQSR) was performed using sites from dbSNP 139 and the Illumina CanineHD BeadChip as training resources.
Reference panel development
In order to select only the highest quality variants for our reference panel, we selected a VQSR tranche sensitivity cutoff of 90 % to eliminate the top 10 % most likely false positive sites. Using both GATK and VCFtools (Danecek et al. 2011), we then set any genotype call with a Phred-scaled quality score <20 to missing, and further filtered the variant sites to include only bi-allelic SNPs with a minimum call rate across all samples of 95 %, minimum minor allele frequency (MAF) of 0.05, and Hardy–Weinberg equilibrium (HWE) p value >1 × 10−7. For certain downstream analyses, reference panels were divided into breed or sample subsets using VCFtools or GATK’s SelectVariants walker.
SNP genotyping
SNP genotyping was performed on 180 Standard Poodles, 48 Boxers, and 48 Great Danes. Genotypes derived from this population of 276 dogs were used to create the target panel for SNP imputation. Approximately 0.4 μg of DNA was submitted to Neogen/GeneSeek (Lincoln, NE) for processing and genotyping using the Illumina Canine HD BeadChip. Genotyping and variant calling were carried out per manufacturer recommendations.
Target panel development
SNP genotypes were filtered for per-sample call rate >95 %, per SNP call rate >90 %, MAF >0.05, and HWE p value >1 × 10−7. Prior to imputation, the target panel was compared to the reference panel using conform-gt (Browning and Browning 2007) to exclude target records without a corresponding reference panel record and to adjust target records to match the allele order and chromosome strand in the reference panel.
Genotype imputation
Genotype imputation was performed with both Beagle 4.0 (Browning and Browning 2007) and IMPUTE2 2.3.2 (Marchini et al. 2007; Howie et al. 2009). Beagle was used for all analyses; IMPUTE2 was used secondarily to compare against the Beagle output.
Prior to imputation, the reference panel was pre-phased. For imputation with Beagle, the reference panel was split by chromosome and phased using Beagle 4.0 with available pedigree data. For imputation with IMPUTE2, the reference panel was split by chromosome and phased using SHAPEIT 2.2 (Delaneau et al. 2012) with an effective population size of 200 and a window size of 0.5 Mb. Publicly available canine genetic maps (Wong et al. 2010) were converted from canFam2 to canFam3 coordinates using LiftOver (Kent et al. 2002) for phasing with SHAPEIT.
For genotype imputation using Beagle, data were imputed on a per chromosome basis using mostly default settings; however, the overlap setting was decreased to 2000 markers and the window setting was decreased to 25,000 markers given the density of our reference panel. All available pedigree information from the target panel samples was incorporated into the input parameters using the “ped” option.
For genotype imputation using IMPUTE2, the target panel was pre-phased using SHAPEIT 2.2 with an effective population size of 200 and a window size of 0.5 Mb. Imputation was then performed using phased target and reference panels with an effective population size of 200 and the “allow_large_regions” flag to enable imputation on a per chromosome basis. We used a low effective population size given the very small number of founders within dog breeds, as well as estimates of N e that have been made in other breeds (Calboli et al. 2008). For imputation on the X chromosome, the “chrX” flag was used and the sample genders were added to the appropriate input files. IMPUTE2 genotype probabilities were converted into VCF files with no minimum genotype likelihood for called genotypes, thereby disallowing unknown genotype calls. This was done in order to allow a direct comparison between Beagle and IMPUTE2 outputs.
Analysis of imputed genotypes
We performed analyses of the imputed data to evaluate breed clustering before and after imputation, determine an optimal reference panel, and compare the imputation accuracy of Beagle vs. IMPUTE2.
Breed clustering
Breed clustering was performed using principal components analysis to compare the actual vs. imputed genotype calls for all dogs in the target panel (175 Standard Poodles, 46 Boxers, 47 Great Danes). Actual genotypes were determined from the Illumina Canine HD BeadChip as described above. Imputed genotypes were determined using Beagle with all 83 dogs included in the reference panel. Principal components were calculated using PLINK 1.9 (Chang et al. 2015) and visually compared along the top four eigenvalues.
Reference panel development
An optimal reference panel was determined in three experiments by evaluating imputation accuracy using data from 18 Standard Poodles with genotype data in both the reference and target panels (two Standard Poodles in the reference panel were not genotyped on the SNP array). The first experiment was designed to evaluate imputation accuracy using the entire multi-breed reference panel. Five unrelated Standard Poodles that had no other relatives in the reference panel were randomly chosen and masked from the reference panel one-at-a-time. The remaining 82 dogs were then used as a reference panel to impute each masked dog’s genotypes, leaving only one dog in the target panel at a time. The second experiment was designed to evaluate the imputation accuracy of a breed-specific reference panel. All non-Poodles were removed from the reference panel, and the same five Standard Poodles were masked one-at-a-time from the remaining 20 dogs, leaving 19 Standard Poodles as the reference panel and one dog as the target panel for each imputation trial. The third experiment was designed to evaluate the imputation accuracy of a reference panel with no breed overlap with the target panel. All Standard Poodles were removed from the reference panel (leaving 63 dogs) and the genotypes of all 18 Standard Poodles with WGS data were imputed. For this experiment, all 18 Standard Poodles comprised the target panel. In the experiments described above, imputed genotypes were compared to actual genotypes using GATK’s GenotypeConcordance walker using actual genotypes as the truth set; known genotypes from the HD array were masked when calculating genotype concordance.
We validated our findings in Standard Poodles by evaluating similar reference panels in both Boxers and Great Danes. As none of the Boxers or Great Danes overlapped in the original target and reference panels, we created a target panel by selecting the SNPs on the Illumina Canine HD array from five unrelated Boxers and five unrelated Great Danes in the reference panel. We then masked these dogs from the reference panel one-at-a-time and imputed each dog’s genotypes using either (1) the 82-dog reference panel or (2) a breed-specific reference panel (19 dogs for the Boxer-only panel, and 5 dogs for the Great Dane-only reference panel). Imputation was performed using Beagle and imputed genotypes were compared to the actual genotypes using GATK’s GenotypeConcordance walker as described above.
Comparison of imputation software
The imputation accuracy of Beagle vs. IMPUTE2 was compared using the same five Standard Poodles from the previous analysis. Each dog was masked one-at-a-time from the complete multi-breed reference panel, imputation was performed using each software algorithm, and imputed and actual genotypes were compared using GATK’s GenotypeConcordance walker. We subdivided the genotype concordance results into deciles based upon either Beagle’s allelic R 2 parameter or IMPUTE2’s Info parameter (Browning and Browning 2007; Marchini and Howie 2010); these parameters are calculated differently, but are provided by the software developers in order to evaluate the certainty of the imputed genotypes (Marchini and Howie 2010). Within each R 2 or Info group, we examined the concordance of heterozygous and homozygous calls separately.
As part of this analysis, we also compared the actual to the imputed genotypes derived from both Beagle and IMPUTE2 by calculating the Pearson correlation coefficient. Actual genotypes were converted to genotype dosages using the “–recode A” option in PLINK 1.9 (Chang et al. 2015), and the correlation was calculated using the genotype probabilities determined by both Beagle and IMPUTE2 (also converted to dosages). Correlation analysis was performed using custom scripting in R version 3.2.3 (Team 2015) using the “cor” function and “method = ‘pearson’” option.
Results
Reference and target panel development
Average coverage for whole genome sequences ranged from 18–51× . After variant calling and filtration of the reference panel, 4,885,283 high-quality SNPs remained, representing one SNP every 476 bases on average. The distribution of SNPs by chromosome in the reference panel is provided in Supplemental Table 1.
After filtering the target panel samples, 175 Standard Poodles, 46 Boxers, and 47 Great Danes remained. Among these dogs, 18 Standard Poodles but none of the Boxers or Great Danes, were contained in the reference panel. The target panel contained 124,595 SNPs after filtering for call rate, MAF, and HWE p value; this number was reduced to 84,193 after application of conform-gt. Imputation of the target panel from the reference panel therefore represented a 58-fold increase in SNP count.
Breed clustering
We used principal components analysis to visualize breed clustering in the actual (SNP array-derived) and imputed genotypes for all dogs in the target panel. A scree plot of both the actual and imputed genotypes (Figure S1) shows that nearly all of the variation is contained in the first four principal components, with the vast majority of the variation present in the first two principal components (actual genotypes: EV1 = 45.2 %, EV2 = 27.6 %; imputed genotypes: EV1 = 46.3 %, EV2 = 29.7 %).
A comparison of the first two principal components between the reference and imputed genotypes shows that the breed clustering in the imputed genotypes is highly consistent with the actual genotypes (Fig. 1). This suggests that the major breed-specific haplotypes were correctly identified in the imputation process. In contrast, breed clustering was slightly less accurate for the third and fourth principal components (Figure S2, panels B–F); however, these represent a much smaller portion of the variation in the imputed genotypes.

Principal components (PC) plots comparing the first and second eigenvalues for the actual (a) vs. the imputed (b) genotypes of 175 Standard Poodles, 46 Boxers, and 47 Great Danes. Panel a shows PC data derived from actual genotype calls at 124,595 sites using the Illumina Canine HD BeadChip. Panel b shows PC data derived from imputed genotype calls at 4,885,283 sites using a reference panel of SNPs obtained from whole genome sequencing of 83 dogs across 15 different breeds. Imputation of the data in Panel b was performed using Beagle as described in the manuscript text. Scree plots and additional eigenvalue comparisons are shown in Figures S1 and S2, respectively
Reference panel optimization
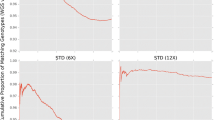
Using data from the 18 Standard Poodles with genotypes in both the target and reference panels, we used Beagle to evaluate which of three reference panel subsets (a multi-breed panel of 82 dogs including 19 Standard Poodles, a single-breed panel containing 19 Standard Poodles only, or a multi-breed panel of 63 dogs containing no Standard Poodles) would allow imputation with the highest degree of genotype concordance (Fig. 2). On average across all chromosomes, concordance was highest using the multi-breed reference panel at 92.4 %; with 19 Standard Poodles only as the reference panel, the average imputation accuracy was 87.0 %; and with 63 non-Poodles the imputation accuracy was 74.9 %. There was also variability observed in imputation accuracy by chromosome. Using the multi-breed reference panel, for example, the highest concordance was observed on chromosome 20 (95.3 %) and the lowest on the X chromosome (87 %).

Genotype concordance by chromosome for imputed vs. actual genotypes using three different reference panels. Boxes are demarcated by the first and third quartiles for each dataset, and whiskers represent 1.5 × IQR, with outliers represented as dots above or below the whiskers; lines within each box represent median values. The dotted line colored by reference panel represents the overall genotype concordance for that reference panel
Our findings were directionally similar using the multi-breed or breed-specific reference panels in Boxers and Great Danes, with a higher genotype concordance using the multi-breed vs. the breed-specific reference panels. On average across all chromosomes, for Boxers the average genotype concordance was 97.8 % using the entire multi-breed reference panel and 94.9 % for the Boxer-specific reference panel; for Great Danes, the concordance was 86.8 % using the entire multi-breed reference panel and 62.8 % for the Great Dane-specific reference panel. The overall genotype concordance was much lower for the Great Danes compared to either the Boxers or the Standard Poodles, regardless of the reference panel used. This observation is likely a result of the much smaller number of Great Danes in the reference panel compared to either the Boxers or the Standard Poodles. Genotype concordance data on a per chromosome basis for each breed are shown in Supplemental Figure 3 panels A (Boxers) and B (Great Danes).
Comparison of imputation software
We used the multi-breed reference panel of 83 dogs to compare the accuracy of two imputation software packages: Beagle 4.0 and IMPUTE2 2.3.2. Imputed sites were grouped into deciles using either Beagle’s allelic R 2 or IMPUTE2’s Info metric, and genotype concordance was determined within each decile for five Standard Poodles with both imputed and reference genotypes (Fig. 3, top panels). None of the sites were filtered by genotype likelihood prior to evaluating genotype concordance. The average genotype concordance was 92.4 % using Beagle and 94.1 % using IMPUTE2; data for each individual dog are shown in Supplemental Table 2. For both software algorithms, genotype concordance was higher for homozygous imputation calls compared to heterozygous calls across most of the genotype certainty bins. We also determined the fraction of imputed calls within each decile (Fig. 3, bottom panels). The distribution of genotype calls by certainty metric was significantly more left-skewed with IMPUTE2 vs. Beagle (skewness −3.1 vs. −0.87).

Genotype concordance results using Beagle (a) and IMPUTE2 (b). In (a), the top panel shows the concordance grouped by allelic R 2 and the type of actual genotype call; the dashed line represents the overall genotype concordance. The bottom panel shows a histogram of the percentage of imputed calls within each allelic R 2 bin. In (b), the data are similarly represented but concordance is grouped by the IMPUTE2 Info parameter instead
IMPUTE2 also performs an internal cross-validation step, calculating concordance metrics by masking one variant at a time and comparing the actual and imputed genotype for that variant for all samples contained in both the target and reference panels. In calculating the genotype concordance using this method, the algorithm only considers variant calls with a genotype likelihood of ≥0.9. Across all chromosomes, the average genotype concordance determined in this manner was 95.1 %, with a range of 91.7 % on the X chromosome to 96.6 % on chromosome 13.
We also compared the imputation accuracy of Beagle vs. IMPUTE2 using Pearson’s correlation coefficient between the actual and imputed genotype dosages. The average correlation across all chromosomes was 0.946 for Beagle and 0.961 for IMPUTE2, which is slightly higher than the respective genotype concordance values. Further detail on the genotype correlation data broken out by dog is shown in Supplemental Table 2, and by chromosome in Supplemental Table 3.
Discussion
In this study, we describe the feasibility of genotype imputation in the dog using genotypes derived from the Illumina CanineHD BeadChip to nearly 5 million SNPs derived from whole genome sequencing. SNP array-derived data were available from 175 Standard Poodles, 46 Boxers, and 47 Great Danes, and whole genome data were available from 83 dogs in 15 breeds. Genotypes from 18 Standard Poodles were present in both datasets, and a subset of these dogs was used to compare the genotype concordance between actual and imputed genotypes with three different reference panel breed combinations and two linkage disequilibrium-based imputation software packages. We also validated these findings in both Boxers and Great Danes using a target panel derived from the whole genome sequencing data.
As has been demonstrated in other species, we showed that direct imputation from SNP array-derived data to whole genome-derived data is both accurate and feasible. Our imputation represents a 58-fold increase in the number of available SNPs. We suspect that the high level of accuracy achievable here with a relatively small reference panel is largely due to the extensive linkage disequilibrium (Sutter et al. 2004; Stern et al. 2013), high degree of within-breed relatedness, and low effective population size in the dog (Calboli et al. 2008).
In terms of reference panel design, we showed the benefits of a multi-breed reference panel compared to a breed-specific reference panel in three dog breeds, and demonstrated the importance of breed overlap between the target and reference panels. The added benefit of multiple breeds has been shown in cows and horses as well (Frischknecht et al. 2014; Daetwyler et al. 2014; Bouwman and Veerkamp 2014). Here, it is likely that some variants with a low minor allele frequency in one breed have a higher minor allele frequency (MAF) in other breeds, which could aid imputation accuracy given the difficulty of accurate imputation at sites with low MAF (Li et al. 2011; Kreiner-Moller et al. 2015). Furthermore, studies in cows have shown that the addition of multiple breeds to a reference panel is particularly beneficial when the reference population of a particular breed is small (Larmer et al. 2014), as is the case in this study where the greatest number of dogs in any one breed was 20. We also demonstrated that even for a highly inbred species like dogs, having a relatively small number breed-matched animals in a reference panel is likely to yield poor imputation quality given the low genotype concordance we observed in imputing Great Dane genotypes (up to 86.8 %) with only 5 breed-matched dogs.
In our analysis of the optimal reference panel design, we noted small differences in imputation accuracy across each chromosome (Fig. 2). While we did not evaluate the reasons for these differences, we would suspect that those chromosomes or regions of the genome with low imputation accuracy may correlate with regions of high heterozygosity or shorter stretches of linkage disequilibrium, as both of these factors can present challenges in reconstructing haplotypes. Further investigation into the cause of these differences across the genome may also yield information regarding potential errors in misplaced SNPs on the Canine SNP array or in the assembly of the reference genome, as has been demonstrated in other species (Pausch et al. 2013; Daetwyler et al. 2014).
We also found that both the genotype concordance and correlation between the reference and target panels was slightly higher using IMPUTE2 compared to Beagle. This is consistent with findings in other species (Frischknecht et al. 2014; Daetwyler et al. 2014). Both Beagle and IMPUTE2 use a hidden Markov model to calculate missing genotypes; however, Beagle reconstructs haplotypes present in the reference population only and IMPUTE2 reconstructs haplotypes present in both the reference and test populations. This may improve imputation accuracy with small reference populations. Others have suggested that differences in software performance may be due to differences in the methods of haplotype reconstruction or the ability of each software algorithm to define haplotypes over long distances (Frischknecht et al. 2014). Interestingly, the difference in imputation accuracy between the two software algorithms was lower in our study than has been reported in the studies cited above; this may be due to the extensive linkage disequilibrium in the dog, which could allow for improved prediction over greater distances or the better performance of imputation algorithms broadly when imputing genotypes within a single breed.
When comparing the genotyped to the imputed data, we found that the correlation values were generally higher than the concordance values for both Beagle and IMPUTE2 (although still on average higher in IMPUTE2 compared to Beagle). The higher correlation values are likely the result of the underlying methodology: on a per SNP basis, concordance yields in a binary measure (matches/does not match), whereas correlation accounts for the imputed genotype probabilities and genotype dosages on a continuous scale. Additionally, some authors have also suggested that correlation is a better method of assessing imputation accuracy as this method better accounts for the correct imputation of rare alleles (Calus et al. 2014).
In our comparison of Beagle and IMPUTE2, we also evaluated the accuracy and distribution of imputed genotype calls by allelic R 2 (Beagle) or Info (IMPUTE2) parameters. As expected, the overall genotype concordance is higher as each parameter approaches its maximum value of one. Our data show that heterozygous genotypes are much less likely than homozygous genotypes to be called accurately at lower R 2 or Info values, which is consistent with the way in which these parameters are calculated (Marchini and Howie 2010). Post-imputation filtering by genotype likelihood might have eliminated many of these inaccurately called heterozygous genotypes, and this type of post-imputation filtering could certainly be considered prior to using imputed genotype calls for association studies or genomic prediction analyses.
Our data also demonstrated a marked left skewness of the distribution of genotype calls by IMPUTE2’s Info metric compared to Beagle’s allelic R 2 metric. This left skew was also noted in imputed human genotype calls using IMPUTE2 (Verma et al. 2014). Because these parameters are generally used as quality metrics to establish a cutoff for downstream analysis of imputed genotypes, our findings suggest that examining the distribution of genotype calls by quality metric may be useful prior to setting a specific cutoff value for any particular experiment. Additionally, because the distribution of the quality metrics differs for the software packages we evaluated (likely due to differences in the underlying calculation methods), using a different cutoff value for each package may be indicated when incorporating imputed genotypes into downstream analyses.
One major limitation of this study is the small number of animals contained within both the reference and target panels that could be used for comparing the accuracy of genotype calls. All of these dogs were also Standard Poodles, and as a result the per-animal genotype concordance could only be calculated for this breed. We attempted to mitigate this limitation by developing a target panel in both Boxers and Great Danes using reference panel data; however, this method cannot account for differences in genotyping accuracy between SNP arrays and next-generation whole genome sequencing. Future studies with a greater overlap between target and reference panels could improve our ability to evaluate genotype concordance more broadly in the dog. Additionally, the number of dogs in our reference panel (83) was relatively small to what is commonly used in both human and bovine studies. We expect that increasing the number of animals (particularly within a breed) would increase the imputation accuracy that we achieved in this study, in particular for low MAF sites that are difficult to impute with confidence.
In summary, our findings demonstrate that genotype imputation from SNP array-derived data to whole genome-level genotypes is both feasible and accurate in the dog with appropriate breed overlap between the target and reference panels. Using only 83 dogs as a reference panel, we achieved >92 % genotype concordance using Beagle and >94 % genotype concordance using IMPUTE2 in Standard Poodles, with even higher values observed in Boxers. The IMPUTE2 concordance was further increased to 95.1 % considering only those sites with a genotype likelihood above 0.9. Genome-wide genotype correlations were similarly high at 0.946 for Beagle and 0.961 for IMPUTE2. Further studies are warranted, which are aimed at evaluating the usefulness of imputed genotypes in downstream analyses such as genome-wide association studies or the development of genomic prediction models.
References
Abecasis GR, Altshuler D, Auton A et al (2010) A map of human genome variation from population-scale sequencing. Nature 467:1061–1073. doi:10.1038/nature09534
Ahonen SJ, Pietilä E, Mellersh CS et al (2013) Genome-wide association study identifies a novel canine glaucoma locus. PLoS ONE 8:e70903–e70903. doi:10.1371/journal.pone.0070903
Axelsson E, Ratnakumar A, Arendt M-L et al (2013) The genomic signature of dog domestication reveals adaptation to a starch-rich diet. Nature 495:360–364. doi:10.1038/nature11837
Bannasch D, Young A, Myers J et al (2010) Localization of canine brachycephaly using an across breed mapping approach. PLoS ONE 5:e9632. doi:10.1371/journal.pone.0009632
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. doi:10.1093/bioinformatics/btu170
Bolormaa S, Gore K, van der Werf JHJ et al (2015) Design of a low-density SNP chip for the main Australian sheep breeds and its effect on imputation and genomic prediction accuracy. Anim Genet 46:544–556. doi:10.1111/age.12340
Bouwman AC, Veerkamp RF (2014) Consequences of splitting whole-genome sequencing effort over multiple breeds on imputation accuracy. BMC Genet 15:105. doi:10.1186/s12863-014-0105-8
Brøndum RF, Guldbrandtsen B, Sahana G et al (2014) Strategies for imputation to whole genome sequence using a single or multi-breed reference population in cattle. BMC Genom 15:728. doi:10.1186/1471-2164-15-728
Browning SR, Browning BL (2007) Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am J Hum Genet 81:1084–1097. doi:10.1086/521987
Calboli FCF, Sampson J, Fretwell N, Balding DJ (2008) Population structure and inbreeding from pedigree analysis of purebred dogs. Genetics 179:593–601. doi:10.1534/genetics.107.084954
Calus MPL, Bouwman AC, Hickey JM et al (2014) Evaluation of measures of correctness of genotype imputation in the context of genomic prediction: a review of livestock applications. Animal 8:1743–1753. doi:10.1017/S1751731114001803
Chang CC, Chow CC, Tellier LC et al (2015) Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4:7. doi:10.1186/s13742-015-0047-8
Check Hayden E (2014) Is the $1000 genome for real? Nature. doi:10.1038/nature.2014.14530
Daetwyler HD, Capitan A, Pausch H et al (2014) Whole-genome sequencing of 234 bulls facilitates mapping of monogenic and complex traits in cattle. Nat Genet 46:858–865. doi:10.1038/ng.3034
Danecek P, Auton A, Abecasis G et al (2011) The variant call format and VCFtools. Bioinformatics 27:2156–2158. doi:10.1093/bioinformatics/btr330
Delaneau O, Marchini J, Zagury J-F (2012) A linear complexity phasing method for thousands of genomes. Nat Methods 9:179–181. doi:10.1038/nmeth.1785
DePristo MA, Banks E, Poplin R et al (2011) A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43:491–498. doi:10.1038/ng.806
Frischknecht M, Neuditschko M, Jagannathan V et al (2014) Imputation of sequence level genotypes in the Franches-Montagnes horse breed. Genet Sel Evol 46:63. doi:10.1186/s12711-014-0063-7
Guo G, Zhou Z, Wang Y et al (2011) Canine hip dysplasia is predictable by genotyping. Osteoarthr Cartil 19:420–429. doi:10.1016/j.joca.2010.12.011
Heidaritabar M, Calus MPL, Vereijken A et al (2015) Accuracy of imputation using the most common sires as reference population in layer chickens. BMC Genet 16:101. doi:10.1186/s12863-015-0253-5
Howie BN, Donnelly P, Marchini J (2009) A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet 5:e1000529–e1000529. doi:10.1371/journal.pgen.1000529
Howie B, Fuchsberger C, Stephens M et al (2012) Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat Genet 44:955–959. doi:10.1038/ng.2354
Huang Y, Hickey JM, Cleveland MA, Maltecca C (2012) Assessment of alternative genotyping strategies to maximize imputation accuracy at minimal cost. Genet Sel Evol 44:25. doi:10.1186/1297-9686-44-25
Kent WJ, Sugnet CW, Furey TS et al (2002) The human genome browser at UCSC. Genome Res 12:996–1006. doi:10.1101/gr.229102
Kreiner-Moller E, Medina-Gomez C, Uitterlinden AG et al (2015) Improving accuracy of rare variant imputation with a two-step imputation approach. Eur J Hum Genet 23:395–400. doi:10.1038/ejhg.2014.91
Larmer SG, Sargolzaei M, Schenkel FS (2014) Extent of linkage disequilibrium, consistency of gametic phase, and imputation accuracy within and across Canadian dairy breeds. J Dairy Sci 97:3128–3141. doi:10.3168/jds.2013-6826
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. doi:10.1093/bioinformatics/btp324
Li Y, Willer C, Sanna S, Abecasis G (2009) Genotype imputation. Annu Rev Genom Human Genet 10:387–406. doi:10.1146/annurev.genom.9.081307.164242
Li L, Li Y, Browning SR et al (2011) Performance of genotype imputation for rare variants identified in exons and flanking regions of genes. PLoS ONE 6:e24945–e24945. doi:10.1371/journal.pone.0024945
Lindblad-Toh K, Wade CM, Mikkelsen TS et al (2005) Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature 438:803–819. doi:10.1038/nature04338
Marchini J, Howie B (2010) Genotype imputation for genome-wide association studies. Nat Rev Genet 11:499–511. doi:10.1038/nrg2796
Marchini J, Howie B, Myers S et al (2007) A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet 39:906–913. doi:10.1038/ng2088
McKenna A, Hanna M, Banks E et al (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20:1297–1303. doi:10.1101/gr.107524.110
Meurs KM, Mauceli E, Lahmers S et al (2010) Genome-wide association identifies a deletion in the 3′ untranslated region of striatin in a canine model of arrhythmogenic right ventricular cardiomyopathy. Hum Genet 128:315–324. doi:10.1007/s00439-010-0855-y
Pausch H, Aigner B, Emmerling R et al (2013) Imputation of high-density genotypes in the Fleckvieh cattle population. Genet Sel Evol 45:3. doi:10.1186/1297-9686-45-3
Stern JA, White SN, Meurs KM (2013) Extent of linkage disequilibrium in large-breed dogs: chromosomal and breed variation. Mamm Genome 24:409–415. doi:10.1007/s00335-013-9474-y
Sutter NB, Eberle MA, Parker HG et al (2004) Extensive and breed-specific linkage disequilibrium in Canis familiaris. Genome Res 14:2388–2396. doi:10.1101/gr.3147604
Team RC (2015) R: A language and environment for statistical computing
Van der Auwera GA, Carneiro MO, Hartl C, et al (2013) From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinform 11:11.10.1–11.10.33. doi: 10.1002/0471250953.bi1110s43
Verma SS, de Andrade M, Tromp G et al (2014) Imputation and quality control steps for combining multiple genome-wide datasets. Front Genet 5:370. doi:10.3389/fgene.2014.00370
Wilbe M, Jokinen P, Truvé K et al (2010) Genome-wide association mapping identifies multiple loci for a canine SLE-related disease complex. Nat Genet 42:250–254. doi:10.1038/ng.525
Wong AK, Ruhe AL, Dumont BL et al (2010) A comprehensive linkage map of the dog genome. Genetics 184:595–605. doi:10.1534/genetics.109.106831
Acknowledgments
SGF is supported by a National Institutes of Health T32 training award (5T32OD011130-07). Funding for whole genome sequencing was provided in part by the Poodle Club of America Foundation and the American Kennel Club Canine Health Foundation. Some whole genome sequencing data were graciously contributed by Drs. Leigh Anne Clark (13 dogs), Natasha J. Olby and Theirry Olivry (11 dogs), and Joshua A. Stern (2 dogs).
Authors contributions
SGF collected samples, designed the study, analyzed the data, and wrote the manuscript. KMM collected samples and supervised the study. All authors have read and edited the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Friedenberg, S.G., Meurs, K.M. Genotype imputation in the domestic dog. Mamm Genome 27, 485–494 (2016). https://doi.org/10.1007/s00335-016-9636-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00335-016-9636-9