
Abstract
Familial dilated cardiomyopathy is a primary myocardial disease that can result in the development of congestive heart failure and sudden cardiac death. Spontaneous animal models of familial dilated cardiomyopathy exist and the Doberman pinscher dog is one of the most commonly reported canine breeds. The objective of this study was to evaluate familial dilated cardiomyopathy in the Doberman pinscher dog using a genome-wide association study for a genetic alteration(s) associated with the development of this disease in this canine model. Genome-wide association analysis identified an area of statistical significance on canine chromosome 14 (p raw = 9.999e−05 corrected for genome-wide significance), fine-mapping of additional SNPs flanking this region localized a signal to 23,774,190–23,781,919 (p = 0.001) and DNA sequencing identified a 16-base pair deletion in the 5′ donor splice site of intron 10 of the pyruvate dehydrogenase kinase 4 gene in affected dogs (p < 0.0001). Electron microscopy of myocardium from affected dogs demonstrated disorganization of the Z line, mild to moderate T tubule and sarcoplasmic reticulum dilation, marked pleomorphic mitochondrial alterations with megamitochondria, scattered mitochondria with whorling and vacuolization and mild aggregates of lipofuscin granules. In conclusion, we report the identification of a splice site deletion in the PDK4 gene that is associated with the development of familial dilated cardiomyopathy in the Doberman pinscher dog.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Dilated cardiomyopathy is a primary myocardial disease that can result in the development of congestive heart failure and sudden cardiac death (Hershberger et al. 2010). It is a familial disease in 20–50 % of the cases in human beings and is most commonly inherited as an autosomal dominant trait (Hershberger et al. 2009, 2010). Genetic heterogeneity is a well-known phenomenon and causative mutations have now been identified in at least 30 genes (Hershberger et al. 2010).
Spontaneous animal models of familial dilated cardiomyopathy exist as well. The canine form is particularly well described and has been suggested as a large animal model of this familial human disease because of clinical and pathological similarities (Dukes-McEwan et al. 2003; Meurs et al. 2007; Smucker et al. 1990). The Doberman pinscher dog is one of the most commonly reported canine breeds with familial dilated cardiomyopathy. We have previously shown it to be inherited as an autosomal dominant trait with incomplete penetrance (Meurs et al. 2007). Similarities between the canine and human forms of the disease have resulted in the study of several genes known to cause the human form of the disease in hopes of finding the molecular cause of the disease in species. To date, several genes have been evaluated including desmin, delta sarcoglycan, phospholamban, actin, lamin A/C, cardiac beta-myosin heavy chain, troponin T, troponin C and the cysteine-rich protein 3 gene (Meurs et al. 2001, 2010; Stabej et al. 2004, 2005a, b). However, a causative mutation has not been identified.
The objective of this study was to evaluate familial dilated cardiomyopathy in the Doberman pinscher using a genome-wide association study for a genetic alteration(s) associated with the development of this disease in this canine model.
Materials and methods
Selection of animal subjects
This study was conducted in accordance with the guidelines of the Animal Care and Use Committee at The Ohio State University. Written consent authorizing study participation was obtained from each client.
Sixty-six client-owned pet Doberman pinscher dogs with dilated cardiomyopathy as diagnosed by a left ventricular diastolic dimension of at least 4.8 cm and a fractional shortening less than 20 % were recruited for participation in an ongoing study of familial canine dilated cardiomyopathy. Additionally, 66 apparently healthy Doberman pinscher dogs at least 10 years of age with a normal physical examination and absence of cardiac disease were recruited for participation. Pedigrees were collected when available from both affected and unaffected dogs.
One hundred canine DNA samples were also obtained from Washington State University’s Veterinary Cardiac Genetics Laboratory bank of canine DNA to be used as controls. Samples were selected from eleven different breeds to make up the 100 samples. Represented breeds included Boxer (4), English Bulldog (2), Cavalier king Charles Spaniel (4), golden Retreiver (25), Great Dane (25), Mastiff (1), Malamute (2), Newfoundland (4), Standard Poodle (11), Rottweiler (14) and yellow Labrador (8).
Myocardial tissue was obtained immediately postmortem from selected DCM-affected Doberman pinschers (n = 2) and from non-Doberman pinscher healthy dogs (n = 2), and frozen at −80 °C. Myocardial samples from the left ventricle of the affected Doberman pinschers were also placed in 10 % phosphate-buffered formalin for evaluation by light microscopy and glutaraldehyde for electron microscopy.
Genome-wide association analysis and fine-mapping
Five to seven milliliters of blood was collected from the jugular vein from each dog for genomic DNA extraction as previously described (Meurs et al. 2000).
Genome-wide analysis (GWA) was performed with the Affymetrix Canine Genome 2.0 Array “Platinum Panel” (Affymetrix, Santa Clara, CA, USA) containing 49,663 single nucleotide polymorphisms (SNP) markers with DNA from 48 Doberman pinscher DCM cases and 48 Doberman pinscher controls. Only affected and control dogs with pedigrees were used for the GWA and participating dogs were specifically selected so that no dogs were related within a three generational pedigree. SNP genotypes were obtained following the human 500 K array protocol, but with a smaller hybridization volume to allow for the smaller surface area of the canine array as previously described (Karlsson et al. 2007; Butcher et al. 2008). Case–control GWA mapping was evaluated with PLINK (Purcell et al. 2007). Haplotype analysis was performed with Haploview (Barrett et al. 2004).
Nineteen additional canine SNPs (http://www.broad.mit.edu/node/459) surrounding the identified region of interest (Chromosome 14: 23,698,921–23,931,510) were selected for fine-mapping in a subset of 20 DCM cases and 20 controls selected from the original group of dogs as described above. Polymerase chain reaction (PCR) amplification was used to amplify each SNP. Standard PCR amplifications were carried out with a cocktail of NH4SO4 amplification buffer, Taq DNA polymerase (0.1 units/μL of reaction volume), 2.5 mM MgCl2, 12.5 μM of each dNTP, 2.5 mM of each primer, and 100 ng of template DNA. Samples were denatured for 5 min at 94°C followed by 40 cycles of 94°C for 20 s; 58°C for 30 s; 72°C for 30 s; and 72°C for 7 min. Annealing temperature was optimized to accommodate the respective primer requirement. Amplicons were analyzed on an ABI Prism 377 Sequencer (Applied Biosciences, Foster City, CA, USA). Sequences from cases and controls were scored for alleles at each SNP. Genotype frequencies were compared between case and control groups using Fischer’s exact test with p < 0.05 considered significant.
Mutation detection
PCR primers (Supplemental Table 1) were designed for exons and splice site regions of the pyruvate dehydrogenase kinase 4 (PDK4) gene as well as strongly conserved regions at the 5′ and 3′ untranslated regions (UTR) with Primer 3 software and the canine UCSC database (canFam 2.0; http://genome.ucsc.edu/) (Rozen and Skaletsky 2003). Standard PCR amplifications and sequencing was performed as described above with a negative control run for each amplification. Sequences from cases and controls were compared to each other as well as to the published reference CanFam2.0 canine sequence (http://genome.ucsc.edu/) to identify nucleotide sequence changes.
Subsequently, sequences from 66 cases (38/48 samples from the GWA that had sufficient amount of remaining DNA), 66 controls (36/48 from the GWA) and 100 non-Doberman pinscher dog controls were genotyped for the splice site deletion of PDK4.
Computer evaluation of the splice site region
Human splicing finder (http://www.umd.be/HSF/) was used to assess the likely impact of the deletion mutation on exonic splicing. Additional evaluation using MaxEnt (http://genes.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq.html) was also performed (Eng et al. 2004; Yeo and Burge 2004).
Expression analysis
Due to the difficulty of rapid collection of myocardial tissue immediately at the time of death in client-owned pet dogs that frequently die of sudden cardiac death associated with their disease, only samples from two DCM-affected Doberman pinschers were of the quality needed for expression analysis. Myocardial tissues were also available from two non-Doberman pinscher healthy dogs (controls), and frozen at −80 °C.
Quantitative real-time PCR was performed with left ventricular myocardial sections from one homozygous mutant Doberman pinscher, one heterozygous Doberman, and two non-Dobermans homozygous wild-type controls. Approximately 50 mg of myocardium was pulverized for total RNA extraction with the RNeasy Fibrous Tissue Mini Kit (Qiagen, Valencia, CA, USA). Reverse transcription was performed using Superscript II Reverse Transcriptase for cDNA synthesis (Invitrogen, Carlsbad, CA, USA). Real-time PCR primers were designed for exonic regions 9, 10 and 11 within PDK4. Primers were also designed for regions spanning exons 6 and 7 of hypoxanthine phosphoribosyltransferase (HPRT) to be used as a housekeeping gene. TaqMan Gene Expression Assays (Applied Biosystems) and real-time PCR protocols were used to amplify these targets using the Applied Biosystems 7500 Fast Real-Time PCR System. Samples were evaluated in triplicate. The triplicate C T values for each sample were averaged resulting in mean C T values for PDK4 and HPRT. The PDK4 C t values were standardized to the housekeeping gene by taking the difference ΔC T = C t[PDK4] − C t[HPRT]. Data was compared between control and affected dogs with a T test and are given as mean ± SEM of normalized gene expression levels. A p < 0.05 was considered significant.
Western blot
Frozen myocardial samples from the left ventricle of two non-Doberman controls and one heterozygous and one homozygous Doberman pinscher dogs were prepared using a mitochondrial isolation kit (Pierce Biotechnology, Rockford, IL, USA). Briefly, approximately 50 mg of tissue was homogenized in a cooled dounce homogenizer in homogenization reagent with BSA and protease inhibitors. The mitochondrial pellet was obtained after centrifugation of the sample in mitochondrial isolation reagent at 3,000g for 15 min at 4 °C. For protein concentration the mitochondrial pellet was lysed with 2 % CHAPS in TBS and the concentration was determined with the Pierce 660 protein assay (Pierce Biotechnology, Rockford, IL, USA). For western blot, the mitochondrial pellet was boiled in the SDS-PAGE sample buffer. Twenty micrograms of protein extract for each dog was separated on a 4–20 % gradient polyacrylamide gel and transferred to a polyvinylidene fluoride membrane. Membranes were blocked with 5 % milk and PDK4 epitopes were probed with a rabbit polyclonal antibody generated against the C-terminal region (amino acid 300–400) of PDK4 (1:50) (Abcam Transduction Laboratories, Cambridge, MA, USA). Blots were stripped and also probed with VDAC/porin monoclonal antibody (1:750) as a loading control. The species appropriate secondary IgG–HRP (1:3,500 and 1:4,000, respectively) (Santa Cruz Biotechnology Inc, Santa Cruz, CA, USA) was used followed by chemiluminescence detection and optical density determination using Quantity One software (BioRad, Hercules, CA, USA). Molecular weight was estimated using a standard curve generated from the Precision Protein Plus Western C standard (BioRad). Data was compared between control and affected dogs with a T test and are given as mean ± SEM of normalized gene expression levels. A p < 0.05 was considered significant.
Electron microscopy
Sections of the left ventricle from two affected Doberman pinscher dogs were evaluated by both light microscopy and electron microscopy.
Results
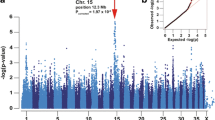
Genome-wide association analysis identified an area of statistical significance on canine chromosome 14 (p raw = 9.999e−05 and p genome < 0.009 corrected for genome-wide significance based on 100,000 permutations using the software package PLINK) at 14:23,781,919 (Purcell et al. 2007) (Fig. 1). Fine-mapping of 13 additional SNPs flanking this region localized a signal to 23,774,190–23,781,919 (p = 0.001). There was one gene within this region, PDK4 (23,774,985–23,785,987).

Genome-wide association mapping of canine dilated cardiomyopathy. Genome-wide association analysis identified an area of statistical significance on canine chromosome 14 (p raw = 9.999e−05 corrected for genome-wide significance, p genome < 0.009 based on 100,000 permutations)
DNA sequencing of the exonic and splice site regions of the PDK4 gene as well as strongly conserved regions at the 5′ and 3′ ends (23,774,058–23,774,985; 23,785,987–23,786,269) identified only one alteration, a 16-base pair deletion in the 5′ donor splice site of intron 10 in affected dogs (23,776,713–23,776,728) (Fig. 2). The 16-base pair deletion removed the 5′ donor site gt plus an additional 14 bases downstream to the next gt in the intron creating a presumed cryptic splice site (Fig. 2). The cryptic gt site had a lower Max Ent score (6.33) than the accepted splice site (8.33).

DNA sequencing of the exonic and splice site regions of the PDK4 gene as well as strongly conserved regions at the 5′ and 3′ ends identified a 16-base pair deletion in the 5′ donor splice site of intron 10 in affected dogs. The black bar above the sequence indicates the sequence deleted in affected dogs. The deletion removed the 5′ donor site gt plus an additional 14 bases downstream to the next gt in the intron creating a presumed cryptic splice site
Evaluation of 132 Doberman pinschers (66 affected, 66 unaffected) identified a strong statistical association of the deletion to the disease (p < 0.0001). The deletion was identified in 54 (45 positive heterozygotes, 9 positive homozygotes) of 66 (82 %) affected dogs and 26 (18 positive heterozygotes, 8 positive homozygotes) of 66 (39 %) unaffected dogs. The deletion was not observed in any of 100 unaffected dogs of 11 other breeds.
Expression analysis
A significant difference was not noted between the affected dogs and the unaffected dogs for expression of mRNA levels in cardiac tissue. However, a 14-fold reduction in expression of PDK for exons 10 and 11 in comparison to the normal dogs was noted for the homozygous dog and a twofold reduction was noted for the heterozygous dog (Fig. 3).

A significant difference was not noted between the affected dogs and the unaffected dogs for expression of mRNA levels in cardiac tissue. However, a 14-fold reduction in expression of PDK for exons 10 and 11 was noted for the homozygous dog (solid black) and a twofold reduction was noted for the heterozygous dog (solid white) in comparison to expression in normal dogs
Western blot
Western blot analysis did not identify a significant difference in PDK4 or VDAC, the mitochondrial control protein, between the non-Doberman controls and the Doberman DCM dogs.
Pathologic analysis
Light microscopic findings included multifocal to confluent myocyte hypertrophy and a mild increase in intramyocardial interstitial collagen.
Electron microscopy of myocardium from affected dogs demonstrated disorganization of the Z line, mild to moderate T tubule and sarcoplasmic reticulum dilation, marked pleomorphic mitochondrial alterations with megamitochondria (Fig. 4), scattered mitochondria with whorling (Fig. 4) and vacuolization disruption and mild aggregates of lipofuscin granules. Focal areas of deposits of collagen were also observed.

Electron microscopy of myocardium from affected dogs demonstrated disorganization of the Z line, mild to moderate T tubule and sarcoplasmic reticulum dilation, marked pleomorphic mitochondrial alterations with megamitochondria (A), scattered mitochondria with whorling (B) and vacuolization disruption and mild aggregates of lipofuscin granules
Discussion
In the study described here we identified a 16-base pair deletion at the donor splice site region of intron 10 of the PDK4 gene that is associated with the development of dilated cardiomyopathy in the Doberman pinscher. Expression analysis showed a reduction in expression of exons 10 and 11, particularly in the homozygous dog. Exon 11 is known to contain a highly conserved DW motif (Asp 394, Trp395) and deletion or alteration of the DW motif has been shown to result in decreased activity of the PDK4 protein (Wynn et al. 2010). Regrettably, in this study the quantity and quality of our myocardial samples were not sufficient to adequately assess PDK4 activity. These evaluations will serve as important aspects of future studies.
PDK4 has an important regulatory role in cardiac energy metabolism. Fatty acid oxidation is the preferential form of energy metabolism used by the healthy heart in the non-postprandial state. When glucose levels are high or when fatty acid oxidation is reduced as in an exercising state, circulating fatty acids are suppressed and glucose oxidation, a less efficient source of high energy phosphate generation than fatty acid metabolism, becomes the predominant source of energy. PDK4 plays a pivotal role in regulating energy metabolism by phosphorylating pyruvate dehydrogenase (PDH), and thus decreasing glucose oxidation during periods when fatty acid oxidation can more efficiently generate energy.
The location of this deletion in the affected dogs in this study would be expected to produce a protein with reduced PDK4 activity and a decreased ability to phosphorylate and negatively regulate glucose oxidation (Lopaschuk et al. 2010). One would expect that dogs with this PDK4 defect would then have glucose oxidation as their predominant metabolic pathway, since as glucose oxidation increases, a parallel decrease in fatty acid oxidation has been noted (Saddik et al. 1993). Ultimately, this change could result in an energy starved state, since glucose oxidation is a much less efficient source of high energy phosphate production than fatty acid oxidation. This is consistent with previous studies that evaluated myocardial samples from Doberman pinschers with dilated cardiomyopathy and found that affected dogs had markedly reduced mitochondrial electron transport activity and impaired oxidative production of ATP as well as decreased expression of PDH and malate dehydrogenase (Lopes et al. 2006; McCutcheon et al. 1992). The reduction in expression of PDH and malate dehydrogenase are consistent with negative feedback associated with an increased rate of glucose oxidation due to an inability of PDK4 to negatively regulate PDH (Lei et al. 2011). Mitochondrial fuel selection can be altered by differential expression of the enzymes of the fatty acid oxidation pathway relative to the enzymes that oxidize pyruvate or acetyl Co-A (i.e., the glucose oxidation pathway).
The pathologic changes observed in the mitochondria in these dogs are also consistent with reduced high-energy phosphate production associated with decreased fatty acid oxidation, and megamitochondria have been described in some human cardiomyopathies associated with respiratory chain defects (Arbustini et al. 1998; Guenthard et al. 1995; Hoppel et al. 2009). It has been suggested that defects of electron transport and energy production may represent one common pathway to structural mitochondrial abnormalities. In a study of ultrastructural abnormalities observed in human dilated cardiomyopathy patients, approximately 14 % had a combination of findings similar to those described in the dogs studied here, including giant mitochondria, crystalloid and osmiophilic inclusion bodies and glycogen or lipid inclusions (Arbustini et al. 1998). Some, but not all, of these patients had mitochondrial mutations; suggesting that there may be other causative etiologies for these findings in addition to those observed in this PDK4 abnormality (Guenthard et al. 1995).
The mutation identified here is a splice site deletion that would appear to result in the use of a cryptic gt site. Splicing at the 5′ end of an intron is dependent on the recognition of the donor gt by a U1 snRNP; however, only about half of the information needed to accurately define exon/intron boundaries is defined by that core sequence motif (Stanley et al. 2005). Additional information on site recognition is believed to be related to both exonic and intronic splicing enhancers and silencers which are often found within the sequences of the surrounding splice site regions. In this case the flanking sequence of the cryptic gt site is quite different from the original splice site. The strengths of both the original 5′ splicing site and the cryptic site were compared with MaxEntScan, a program developed to predict true splice sites from decoys using a Maximum Entropy model (Yeo and Burge 2004). The original splice site had a higher score than that assigned to the cryptic site, consistent with the preference for the original site (Eng et al. 2004). Therefore, although it is likely that the cryptic site may be used, it would be used with lower preference and possibly decreased efficiency.
PDK4 is one of four isoenzymes of PDK. PDK4 is predominantly expressed in the heart and skeletal muscle (Matkovich et al. 2010) and it is not known whether the other PDK isoenzymes could compensate for PDK4 dysfunction in these tissues. It is noteworthy that the PDK isoenzymes do not all have similar activities or responses to the intermediaries of glucose oxidation (Bowker-Kinley et al. 1998). Although it is tempting to speculate that a dysfunctional PDK4 that is unable to negatively regulate PDH could be compensated for by one of the other PDK isoenzymes, it is possible that the differences in activity and response to metabolites would prevent effective compensation (Bowker-Kinley et al. 1998).
Although this mutation had a strong association with disease, some of the affected dogs (18 % of the population) did not have the mutation. This finding is consistent with the genetic heterogeneity observed with dilated cardiomyopathy in human beings (Hershberger et al. 2010). At this time, more than 30 genes have been associated with the development of familial dilated cardiomyopathy in human beings (Hershberger et al. 2010). Additionally, it has been estimated that the genetic etiology of familial DCM has only been identified in about 30–35 % of the human cases, therefore, other genetic causes have yet to be identified (Hershberger et al. 2010). The gene reported here, PDK4, has not yet been reported as a causative gene for dilated cardiomyopathy in human beings. It might be assumed that a pure breed domestic animal with a familial mutation would have less genetic heterogeneity than the diverse population of human beings. However, other examples of cardiomyopathy in domestic animals have refuted this assumption, including familial hypertrophic cardiomyopathy in the Maine Coon cat, where there are at least two separate mutations (Mary et al. 2010).
The penetrance of this deletion mutation in the population studied was approximately 68 %. Familial dilated cardiomyopathy has an incomplete age-dependent penetrance with variable expression in human beings (Burkett and Hershberger 2005). Particular mutations may have a wide variability in effect and severity, even within the same family. The incomplete penetrance and variability of expression in human beings and dogs suggests a possible role for either environmental factors, genetic modifiers or both in influencing the natural history of the disease.
In conclusion, we report the identification of a splice site deletion in the PDK4 gene that is associated with the development of familial dilated cardiomyopathy in the Doberman pinscher dog.
References
Arbustini E, Diegoli M, Fasani R, Grasso M, Morbini P, Banchieri N, Bellini O, DalBello B, Pilotto A, Magrini G, Campana C, Fortina P, Gavazzi A, Narula J, Vigano M (1998) Mitochondrial DNA mutations and mitochondrial abnormalities in dilated cardiomyopathy. Am J Path 153:1501–1510
Barrett JC, Fry B, Maller J, Daly MJ (2004) HalpoviewL analysis and visualization of LD and haploview maps. Bioinformatics 21:263–265
Bowker-Kinley MM, Davis WI, Wu P, Harris RA, Popov KM (1998) Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenases complex. Biochem J 329:191–196
Burkett EL, Hershberger RE (2005) Clinical and genetic issues in familial dilated cardiomyopathy. J Am Coll Cardiol 45:969–981
Butcher LM, Davis OSP, Craig IW, Plomin R (2008) Genome-wide quantitative trait locus association scan of general cognitive ability using pooled DNA and 500 K single nucleotide polymorphism microarrays. Genes Brain Behav 7:435–446
Dukes-McEwan J, Borgarelli M, Tidholm A, Vollmar AC, Haggstrom J (2003) Proposed guidelines for the diagnosis of canine idiopathic dilated cardiomyopathy. J Vet Cardiol 5:7–19
Eng L, Coutinho G, Nahas S, Yeo G, Tanouye R, Babaei M, Dork T, Burge C, Gatti RA (2004) Nonclassical splicing mutations in the coding and noncoding regions of the ATM gene: Maximum entropy estimates of splice junction strengths. Hum Mut 23:67–76
Guenthard J, Wyler F, Fowler B, Baumgartner R (1995) Cardiomyopathy in respiratory chain disorders. Arch Dis Child 72:223–226
Hershberger RE, Cowan J, Morales A, Siegfried JD (2009) Progress with genetic cardiomyopathies: screening, counseling, and testing in dilated hypertrophic and arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ Heart Fail 2:253–261
Hershberger RE, Morales A, Siegfried JD (2010) Clinical and genetic issues in dilated cardiomyopathy: a review for genetics professionals. Genet Med 12:655–667
Hoppel CL, Tandler B, Fujioka H, Riva A (2009) Dynamic organization of mitochondria in human heart and in myocardial disease. Int J Biochem Cell Biol 41:1949–1956
Karlsson EK, Baranowska I, Wade CM, Salmon Hillbertz NH, Zody MC, Anderson N, Biagi TM, Patterson N, Pielberg GR et al (2007) Efficient mapping of Mendelian traits in dogs through genome-wide association. Nat Genet 39:1321–1328
Lei B, Lionetti V, Young ME, Chandler MP, d’Agostino C, Kang E, Altarejos M, Matsuo K, Hintze TH, Stanley WC, Recchia FA (2011) Paradoxical downregulation of the glucose oxidation pathway despite enhanced flux in severe heart failure. J Mol and Cell Cardiol 36:567–576
Lopaschuk GD, Ussher JR, Folmes CDL, Jaswal JS, Stanley WC (2010) Myocardial fatty acid metabolism in health and disease. Physiol Rev 90:207–258
Lopes R, Solter PF, Sisson D, Oyama MA, Prosek R (2006) Characterization of canine mitochondrial protein expression in natural and induced forms of idiopathic dilated cardiomyopathy. Am J Vet Res 67:963–970
Mary J, Chetboul V, Sampedrano CC, Abitbol M, Gouni V, Trehiou-Sechi E, Tissier R, Queney G, Pouchelon JL, Thomas A (2010) Prevalence of the MYBPC3-A31P mutation in a large European feline population and association with hypertrophic cardiomyopathy in the Maine Coon breed. J Vet Cardiol 12:155–161
Matkovich SJ, Van Booven DJ, Hindes A, Kang MY, Druley TE, Vallania FL, Mitra RD, Reilly MP, Cappola TP, Dorn GW 2nd (2010) Cardiac signaling genes exhibit unexpected sequence diversity in sporadic cardiomyopathy revealing HSPB7 polymorphisms associated with disease. J Clin Invest 120:280–289
McCutcheon LJ, Cory CR, Nowack L (1992) Respiratory chain defect of myocardial mitochondria in idiopathic dilated cardiomyopathy of Doberman pinscher dogs. Can J Phys Pharm 70:1529–1533
Meurs KM, Kittleson M, Spangler E, Ware WA, Womack JE, Towbin JA (2000) Nine polymorphisms within head and hinge region of the feline cardiac B-myosin heavy chain gene. Anim Genet 31:231
Meurs KM, Magnon AL, Spier AW, Miller MW, Lehmkuhl LB, Towbin JA (2001) Evaluation of the cardiac actin gene in Doberman pinschers with dilated cardiomyopathy. Am J Vet Res 62:33–36
Meurs KM, Fox PR, Norgard MM, Spier AW, Lamb A, Koplitz SL, Baunwart RD (2007) A prospective genetic evaluation of familial dilated cardiomyopathy in the Doberman pinscher. J Vet Intern Med 21:1016–1020
Meurs KM, Hendrix KP, Norgard MM (2010) Molecular evaluation of five cardiac genes in Doberman pinschers with dilated cardiomyopathy. Am J Vet Res 68:1050–1063
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC (2007) PLINK: a tool set for whole genome association and population-based linkage analysis. Am J Hum Genet 15:263–265
Rozen S, Skaletsky HJ (2003) Primer 3 on the WWW for general users and for biologist programmers. In: Krawetz SA, Misener S (eds) Bioinformatics methods and protocols: methods in molecular biology. Humana Press, New Jersey, pp 365–386
Saddik M, Gamble J, Witters LA, Lopaschuk GD (1993) Acetyl-CoA carboxylase regulation of fatty acid oxidation in the heart. J Biol Chem 268:25836–25845
Smucker ML, Kaul S, Woodfield JA (1990) Naturally occurring cardiomyopathy in the Doberman pinscher: A possible large animal model of human cardiomyopathy? J Am Coll Card 16:200–206
Stabej P, Imholz S, Veersteeg SA, Zilstra C, Stokhof AA, Domanjko-Petric A, Leegwater PA, van Oost BA (2004) Characterization of the canine desmin gene and evaluation as a candidate gene for dilated cardiomyopathy in the Doberman. Gene 340:241–249
Stabej P, Leegwater PA, Imholz S, Veersteeg SA, Zilstra C, Stokhof AA, Domanjko-Petric A, van Oost BA (2005a) The canine sarcoglycan delta gene: BAC clone contig assemby, chromosome assignment and interrogation as a candidate gene for dilated cardiomyopathy in Dobermann dogs. Cytogenetics and Genome Research 111:140–146
Stabej P, Leegwater PA, Stokhof AA, Domanjko-Petric A, van Oost BA (2005b) Evaluation of the phospholamban gene in purebred large-breed dogs with dilated cardiomyopathy. Am J Vet Res 66:432–436
Stanley WC, Recchia FA, Lopaschuk GD (2005) Myocardial substrate metabolism in the normal and failing heart. Physiol Rev 85:1093–1129
Wynn RM, Kato M, Chuang JL, Tso SC, Li J, Chuang DT (2010) Pyruvate dehydrogenase kinase-4 structure reveal a metastable open conformation fostering robust core-free basal activity. J Biol Chem 12:25305–25315
Yeo G, Burge CB (2004) Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol 11:377–394
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Meurs, K.M., Lahmers, S., Keene, B.W. et al. A splice site mutation in a gene encoding for PDK4, a mitochondrial protein, is associated with the development of dilated cardiomyopathy in the Doberman pinscher. Hum Genet 131, 1319–1325 (2012). https://doi.org/10.1007/s00439-012-1158-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-012-1158-2