
Abstract
Epilepsy and migraine are both episodic disorders and share clinical as well as pathophysiological mechanisms. The prevalence of epilepsy in migraine patients is generally higher than normal as compared to general population and vice versa. Various environmental risk factors and genetic factors have been reported to be associated with susceptibility of these comorbid diseases. Specific genes have been implicated in the pathogenesis of the two diseases. However, the shared genetic susceptibility has not been explored extensively. Previous studies have reported that the alterations in the genes encoding ion channel proteins are common risk factors for both the diseases. The alterations in ion channel-encoding genes CACNAIA (T666M) and SCNIA (Q1489K and L1649Q) have been found to be involved in the development of familial hemiplegic migraine (FHM) as well as generalized epilepsy and some cases of focal epilepsy as well. The fact that both these disorders are treated with anti-epileptic drugs (AEDs) strongly supports common underlying mechanisms. This review has been compiled with an aim to explore the alterations in common genes involved in various pathways regulating neuronal hyperexcitability, a common risk factor for both these conditions. The avenue for future treatment strategies targeting common genes and molecular mechanisms has also been discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Migraine and epilepsy are both episodic neurological disorders having similar clinical features as well as pathophysiological mechanisms [1]. The prevalence of epilepsy in migraineurs substantially exceeds that of the general population [2], and the incidence of migraine headaches in patients with epilepsy is nearly twice in comparison with others without epilepsy [1]. There are evidences indicating that risk of migraine is increased significantly in patients with epilepsy as compared to general population and vice versa [3]. Patients with migraine are at a higher risk of developing epilepsy almost sixfold as compared to general population. Migraine is characterized by periodic attacks of throbbing pain on one side of brain along with symptoms including fatigue, nausea, hypersensitivity to light and sound, and temporary visual disturbances in some cases [4]. On the other hand, epilepsy is characterized by recurrent seizures due to abnormal brain activity, loss of consciousness, fear, anxiety, and severe headaches. Two main types of migraine are migraine without aura (MO) and migraine with aura (MA). Other subtypes of migraine include hemiplegic migraine and familial hemiplegic migraine (FHM). Epilepsies are classified as focal, generalized, combined generalized and focal epilepsy, and idiopathic.
Migraine aura-triggered seizure (migralepsy) is a complication and a rare condition in which an epileptic seizure is preceded by a migraine with aura attack. Conventional risk factors for migraine involve stress, tiredness, anger, smoking, and not getting enough exercise or sleep [5], whereas risk factors for epilepsy involve head trauma, brain conditions (brain tumors or stroke), infectious diseases, prenatal injury, and developmental disorders [6]. Most of these risk factors lead to the hyperexcitability of the brain.
Based on the outcome of epidemiological studies, epilepsy and migraine have been found to be comorbid conditions. The environmental and genetic factors have been reported to be significant risk factors for brain hyperexcitability. Although both epilepsy and migraine are individually influenced by genetic factors, the contribution of a shared genetic susceptibility has been evaluated to some extent. However, the mechanisms leading to co-existence of these two diseases remains unclear [1, 7,8,9]. Variations in genes encoding ion channel proteins have been found to be associated with the development of both the disorders. The fact that both these disorders are treated with anti-epileptic drugs (AEDs) strongly supports common underlying mechanisms [10]. Strong support for a shared genetic basis leading to FHM, an autosomal dominant syndrome, has been reported in severe migraine that occurs because of alterations in genes encoding ion channel proteins CACNAIA (P/Q-type voltage-gated calcium channel, T666M), SCN1A (voltage-gated sodium channel, Q1489K and L1649Q), and ATP1A2 (Na+-K+ ATPase, M721T and R689Q). T666M mutation in CACNA1A gene disrupts the Ca2+-binding sites in the pore [11], whereas Q1489K and L1649Q mutations in SCNA1A gene lead to the folding defects and thereby interfere with hydrophobic latch which block the ion pore [12]. The R689Q mutation in ATP1A2 gene affects the large intracellular loop between transmembrane domains M4 and M5, which harbors the ATP-binding and hydrolase domains and is implicated in several functions [13]. Alterations in all the three genes are also implicated in generalized and in some cases of focal epilepsy [14]. In epilepsy, neuronal hyperexcitability causes unusual hypersynchronous electrical discharge in neurons and further modifications in ion exchange process across membrane leading to recurrent seizures, while in migraine, hyperexcitability increases the extracellular concentration of glutamate (excitatory neurotransmitter) and thereby leads to cortical spreading depression (CSD), which further blocks the activity of neurons. There is tremendous efflux of K+ ions to extracellular compartments causing sustained depolarization followed by neuronal suppression. CSD activates trigeminal system releasing inflammatory molecules from the neurons and also suppresses the inhibitory function of GABA. Occipital lobe of brain is considered to be responsible for both the conditions because it is more prone to CSD. The threshold for onset of CSD in migraine is found to be lower than CSD in seizure. In addition the mutations in ion channels such as Na+-K+ ATPase pump have also been reported to be associated with both the disorders together [15]. Normally Na+-K+ pump regulates the extracellular concentration of accumulated K+ ions by removing increasing glial Na+-K+ ATPase activity. The mutation in the sequence of α2 subunit of ATP1A2 gene has been found to be associated with FHM and benign familial convulsions in infants [16]. Mantegazza and Cestele (2018) reviewed extensively the pathophysiological similarities and differences between migraine and epilepsy [17]. They have concluded that these comorbidities might be generated on account of dysfunction in the common neuronal networks. The pathogenic mutations might target the same proteins but the dysfunction might be disease specific. There is a need to decipher the common pathological mechanisms leading to migraine and epilepsy. Therefore, the current review has been compiled with an aim to explain the common molecular mechanisms and pathways implicated in the pathogenesis of epilepsy as well as migraine. The genes involved in specific pathways altering molecular mechanistic aspects have also been discussed in detail.
Common Channel and CSD Mechanism
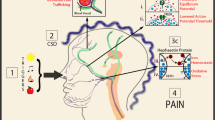
Migraine and epilepsy association is not a cause-and-effect relationship. Most likely they both are result of cortical neuron over-excitation. Epilepsy results from the synchronized discharge of neurons whereas migraine is associated with CSD, i.e., the strong depolarization of a large group of nerve cells or neuroglia that spreads to adjacent areas and inhibits neural activity [8]. Epilepsy and migraine can spontaneously induce CSD [18]. The results of the studies using animal models have suggested that many changes that occur during CSD (e.g., the increased release of glutamate, the increased concentrations of extracellular potassium ions, and the inhibition of the Na+/K+ ATPase) are also associated with the sense of prior prediction by some migraine patients [19,20,21]. Collectively these results indicate that CSD may be the basis for migraine with aura (Fig. 1). Further neocortical hyperexcitability is supposed to get transformed to abnormal hypersynchronous electrical discharges in epilepsy. Subsequent alterations in permeability of ion or ion exchange activity lead to recurrent seizures. However, in case of migraine, the neocortical hyperexcitability transits to CSD rather than the hyper synchronous activity in case of epilepsy. Action potential happening in neurons regulates all the activities of brain including epilepsy, which is a typical ion channel disease. Balance of synaptic excitation and inhibition controls the action potential in a neuron or group of neurons. This balance can be broken by GABA a principal inhibitor neurotransmitter and glutamate anomalies which may be the core of an excitatory neurotransmitter. Many mutations occurring in GABA receptor encoding gene or in calcium channel encoding genes result in idiopathic epilepsy that is generally inherited. These GABA and other channels regulate abnormal synchronization between cortex and thalamus and thereby lead to generalized spike-wave discharges. In addition, one of the significant common pathways of action potentials requires opening and closing of the voltage-gated sodium channels. Therefore, some idiopathic seizures result because of the alterations in sodium channel. The frequently used AEDs like phenytoin overpower the quick opening and closing of sodium channels. Another drug, topiramate, an antagonist at the AMPA-type glutamate receptor, weakens the carbonic anhydrase inhibitor, enhances the activity of GABA on chloride channel, and controls the opening of L-type calcium channel. These anti-epileptic drugs are also found to be effective for migraine indicating that migraine might also result on account of changes in the ion channels [22].

Common molecular mechanisms shared by epilepsy and migraine: Both epilepsy and migraine are influenced by genetic factors, and these genetic factors are significant risk factors for brain hyperexcitability via the alterations in genes (shown in green) coding ion channel protein. Alteration in genes leads to unusual hypersynchronous electric discharge in neurons. Neuronal hyperexcitability causes modifications in ion exchange process across membrane leading to recurrent seizures in epilepsy whereas in migraine extracellular concentration of glutamate increases resulting a tremendous efflux of K+ ions, which further blocks the neuronal activity and thereby leads to brain hyperexcitability. Both these diseases can spontaneously induce CSD, making these as comorbid condition. CSD also activates trigeminal system and suppresses the inhibitory function of GABA. Balance of synaptic excitation and inhibition controls the action potential in a neuron or group of neurons. This balance can be disturbed by inhibition of GABA and glutamate anomalies (which may be the core of an excitatory neurotransmitter) or by mutations that occur in GABA receptor encoding gene. So, changes that occur during CSD lead to disturbance in balance
Shared Genetic Factors Contributing to Migraine and Epilepsy
Both migraine and epilepsy are highly heritable especially idiopathic epilepsy and migraine with aura. The risk of patients with idiopathic epilepsy getting migraine with aura is approximately double [1]. Although there are hundreds of mutations causing monogenic type of both the diseases, but few shared genes involved in ion channel functions are responsible for co-occurrence of these (Tables 1 and 2). Mutations in the genes like CACNA1A (FHM1), ATP1A2 (FHM2), and SCN1A (FHM3) that causes familial hemiplegic migraine are also associated with epileptic seizures (Table 3). These three genes are involved in functioning of ion channels [42, 43]. The alterations in these cause dysfunction and hyperexcitability of neurons. CACNA1A gene codes for α-subunit of voltage-gated P/Q channels. Severe myoclonic epilepsy in infants is caused due to mutations in SCN1A gene [44]. SCN1A codes for the α-subunit of voltage-gated sodium channel Nav1.1. Another gene proline-rich transmembrane protein 2 (PRRT2) is responsible for modulating the release of neurotransmitter at synapses [45]. Alterations in this gene affect the neurotransmitter release and dysregulation of neuronal excitability involved in migraine, benign familial infantile seizures, and paroxysmal kinesigenic dyskinesia [46].
Genetically determined dysfunction of these ion channels and the associated proteins cause changes in neuronal ion concentration, leading to cortical excitability. Imbalance between inhibitory and excitatory factors has been hypothesized to play a key role in epilepsy as well as migraine (Fig. 1) [18]. One of the clear genetic contributions to epilepsy and migraine is SCN1A gene, located on chromosome 2 that encodes for the α-1 subunit of the voltage-gated sodium channels. Sodium channels are mostly located in the cerebral cortex and spinal cord that is closely related to the regulation of action potential. SCN1A gene mutations have been reported to result in seizures and occurrence of FHM3. Mutations in this gene have been commonly observed in epileptic patients [37, 47]. The Dravet syndrome (DS) and infant idiopathic comprehensive seizures and generalized seizures with febrile seizures plus (GEFSþ) and partial seizures with febrile seizures plus (PEFSþ) have been reported to be associated with mutations in this gene [48]. With about 650 heterozygous SCN1A mutations, an average mutation rate of about 85% in DS patients has been observed [49, 50]. About half of these mutations were missense, and half were nonsense. These either increase or decrease the sodium channel function. SCN1A mutations have also been reported to be associated with FHM3 [51]. Some mutations including Q1489K, L1649Q, I1498M, F1661L, and L1624P caused FHM3 but not seizures [12, 37, 52, 53]. However, mutations like L263Q, T1174S, Q1489H, and L263V were found to be associated with both FHM and epilepsy [54, 55]. Out of these Q1489H and F1499L were also associated with elicited repetitive daily blindness (ERDB) [56, 57]. Different types of SCN1A mutations affect the functioning of channels in various ways (Fig. 2). The Q1489K and L1649Q mutations lead to pure FHM3 inhibiting neuronal function, especially the GABA intermediate neurons [58, 59]. On contrary, some studies involving members of Portuguese family bearing L263V mutation in FHM had complex partial seizures or generalized seizures [50, 58, 59]. The L263V alteration leads to the enhancement of channel function responsible for recovery of sodium channel inactivation, thereby prolonging the duration of action and increasing the neuronal excitability. Therefore, this gene variant may lead to epilepsy and FHM in the same individual [60]. CACNA1A gene present on chromosome number 19 encodes the α-1 subunit of the voltage-dependent P/Q calcium channel. The P/Q calcium channel regulates the release of glutamate and serotonin by increasing the flow of calcium to stimulate the presynaptic membrane. CACNA1A gene alterations may impair calcium channel function, causing generalized epilepsy [61]. CACNA1A gene mutations may occur either in epileptics or FHM. However, at the same time CACNA1A mutations may also lead to FHM1 by affecting CSD, a study on mutant mice (R192Q) showed imbalance in excitation and inhibition of calcium channel in cortical neurons, thereby reducing the threshold for CSD and accelerating its propagation [23]. Another study showed that S218L mutant mice are highly sensitive to CSD [24]. ATP1A2 present on chromosome 1 encodes α-2 subunit of Na+/K+ ATPase. Alpha2 subunit has been reported to be expressed in neurons and astrocytes. Na+/K+ ATPase controls the K+ extracellular concentration in astrocytes, and increased K+ concentration is associated with CSD. This enhances the excitability of neurons and results in a threshold triggering CSD. Abnormal function of Na+/K+ ATPase system on account of ATP1A2 gene mutations results in destruction of K+ gradient thereby influencing glutamate clearance, which may cause seizures, CSD, and FHM. Mutations in ATP1A2 have also been found to cause epilepsy [25].

Schematic representation of a tripartite synapse and the proteins encoded by the different genes involved in migraine and epilepsy: (a) CACN1A gene, encodes for the pore-forming α1A-subunit (CaV2.1) of P/Q-type calcium channels. Alterations in CACN1A gene lead to the alteration in important functional region of CaV2.1 channel especially as pore lining. Changes in biophysical properties of channel lead to greater Ca2+ influx, mainly due to the hyperpolarizing shifts. Gain of function of excitatory neurotransmission leads to increase synaptic strength mainly due to increased action potential-evoked Ca2+ influx via altered P/Q calcium channels. (b) ATP1A2 gene, encodes α2 isoform of the main catalytic subunit of Na+-K+ transporter (here, Na+-K+-ATPase). This α2 isoform is mainly expressed in astrocytes. The role of Na+-K+-ATPase is to maintain the resting potential. Na+-K+ transporter. Here Na+ gradient maintained by Na+-K+ transporter is required by astrocytes for the clearance of extracellular glutamate. Glutamate uptake is mediated by the glutamate receptors, i.e., GLAST AND GLT-1 encoded by EAAT1 and EAAT2 genes, respectively. Glutamate uptake is driven by the efflux of three Na+ ions. Altered Na+-K+ transporter reduces the ability of astrocyte to remove K+ ions that accumulates extracellularly during the action potential, thereby promoting therefore promote neuronal hyperexcitability and indirectly CSD. (c) SCN1A gene, encodes for pore-forming α1-subunit of neuronal type I voltage-gated sodium channel Nav1.1. Alterations in this gene cause biophysical changes in Na+ transport like abnormal regulation of the gate. Loss of function mutations in NaV1.1 cause impaired functioning of GABAergic inhibitory neurons, resulting in disturbed neuronal synchronization. Gain of function mutations in NaV1.1 lead to excessive excitability of glutamatergic neurons. As the action potential arrives at the nerve terminal, GABA present in the vesicles gets releases, under the action of Ca2+ influx. Gain of function or loss of function mutations in SCN1A gene cause neuronal hyperexcitability by disturbing the synthesis of GABA from glutamate and further by hampering its binding to GABA receptor.
Treatment Strategies and Future Directions
Migraine and epilepsy have a major socioeconomic impact on patients and their families. Most of the anti-epileptic drugs (AEDs) are prescribed to migraine patients, because of the same pathophysiological mechanisms shared by epilepsy and migraine. It is very crucial to understand the rationale for AEDs in prevention and treatment of migraine, in order to determine the clinical care precisely. As we know, epilepsy and migraine show a twofold average risk for comorbidity [22]. This endorses the use of AEDs in the treatment of both the disorders. However, the mechanisms of these drugs are not clearly understood as far as their use in treatment of migraine is concerned. It is believed that AEDs block excitation of the neurons (by controlling ion channels) and thereby CSD that might be a key player in the development of migraine. Various clinical trial studies have demonstrated that the use of AEDs in treatment of migraine is well known and effective [26, 27]. Although there are only few AEDs like valproate and topiramate which are currently being used as first-line agents in treatment of migraine, many other drugs like zonisamide, acetazolamide, lamotrigine, and oxcarbazepine are under considerations, although not as first-line treatment [22, 28]. The exact mechanism of valproate is not clearly understood in migraine, but it actually increases the GABA levels in brain by activating glutamic acid decarboxylase enzyme and via inhibition of GABA-degradative enzymes. On the other hand, it also inhibits the voltage-gated ion channels (T-type) by targeting SCN1A and CACNA1A genes and thereby reduces the effect of inflammation of neurons in the brainstem (central trigeminal nerve) [29]. Topiramate modifies the nerve excitability by blocking voltage-gated sodium channels (via targeting SCNA1A gene) and L-type voltage-activated calcium channels. It also enhances the inhibitory effect of GABA by blocking carbonic anhydrase activity [30]. Another drug lamotrigine acts as antagonist to high-voltage-activated n-, P-, and Q-type channels encoded by SCN1A and CACNA1A genes [31]. In spite of the good efficacy of these drugs, some patients do not get required effect of these drugs on account of ADRs. So, the adverse effects, compliance, and cost of prolonged treatment are important issues because they are discovered coincidently without taking into account the pathophysiology of the migraine. Both epilepsy and migraine are multifactorial disorder with a genetic component and environmental factors interacting with genome. However, the genetic contribution to epilepsy is well understood, and the pharmacogenetic studies have been identified the alterations in genes responsible for variations in inter-individuals response to AEDs as well as ADRs. More studies are needed to delineate the roles of newer and existing AEDs in migraine prevention. Targeting the shared genes by both the diseases might be helpful in generating new and effective treatments for these comorbid diseases. Three significant genes CACNA1A, SCN1A, and ATP1A2 have been found to be common in pathogenesis of both the disorders. However, there are certain concerns that have been highlighted by Cestele et al. (2013) regarding the T1174S variant of SCN1A gene which was observed to induce two contrasting effects: a loss of function that was caused by modification of activation properties and a gain of function that was reported to be caused by increased persistent current. The later was not intrinsic property of the mutation but, on the contrary, was generated by a modulation. Mutation analysis was carried out by sequencing of the gene whereas functional analysis was carried out by expressing the mutant allele in tsA-201 cells. The switch between gain of function and loss of function was understood using a computational model [32]. Therefore, the modulations might switch the effect of a particular variant from epileptogenic to promigraine. There seem to be complex genotypic-phenotypic relationship of SCN1A mutations. This aspect needs to be expressed for other significant genes contributing to both the disorders. Therefore, if new drug molecules are designed to regulate the expression of these genes, the modulations that might be responsible for phenotypic switch have to be taken into consideration. If no such modulations are observed in other genes, then the novel drug molecules can be designed accordingly. In addition current high-throughput genetic technologies like CRISPR-Cas9 can also be explored in this direction [33, 34]. For example, a study carried out to generate a disease model to explore the pathophysiological mechanisms in epilepsy is caused by SCN1A loss-of-function mutation using CRISPR/Cas9 genome-editing technology [35]. Further, the available data based on transcriptomic profiling addresses miRNAs as treatment strategies, and their status in clinical trials is highly promising. Various transcriptomic-based studies have been carried out to delineate the disease pathophysiology and new possibilities for the identification of molecular targets for future pharmacological strategies in the treatment of these comorbidities [36, 38,39,40,41, 62]. Further proteome and metabolome analysis can also be carried out to understand the common pathophysiological mechanisms shared by epilepsy and migraine.
Availability of Data and Material
Not applicable.
References
Lipton RB, Ottman R, Ehrenberg BL, Hauser WA (1994) Comorbidity of migraine: the connection between migraine and epilepsy. Neurology 44(10 Suppl 7):S28–S32
Téllez-Zenteno JF, Matijevic S, Wiebe S (2005) Somatic comorbidity of epilepsy in the general population in Canada. Epilepsia 46(12):1955–1962
Davies PT, Panayiotopoulos C (2011) Migraine triggered seizures and epilepsy triggered headache and migraine attacks: a need for re-assessment. J Headache Pain 12(3):287–288
Gross EC, Lisicki M, Fischer D, Sándor PS, Schoenen J (2019) The metabolic face of migraine—from pathophysiology to treatment. Nat Rev Neurol 15(11):627–643
May A, Schulte LH (2016) Chronic migraine: risk factors, mechanisms and treatment. Nat Rev Neurol 12(8):455–464
Hauser WA (1999) Risk factors for epilepsy. In: The Epilepsies. Elsevier, pp 1-11
Breslau N, Davis GC (1993) Migraine, physical health and psychiatric disorder: a prospective epidemiologic study in young adults. J Psychiatr Res 27(2):211–221
Zarcone D, Corbetta S (2017) Shared mechanisms of epilepsy, migraine and affective disorders. Neurol Sci 38(1):73–76
Jasim SA, Al-Obaidy AM, Rabeea AK (2019) Relationship of migraine in epileptic patients. Al-Kufa Univ J Biol 11(2):14–19
Rogawski MA (2008) Antiepileptic drugs and migraine. Innov Drug Dev Headache Disorders 16:153–178
Tottene A, Fellin T, Pagnutti S, Luvisetto S, Striessnig J, Fletcher C, Pietrobon D (2002) Familial hemiplegic migraine mutations increase Ca(2+) influx through single human CaV2.1 channels and decrease maximal CaV2.1 current density in neurons. Proc Natl Acad Sci U S A 99(20):13284–13289. https://doi.org/10.1073/pnas.192242399
Dichgans M, Freilinger T, Eckstein G, Babini E, Lorenz-Depiereux B, Biskup S, Ferrari MD, Herzog J et al (2005) Mutation in the neuronal voltage-gated sodium channel SCN1A in familial hemiplegic migraine. Lancet 366(9483):371–377
Vanmolkot KR, Kors EE, Hottenga JJ, Terwindt GM, Haan J, Hoefnagels WA, Black DF, Sandkuijl LA et al (2003) Novel mutations in the Na+, K+-ATPase pump gene ATP1A2 associated with familial hemiplegic migraine and benign familial infantile convulsions. Ann Neurol 54(3):360–366. https://doi.org/10.1002/ana.10674
Perucca P, Perucca E (2019) Identifying mutations in epilepsy genes: impact on treatment selection. Epilepsy Res 152:18–30
Oyrer J, Maljevic S, Scheffer IE, Berkovic SF, Petrou S, Reid CA (2018) Ion channels in genetic epilepsy: from genes and mechanisms to disease-targeted therapies. Pharmacol Rev 70(1):142–173
Prontera P, Sarchielli P, Caproni S, Bedetti C, Cupini L, Calabresi P, Costa C (2018) Epilepsy in hemiplegic migraine: genetic mutations and clinical implications. Cephalalgia 38(2):361–373
Mantegazza M, Cestele S (2018) Pathophysiological mechanisms of migraine and epilepsy: similarities and differences. Neurosci Lett 667:92–102. https://doi.org/10.1016/j.neulet.2017.11.025
Kim DW, Lee SK (2017) Headache and epilepsy. J Epilepsy Res 7(1):7–15
Charles A, Brennan K (2009) Cortical spreading depression—new insights and persistent questions. Cephalalgia 29(10):1115–1124
Borgdorff P (2018) Arguments against the role of cortical spreading depression in migraine. Neurol Res 40(3):173–181
Unekawa M, Ikeda K, Tomita Y, Kawakami K, Suzuki N (2018) Enhanced susceptibility to cortical spreading depression in two types of Na+, K+-ATPase α2 subunit-deficient mice as a model of familial hemiplegic migraine 2. Cephalalgia 38(9):1515–1524
Parikh SK, Silberstein SD (2019) Current status of antiepileptic drugs as preventive migraine therapy. Curr Treat Options Neurol 21(4):16
Inchauspe CG, Pilati N, Di Guilmi MN, Urbano FJ, Ferrari MD, van den Maagdenberg AM, Forsythe ID, Uchitel OD (2015) Familial hemiplegic migraine type-1 mutated cav2. 1 calcium channels alter inhibitory and excitatory synaptic transmission in the lateral superior olive of mice. Hear Res 319:56–68
Zweckberger K, Erös C, Zimmermann R, Kim S-W, Engel D, Plesnila N (2006) Effect of early and delayed decompressive craniectomy on secondary brain damage after controlled cortical impact in mice. J Neurotrauma 23(7):1083–1093
Deprez L, Weckhuysen S, Peeters K, Deconinck T, Claeys KG, Claes LR, Suls A, Van Dyck T et al (2008) Epilepsy as part of the phenotype associated with ATP1A2 mutations. Epilepsia 49(3):500–508
Bagnato F, Good J (2016) The use of antiepileptics in migraine prophylaxis. Headache: The Journal of Head and Face Pain 56(3):603–615
Kacperski J, Green A, Qaiser S (2020) Management of chronic migraine in children and adolescents: a brief discussion on preventive therapies. Pediatr Drugs 22:635–643 1-9
Shahien R, Beiruti K (2012) Preventive agents for migraine: focus on the antiepileptic drugs. Journal of Central Nervous System Disease 4:JCNSD. S9049
Ghodke-Puranik Y, Thorn CF, Lamba JK, Leeder JS, Song W, Birnbaum AK, Altman RB, Klein TE (2013) Valproic acid pathway: pharmacokinetics and pharmacodynamics. Pharmacogenet Genomics 23(4):236–241. https://doi.org/10.1097/FPC.0b013e32835ea0b2
Naegel S, Obermann M (2010) Topiramate in the prevention and treatment of migraine: efficacy, safety and patient preference. Neuropsychiatr Dis Treat 6:17–28. https://doi.org/10.2147/ndt.s6459
Mitra-Ghosh T, Callisto SP, Lamba JK, Remmel RP, Birnbaum AK, Barbarino JM, Klein TE, Altman RB (2020) PharmGKB summary: lamotrigine pathway, pharmacokinetics and pharmacodynamics. Pharmacogenet Genomics 30(4):81–90. https://doi.org/10.1097/FPC.0000000000000397
Cestele S, Labate A, Rusconi R, Tarantino P, Mumoli L, Franceschetti S, Annesi G, Mantegazza M et al (2013) Divergent effects of the T1174S SCN1A mutation associated with seizures and hemiplegic migraine. Epilepsia 54(5):927–935. https://doi.org/10.1111/epi.12123
Sun L, Lutz BM, Tao Y-X (2016) The CRISPR/Cas9 system for gene editing and its potential application in pain research. Transl Perioper Pain Med 1(3):22–33
Fujihara K, Yamada K, Ichitani Y, Kakizaki T, Jiang W, Miyata S, Suto T, Kato D et al (2020) CRISPR/Cas9-engineered Gad1 elimination in rats leads to complex behavioral changes: implications for schizophrenia. Transl Psychiatry 10(1):1–13
Liu J, Gao C, Chen W, Ma W, Li X, Shi Y, Zhang H, Zhang L et al (2016) CRISPR/Cas9 facilitates investigation of neural circuit disease using human iPSCs: mechanism of epilepsy caused by an SCN1A loss-of-function mutation. Transl Psychiatry 6(1):e703–e703
Tafuri E, Santovito D, de Nardis V, Marcantonio P, Paganelli C, Affaitati G, Bucci M, Mezzetti A et al (2015) MicroRNA profiling in migraine without aura: pilot study. Ann Med 47(6):468–473
Fan C, Wolking S, Lehmann-Horn F, Hedrich UB, Freilinger T, Lerche H, Borck G, Kubisch C et al (2016) Early-onset familial hemiplegic migraine due to a novel SCN1A mutation. Cephalalgia 36(13):1238–1247
Tana C, Giamberardino MA, Cipollone F (2017) microRNA profiling in atherosclerosis, diabetes, and migraine. Ann Med 49(2):93–105
Cheng C-Y, Chen S-P, Liao Y-C, Fuh J-L, Wang Y-F, Wang S-J (2018) Elevated circulating endothelial-specific microRNAs in migraine patients: a pilot study. Cephalalgia 38(9):1585–1591
Greco R, De Icco R, Demartini C, Zanaboni AM, Tumelero E, Sances G, Allena M, Tassorelli C (2020) Plasma levels of CGRP and expression of specific microRNAs in blood cells of episodic and chronic migraine subjects: towards the identification of a panel of peripheral biomarkers of migraine? J Headache Pain 21(1):1–12
Martins-Ferreira R, Chaves J, Carvalho C, Bettencourt A, Chorão R, Freitas J, Samões R, Boleixa D et al (2020) Circulating microRNAs as potential biomarkers for genetic generalized epilepsies: a three microRNA panel. Eur J Neurol 27(4):660–666
Carreño O, Corominas R, Serra SA, Sintas C, Fernández-Castillo N, Vila-Pueyo M, Toma C, Gené GG et al (2013) Screening of CACNA1A and ATP1A2 genes in hemiplegic migraine: clinical, genetic, and functional studies. Mol Gen Genomic Med 1(4):206–222
Hiekkala ME, Vuola P, Artto V, Häppölä P, Häppölä E, Vepsäläinen S, Cuenca-Leon E, Lal D et al (2018) The contribution of CACNA1A, ATP1A2 and SCN1A mutations in hemiplegic migraine: a clinical and genetic study in Finnish migraine families. Cephalalgia 38(12):1849–1863
Fujiwara T (2006) Clinical spectrum of mutations in SCN1A gene: severe myoclonic epilepsy in infancy and related epilepsies. Epilepsy Res 70:223–230
Coleman J, Jouannot O, Ramakrishnan SK, Zanetti MN, Wang J, Salpietro V, Houlden H, Rothman JE et al (2018) PRRT2 regulates synaptic fusion by directly modulating SNARE complex assembly. Cell Rep 22(3):820–831
Marini C, Conti V, Mei D, Battaglia D, Lettori D, Losito E, Bruccini G, Tortorella G et al (2012) PRRT2 mutations in familial infantile seizures, paroxysmal dyskinesia, and hemiplegic migraine. Neurology 79(21):2109–2114
Gargus JJ, Tournay A (2007) Novel mutation confirms seizure locus SCN1A is also familial hemiplegic migraine locus FHM3. Pediatr Neurol 37(6):407–410
Dimova PS, Yordanova I, Bojinova V, Jordanova A, Kremenski I (2010) Generalized epilepsy with febrile seizures plus: novel SCN1A mutation. Pediatr Neurol 42(2):137–140
Escayg A, MacDonald BT, Meisler MH, Baulac S, Huberfeld G, An-Gourfinkel I, Brice A, LeGuern E et al (2000) Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+ 2. Nat Genet 24(4):343–345
Escayg A, Goldin AL (2010) Sodium channel SCN1A and epilepsy: mutations and mechanisms. Epilepsia 51(9):1650–1658
Cestèle S, Schiavon E, Rusconi R, Franceschetti S, Mantegazza M (2013) Nonfunctional NaV1. 1 familial hemiplegic migraine mutant transformed into gain of function by partial rescue of folding defects. Proc Natl Acad Sci 110(43):17546–17551
Vanmolkot KR, Babini E, de Vries B, Stam AH, Freilinger T, Terwindt GM, Norris L, Haan J et al (2007) The novel p. L1649Q mutation in the SCN1A epilepsy gene is associated with familial hemiplegic migraine: genetic and functional studies. Hum Mutat 28(5):522–522
Weller CM, Pelzer N, de Vries B, López MA, De Fàbregues O, Pascual J, Arroyo MAR, Koelewijn SC et al (2014) Two novel SCN1A mutations identified in families with familial hemiplegic migraine. Cephalalgia 34(13):1062–1069
Liao J, Tian X, Wang H, Xiao Z (2018) Epilepsy and migraine—are they comorbidity? Genes Diseases 5(2):112–118
Lestari ND, Mutiawati E, Sadewa AH, Sjahrir H, Syahrul DRE, Harapan H (2018) SCN1A exon 26 variants in epilepsy and migraine patients. J Med Sci 50(4):424–430
Vahedi K, Depienne C, Le Fort D, Riant F, Chaine P, Trouillard O, Gaudric A, Morris M et al (2009) Elicited repetitive daily blindness: a new phenotype associated with hemiplegic migraine and SCN1A mutations. Neurology 72(13):1178–1183
Castro M, Stam A, Lemos C, De Vries B, Vanmolkot K, Barros J, Terwindt G, Frants R et al (2009) First mutation in the voltage-gated Nav1. 1 subunit gene SCN1A with co-occurring familial hemiplegic migraine and epilepsy. Cephalalgia 29(3):308–313
Kahlig KM, Lepist I, Leung K, Rajamani S, George AL (2010) Ranolazine selectively blocks persistent current evoked by epilepsy-associated NaV1. 1 mutations. Br J Pharmacol 161(6):1414–1426
Hedrich UB, Liautard C, Kirschenbaum D, Pofahl M, Lavigne J, Liu Y, Theiss S, Slotta J et al (2014) Impaired action potential initiation in GABAergic interneurons causes hyperexcitable networks in an epileptic mouse model carrying a human NaV1. 1 mutation. J Neurosci 34(45):14874–14889
Barros J, Ferreira A, Brandão AF, Lemos C, Correia F, Damásio J, Tuna A, Sequeiros J et al (2014) Familial hemiplegic migraine due to L263V SCN1A mutation: discordance for epilepsy between two kindreds from Douro Valley. Cephalalgia 34(12):1015–1020
Rajakulendran S, Graves TD, Labrum RW, Kotzadimitriou D, Eunson L, Davis MB, Davies R, Wood NW et al (2010) Genetic and functional characterisation of the P/Q calcium channel in episodic ataxia with epilepsy. J Physiol 588(11):1905–1913
Brennan GP, Henshall DC (2020) MicroRNAs as regulators of brain function and targets for treatment of epilepsy. Nat Rev Neurol 16(9):506–519
Acknowledgements
Financial assistance from Council for Scientific and Industrial Research (CSIR) India is highly acknowledged.
Funding
Financial support to Mr. Abhilash Ludhiadch (Award No-09/1051(0029)/2 019-EMR-1) from the Council for Scientific and Industrial Research (CSIR) India is highly acknowledged.
Author information
Authors and Affiliations
Contributions
Prof Anjana Munshi and Prof Gagandeep Singh contributed to the conception and design of the study. Ms. Palvi Gotra and Ms. Nidhi Bhardwaj performed the literature survey and helped in writing the manuscript along with Mr. Abhilash Ludhiadch. Both Ms. Palvi Gotra and Ms. Nidhi Bhardwaj also helped in drawing the figures. Mr. Abhilash Ludhiadch compiled the study and edited the manuscript
Corresponding author
Ethics declarations
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Gotra, P., Bhardwaj, N., Ludhiadch, A. et al. Epilepsy and Migraine Shared Genetic and Molecular Mechanisms: Focus on Therapeutic Strategies. Mol Neurobiol 58, 3874–3883 (2021). https://doi.org/10.1007/s12035-021-02386-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-021-02386-x