
Abstract
Spinal cord sarcoidosis (SCS) is rare, and its diagnosis is challenging. We examined clinical, laboratory, and imaging features in patients with SCS to obtain useful clues for diagnosis and prognosis. Eleven consecutive patients (four males, seven females) at a single Japanese institution were investigated. Median age at onset was 66 years old. The most frequent site affected, other than the nervous system, was the respiratory system. While histological confirmation of non-caseating granulomas was often found there, no patient had respiratory symptoms. Peripheral nerve involvement was detected in 64% of patients. Soluble IL-2 receptor (sIL-2R) levels in serum and cerebrospinal fluid (CSF) were elevated in 64% and 45% of patients, respectively, and this finding was more common than elevation of angiotensin-converting enzyme (ACE). 18F-fluorodeoxyglucose (FDG) positron emission tomography showed abnormally high uptake in spinal lesions of all examined patients. Although corticosteroids were administrated to all patients, and immuno-suppressants were prescribed to six (55%), the modified Rankin Scale was unchanged or worsened in four (36%) patients during the follow-up period. Neurological exacerbation of myelopathy was seen in four (36%) patients. Complete response rate was only seen in 9%. High levels of cell count, protein, ACE, and sIL-2R in CSF were significantly more frequent in patients with a marked improvement after immunotherapy than in the other patients. These results suggest that high serum and CSF sIL-2R, high uptake of FDG, and peripheral nerve involvement are indicative of SCS. Given that SCS is commonly intractable, CSF abnormalities may predict efficacy of immunotherapies.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Sarcoidosis, a granulomatous multi-organ disease of unknown etiology, commonly affects the respiratory system, eyes, and skin, and less commonly the nervous system [1, 2]. Sarcoidosis of the nervous system makes up ~ 3–15% of all sarcoidosis [2,3,4,5,6,7,8]. Frequently affected lesions in the nervous system are brain parenchyma, meninges, and peripheral nerves [3, 4], and the involvement of spinal cord is extremely rare: spinal cord sarcoidosis (SCS) is reported to account for either 0.29% [4] or 0.43% [9] of all cases of sarcoidosis. Definite diagnosis of sarcoidosis requires histological confirmation of a non-caseating epithelioid granuloma, but it is usually difficult to obtain such direct evidence in the spinal cord because an invasive biopsy procedure is required. Therefore, to diagnose SCS, indirect evidences must be evaluated, including neurological findings and pathologic confirmation of sarcoid granulomas outside of the spinal cord.
It is well known that epidemiological features of sarcoidosis differ considerably across geographical and ethnic groups. The incidence of sarcoidosis in Asian patients is lower than in African American, or Caucasian patients [5, 10]. Moreover, patients with sarcoidosis in Asian countries are older at the time of disease onset and are more likely to be female than those in Western countries. Clinical characteristics of sarcoidosis also differ between patients from Asian and Western countries: Asian patients show more frequent ocular involvement and hypercalcemia, and less frequent nervous system involvement [10,11,12]. For SCS specifically, there are several case series from Western countries that have revealed the demographic and clinical features of SCS in these countries [13,14,15,16,17,18,19], but only few papers have been published regarding SCS in Asian patients [20,21,22,23,24].
We conducted a single-center, retrospective study of 11 consecutive Japanese patients presenting with SCS. The aims of this study were to describe the clinical, laboratory, and imaging features; to identify which features, other than biopsy, can be used to predict SCS diagnosis and outcomes; and to find prognostic factors.
Methods
This was a retrospective, single-center observational study including 11 consecutive Japanese patients (seven women, four men) with SCS, all of whom had been admitted to the Department of Neurology, Yamaguchi University Hospital (Ube, Japan) between January 2011 and June 2018. All the patients met our diagnostic criteria for SCS (Supplemental Information 1), which had been developed for the current study by modifying the criteria for sarcoidosis proposed by the Japan Society of Sarcoidosis and other Granulomatous Disorders [25, 26] (Supplemental Information 2). Information concerning clinical course and outcomes after discharge from our institute was collected by asking the patient’s current physician. Degree of neurological disability was evaluated using the modified Rankin Scale (mRS) [27]. All data were collected by one of the authors (M.F.). Written informed consent was obtained from all patients prior to performing any invasive analysis, including trans-bronchial lung biopsy. Differences between group means were examined by the Mann–Whitney U test. Correlation was analyzed by the Pearson product-moment correlation coefficient. Differences were considered significant if P < 0.05 based on a two-sided test.
Results
Characteristics of patients with SCS
Clinical demographics of the 11 patients are shown in Table 1. Eight were classified as the histological diagnosis group (73%, Patient 1–8) and three were classified as the clinical diagnosis group (27%, Patient 9–11). Age at onset of any sarcoidosis ranged from 28 to 77 years old (median, 66) and age at diagnosis of SCS ranged from 41 to 77 years old (median 68). Median duration from the onset to the diagnosis of SCS was five months (range 2–540). The initial manifestation of the sarcoidosis was a neurological deficit in nine (82%) patients, and an extra-neurological manifestation in only two (18%)—and these two patients had a history of uveitis before the onset of myelopathy.
On the patient’s first admission to our institute, the most commonly affected site, other than the nervous system, was the respiratory system (in 10 patients: 91%). Nine patients showed abnormalities including lung and mediastinal lymphadenopathy, and one exhibited nodule lesions in the lung. However, these abnormalities were all radiological and none of the patients reported respiratory symptoms. Ocular (n = 2), skin (n = 1), and cardiac (n = 2) involvement was also evident. Histological confirmation of sarcoidosis was obtained in the eight patients of the histological diagnosis group by lymph node biopsy (n = 4), trans-bronchial lung biopsy (TBLB; n = 2), skin biopsy (n = 1), or endoscopic ileocecal biopsy (n = 1).
Neurological evaluation on admission revealed sensory disturbance in ten patients, motor weakness in eight, sphincter disturbances in four, sensory ataxia in three, and cranial neuropathy in one. Objective sensory deficits often indicated a clear upper level of spinal cord lesion in six patients; two patients (Patient 6 and 8) showed a multiple mono-neuropathy presentation. The distribution of motor weakness reflected the lesion site of the myelopathy [paraparesis (n = 2), quadriparesis (n = 2), monoparesis (n = 1), bilateral paresis (n = 1), hemiparesis (n = 1), and triparesis (n = 1)]. Pyramidal signs (hyperactive tendon reflexes, Babinski/Chaddock signs, and/or spasticity) were seen in six patients, although tendon reflexes were hypoactive in all four extremities of five patients. The median mRS score before treatment was 4 (range 2–5).
Laboratory, electrophysiological, and imaging evaluations
In serum analyses, soluble interleukin 2 receptor (sIL-2R) level was increased in 64% of patients, whereas angiotensin-converting enzyme (ACE) and calcium levels were rarely elevated (in 18% and 9.1% of patients, respectively; Table 2). Bronchoalveolar lavage fluid (BALF) analysis was done in six patients, four of whom (67%) showed a high CD4/8 ratio (> 3.5). Three of the four patients with a high CD4/8 ratio also had high serum sIL-2R. The most common abnormality in cerebrospinal fluid (CSF) was elevated protein level in nine patients (82%), followed by pleocytosis in seven patients (64%), elevated sIL-2R level in five patients (45%), and increased ACE level in three patients (27%).
A nerve conduction study (NCS) was performed in all patients. Seven patients showed abnormal results: axonal damage in Patient 1, 5, 9, 10, and 11 and mixed axonal and demyelinating features in Patient 6 and 8 (Table 2). None of the patients had a primary demyelinating pattern. Degree of nerve involvement was considerably different between each nerve, supporting the diagnosis of multiple mono-neuropathy. Patient 1 and 5 showed abnormal results only in motor nerves, and Patient 6, 8, 9, 10, and 11 showed abnormalities in both motor and sensory nerves.
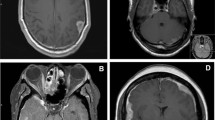
Spinal MRI showed intramedullary T2-elongated lesions in all patients except for Patient 11, in whom only leptomeningeal enhancement was seen. The intramedullary T2-elongated lesions were solitary in 10 patients (Fig. 1a, e) and multifocal in one (Fig. 1i). Five patients showed spinal cord swelling. Spinal lesions were located in the cervical cord (n = 7), thoracic cord (n = 5), and lumbar cord (n = 3). Median length of the intramedullary T2-elonged lesions or the leptomeningeal enhancement was two vertebral segments (0.5 to 3). Five cases (Patient 1, 5, and 7–9) had spinal cord enlargement. The spinal cord enlargement was followed by its atrophy after treatment (Fig. 1c). Abnormal enhancement on the post-contrast T1-weighted image was always seen in the spinal cord lesion; one patient (Patient 11) could not undergo post-contrast examination because of renal dysfunction. Patient 10 showed linear leptomeningeal enhancement (Fig. 1g, h).

Spinal cord MRI and FDG-PET. a–d Sagittal MRI of the cervical cord in Patient 9. a T2-weighted imaging and b T1-weighted post-contrast imaging showing hyper-intensities in the C4-6 vertebral levels with cord swelling and intramedullary gadolinium enhancement before treatment. Three weeks after treatment of prednisolone, the hyper-intensity area of T2-weighted imaging (c) and abnormal contrast enhancement (d) were reduced in size. e Sagittal MRI and f FDG-PET of the cervical cord in Patient 11. e T2-weighted imaging displaying hyper-intensities in the C3-4 vertebral levels. f PET-CT imaging revealing abnormal uptake (SUVmax, 5.4) in the same lesion. g Linear enhancement (arrow) in sagittal T1-weighted post-contrast imaging of the cervical-thoracic cord in Patient 10 and h axial T1-weighted post-contrast imaging showing dorsal linear enhancement (arrowhead). h Sagittal T1-weighted imaging in Patient 3 showing multiple gadolinium-enhanced lesions. CT computed tomography, FDG fluorodeoxyglucose, MRI magnetic resonance imaging, PET positron emission tomography; SUVmax maximum standardized uptake value
Eight patients received 18F-fluorodeoxyglucose positron emission tomography (FDG-PET). All eight showed abnormal uptake in the spinal cord lesion (Fig. 1f). The median of the maximum standardized uptake value (SUVmax) was 4.5 (range 2.7–19.4; Table 2). There was a significant positive correlation between sIL-2R levels in the CSF and SUVmax (correlation coefficient = 0.8, P = 0.03).
Immunotherapies and outcomes
The treatment and the outcome of all cases were shown at Table 3. For their initial immunotherapy, 10 patients received a high-dose of oral prednisolone (PSL; 1.0 mg/kg/day) and one patient was prescribed a moderate-dose of PSL (0.5 mg/kg/day), with or without preceding intravenous steroid pulse therapy [methylprednisolone (mPSL) 1000 mg/day, three consecutive days]. PSL dose was then carefully tapered over several months or years while confirming the objective improvement in neurological deficits as well as the decrease in T2-elongated lesion size and abnormal enhancements based on spinal MRI (Fig. 1b, d) in all patients.
During PSL tapering, neurological exacerbation of myelopathy was seen in four patients (Patient1, 4, 8, and 9). In Patient 1, myelopathy recurred three times when the daily doses of PSL were 15 mg, 20 mg, and 30 mg, and consequently methotrexate (MTX; 8 mg/week) was added. Patient 4 had an exacerbation when the PSL dose was 35 mg/day and low-dose MTX was 4 mg/week, and thus PSL was changed to mPSL 8 mg/day. Patient 8 exacerbated twice: the first time was when the dose of PSL was 15 mg/day, after which MTX (6 mg/week) was initiated, and the second time was when the doses of PSL and MTX were 12.5 mg/day and 8 mg/week, respectively. Then, the dose of MTX was increased to 10 mg/day. In Patient 9, the symptom and the spinal lesion were ameliorated by PSL but gadolinium enhancement in MRI persisted, therefore MTX 4 mg/week was added. Patient 9 experienced an exacerbation in spite of receiving PSL 25 mg/day and MTX 4 mg/week. In six patients who had no neurological exacerbation, immuno-suppressants were also added due to prolonged and intractable sensory deficits (MTX in Patient 5 and MTX + azathioprine in Patient 7). There was no significant difference in laboratory results and mRS scores between patients who required additional immuno-suppressants and patients without immuno-suppressants.
Improvement in mRS was seen in seven (64%) of the 11 patients by the end of the follow-up period (median 48 months; range 1–88 months after the initiation of immunotherapies). In contrast, mRS was unchanged in one patient and became worse in the remaining three patients. Only one (9%) patient showed complete recovery in mRS. There were no significant correlations between the change in mRS and any demographic features, such as sex, age, and duration from onset to the first treatment. However, in the marked-improvement group (n = 3; improvement of 2 or more on mRS), CSF parameters including cell count, protein, ACE, and sIL-2R before treatment were significantly higher than in the other 8 patients (P = 0.02, P = 0.01, P = 0.02, and P = 0.02, respectively). Abnormal findings in spinal MRI (abnormal enhancements, abnormal T2-elonged lesions, or both) disappeared in three patients, and were reduced in seven patients (Fig. 1b, d). NCS was performed again after treatment in six patients, three of whom (Cases 1, 4, and 9) had ameliorated, but still evident, conduction abnormalities.
Discussion
This study showed that peripheral nerves and thoracic organs were frequently affected in SCS, whereas involvements of the eye and skin were relatively rare. Increased serum sIL-2R was the most common finding in blood examinations, and sIL-2R was also elevated in the CSF of five SCS patients. Furthermore, abnormal FDG uptake in the spinal cord was confirmed in all SCS patients who were examined. These data suggest that results from screening of peripheral nerves and thoracic organs, examining of sIL-2R in serum and CSF, and analyzing FDG-PET are important clues that may suggest SCS and are important for the differential diagnosis of myelopathy of unknown origin. mRS scores were unchanged or worsened in 36% of patients, and neurological exacerbation of myelopathy was seen in 36% of patients. Complete response rate of mRS was only 9%. Those patients with a marked improvement after immunotherapy treatment showed significantly higher levels of cell count, protein, ACE, and sIL-2R in the CSF than the other group. These results indicate that prognosis of SCS is generally poor, and abnormalities in cell count, protein, ACE, and sIL-2R in the CSF may predict the efficacy of immunotherapies.
There have been few investigations concerning the involvement of peripheral nerves in SCS. It is noteworthy in our cohort that coexistence of peripheral neuropathy was detected only by NCS in five patients who did not have neurological signs and symptoms suggestive of “neuropathy” (defined as clinically multiple mono-neuropathy presentation which cannot be attributable to spinal cord lesions). The proportion of sarcoid neuropathy was reported to be 2–40% among patients with neuro-sarcoidosis [4, 28,29,30]. However, a study in the United States showed that more than 60% of patients with systemic sarcoidosis, who had no symptoms of peripheral neuropathy, had NCS abnormalities; this suggests that subclinical peripheral nerve lesions are common in systemic sarcoidosis [31]. Although it remains unclear whether the high proportion of peripheral neuropathy in our study was due to ethnic differences between Japan and Western countries or because of differences between SCS and other sarcoidosis, our data indicated that clinicians should conduct tests for peripheral neuropathy to evaluate the likelihood of SCS in myelopathy of unknown origin.
A large proportion of our patients with SCS had elevated sIL-2R levels in the CSF, whereas ACE levels were normal in most patients. In another study, it was shown that a high level of sIL-2R in CSF was more specific and sensitive for neuro-sarcoidosis than that of ACE [32]. In the current study, we confirmed for the first time that this is also true in SCS, indicating that sIL-2R in CSF is a useful biomarker of SCS, although the other causes of high sIL-2R—including infections and central nervous system lymphoma—should be strictly excluded [33]. In addition to sIL-2R in the CSF, serum sIL-2R was also sensitive for systemic sarcoidosis [34]. Particularly in pulmonary sarcoidosis, there was a positive correlation between serum sIL-2R and CD4 + T lymphocyte count in BALF analysis [35]. Our data are compatible with the previous findings [13, 18] that elevation of serum sIL-2R as well as pulmonary involvement are also suggestive of SCS, since all SCS patients in our study had respiratory involvement, despite often lacking pulmonary symptoms and signs.
SCS often accompanies cervical spondylosis. A report showed that 66.7% of SCS in the middle-age had concomitant cervical spondylosis [22]. In our study, 71% of the patients with cervical SCS had cervical spondylosis, and spinal canal stenosis with compression of spinal cord was evident in these patients. It therefore is important to differentiate cervical spondylotic myelopathy in the diagnosis of SCS. Our cohort of SCS patients showed a median SUVmax in FDG-PET of spinal lesions of 4.5, ranging from 2.7 to 19.4. A previous study including six Japanese patients with SCS showed that SUVs of spinal lesions were higher in SCS patients (mean 4.38; range 3.30–4.93) than in those with cervical spondylosis and in those with ossification of the posterior longitudinal ligament [21]. These findings suggest that FDG-PET is useful to evaluate the likelihood of SCS by defining the SUV cut-off value of the spinal lesion of around 4. Persistent enhancement of the lesions is much more suggestive of sarcoidosis [3]. Enhancement lasted more than one month in 36% of our patients. Although these findings are not specific, they can be useful for differential diagnosis.
Patients with systemic sarcoidosis in Asia are older and more likely to be female than those in Western countries [11, 12]. Compared to SCS reports published from Western countries, the onset age of Japanese SCS patients, including of those in the current study and the previous study data, was older (Japan: median 60 years old; Western countries: median 38 years old). In addition, the gender proportion of SCS patients differs between Japan and Western countries: There are twice as many females in Japan, whereas males were predominant in Western countries [15,16,17,18, 20, 21, 24]. The current study confirmed these characteristics (older and predominantly female) in Japanese patients with SCS, indicating that there are common demographic features among SCS and systemic sarcoidosis patients in Asia.
Although all our patients received immunotherapies consisting of a moderate- to high-dose of corticosteroid with or without immuno-suppressants, mRS scores remained unchanged or exacerbated in one-third of patients by the end of the follow-up period, and two of the remaining seven patients experienced neurological exacerbation of myelopathy during the corticosteroid tapering period. In previous reports of SCS, most patients were treated with corticosteroids, but more than half required additional immuno-suppressants because corticosteroids could not sufficiently control the disease activity [13, 17, 19]. After treatment, only 6–33% of patients reached complete recovery [13, 17, 22], and most had neurological sequelae [13, 17, 19, 22], indicating a generally poor prognosis of SCS. Previous studies described that a long interval from the onset to the initiation of therapy was associated with poor recovery in SCS [20, 28]. In the current study, we found that higher levels of cell count, protein, ACE, and sIL-2R in the CSF predicted a marked improvement after immunotherapy treatment. This is partially inconsistent with previous findings reported in France, where high levels of cell count and protein in the CSF were often observed in the SCS patients who had poor outcomes [13]. This discrepancy might be due to the small samples in these analyses, or may be reflection of racial, ethnic, or geographical differences.
There are several limitations in this study. First, this is a retrospective, observational study consisting of a small number of patients. Therefore, availability of clinical, laboratory, and imaging data was limited and a direct comparison to myelopathy of other etiologies has yet to be performed. Second, histological confirmation of non-caseating epithelioid granuloma in the spinal cord was not obtained in our patients, and it is impossible to completely exclude the possibility that myelopathy due to other causes was included in this cohort. Third, the diagnostic criteria we used for SCS was developed for the current study by modifying the criteria for sarcoidosis proposed by the Japan Society of Sarcoidosis and other Granulomatous Disorders. These criteria have yet to be validated and their accuracy should be evaluated in a different cohort.
In conclusion, we should examine peripheral nerves, thoracic organs, sIL-2R in serum and CSF, FDG-PET in patients with myelopathy of unknown origin. The abnormalities in CSF could be prognostic factor but it remains uncertain. To determine the accurate tendency, accumulating more data of patients with SCS was needed.
Data availability
The data that support the findings of this study are available from the corresponding author, TK, upon reasonable request.
Code availability
Not applicable.
References
Valeyre D, Prasse A, Nunes H, Uzunhan Y, Brillet PY, Müller-Quernheim J (2014) Sarcoidosis. Lancet 383:1155–1167. https://doi.org/10.1016/S0140-6736(13)60680-7
Hoitsma E, Faber CG, Drent M, Sharma OP (2004) Neurosarcoidosis: a clinical dilemma. Lancet Neurol 3:397–407. https://doi.org/10.1016/S1474-4422(04)00805-1
Zajicek JP, Scolding NJ, Foster O et al (1999) Central nervous system sarcoidosis–diagnosis and management. QJM 92:103–117. https://doi.org/10.1093/qjmed/92.2.103
Ungprasert P, Crowson CS, Matteson EL (2017) Epidemiology and clinical characteristics of sarcoidosis: an update from a population-based cohort study from Olmsted County, Minnesota. Reumatismo 69:16–22. https://doi.org/10.4081/reumatismo.2017.965
Baughman RP, Field S, Costabel U, Crystal RG, Culver DA, Drent M, Judson MA, Wolff G (2016) Sarcoidosis in America: analysis based on health care use. Ann Am Thorac Soc 13:1244–1252. https://doi.org/10.1513/AnnalsATS.201511-760OC
Morimoto T, Azuma A, Abe S, Usuki J, Kudoh S, Sugisaki K, Oritsu M, Nukiwa T (2008) Epidemiology of sarcoidosis in Japan. Eur Respir J 31:372–379. https://doi.org/10.1183/09031936.00075307
Yamanouchi Y, Sawahata M, Sakamoto N et al (2020) Characteristics of 68 patients with clinically proven sarcoidosis based on the Japan Society of Sarcoidosis and Other Granulomatous Disorders 2015 criteria. Respir Investig 58:102–109. https://doi.org/10.1016/j.resinv.2019.11.003
Sawahata M, Sugiyama Y, Nakamura Y, Nakayama M, Mato N, Yamasawa H, Bando M (2015) Age-related and historical changes in the clinical characteristics of sarcoidosis in Japan. Respir Med 109:272–278. https://doi.org/10.1016/j.rmed.2014.12.012
Bogousslavsky J, Hungerbühler JP, Regli F, Graf HJ (1982) Subacute myelopathy as the presenting manifestation of sarcoidosis. Acta Neurochir 65:193–197. https://doi.org/10.1007/BF01405845
Brito-Zerón P, Kostov B, Superville D, Baughman RP, Ramos-Casals M, Autoimmune Big Data Study Group (2019) Geoepidemiological big data approach to sarcoidosis: geographical and ethnic determinants. Clin Exp Rheumatol 37:1052–1064
Hsieh CW, Chen DY, Lan JL (2006) Late-onset and rare far-advanced pulmonary involvement in patients with sarcoidosis in Taiwan. J Formos Med Assoc 105:269–276. https://doi.org/10.1016/S0929-6646(09)60117-0
Ying Z, Elyse EL, Yinping F, Shanshan D, Huiping L, Robert PB (2017) Clinical characteristics of sarcoidosis patients in the United States versus China. Sarcoidosis Vasc Diffuse Lung Dis 34:209–216. https://doi.org/10.36141/svdld.v34i3.5727
Cohen-Aubart F, Galanaud D, Grabli D et al (2010) Spinal cord sarcoidosis: clinical and laboratory profile and outcome of 31 patients in a case-control study. Medicine 89:133–140. https://doi.org/10.1097/MD.0b013e3181d5c6b4
Durel CA, Marignier R, Maucort-Boulch D et al (2016) Clinical features and prognostic factors of spinal cord sarcoidosis: a multicenter observational study of 20 biopsy-proven patients. J Neurol 263:981–990. https://doi.org/10.1007/s00415-016-8092-5
Junger SS, Stern BJ, Levine SR, Sipos E, Marti-Masso JF (1993) Intramedullary spinal sarcoidosis: clinical and magnetic resonance imaging characteristics. Neurology 43:333–337. https://doi.org/10.1212/wnl.43.2.333
Jallo GI, Zagzag D, Lee M, Deletis V, Morota N, Epstein FJ (1997) Intraspinal sarcoidosis: diagnosis and management. Surg Neurol 48:514–520. https://doi.org/10.1016/s0090-3019(96)00440-5
Varron L, Broussolle C, Candessanche JP, Marignier R, Rousset H, Ninet J, Sève P (2009) Spinal cord sarcoidosis: report of seven cases. Eur J Neurol 16:289–296. https://doi.org/10.1111/j.1468-1331.2008.02409.x
Saleh S, Saw C, Marzouk K, Sharma O (2006) Sarcoidosis of the spinal cord: literature review and report of eight cases. J Natl Med Assoc 98:965–976
Bradley DA, Lower EE, Baughman RP (2006) Diagnosis and management of spinal cord sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 23:58–65
Koike H, Misu K, Yasui K, Kameyama T, Ando T, Yanagi T, Sobue G (2000) Differential response to corticosteroid therapy of MRI findings and clinical manifestations in spinal cord sarcoidosis. J Neurol 247:544–549. https://doi.org/10.1007/s004150070154
Sakushima K, Yabe I, Nakano F, Yoshida K, Tajima Y, Houzen H, Maruo Y, Sasaki H (2011) Clinical features of spinal cord sarcoidosis: analysis of 17 neurosarcoidosis patients. J Neurol 258:2163–2167. https://doi.org/10.1007/s00415-011-6080-3
Kobayashi S, Nakata W, Sugimoto H (2013) Spinal magnetic resonance imaging manifestations at neurological onset in Japanese patients with spinal cord sarcoidosis. Intern Med 52:2041–2050. https://doi.org/10.2169/internalmedicine.52.0186
Sakai Y, Matsuyama Y, Imagama S et al (2010) Is decompressive surgery effective for spinal cord sarcoidosis accompanied with compressive cervical myelopathy? Spine 35:E1290–E1297. https://doi.org/10.1097/BRS.0b013e3181e6d592
Oe K, Doita M, Miyamoto H, Kanda F, Kurosaka M, Sumi M (2009) Is extensive cervical laminoplasty an effective treatment for spinal cord sarcoidosis combined with cervical spondylosis? Eur Spine J 18:570–576. https://doi.org/10.1007/s00586-009-0891-2
The Japan Society of Sarcoidosis and Other Granulomatous Disorders. Diagnostic standard and guideline for sarcoidosis—2015. http://www.jssog.com/www/top/shindan/shindan2-1new.html [in Japanese]
Moriyama N, Ohara T, Kanzaki H, Tsuda E, Ishihara M, Anzai T (2015) Active cardiac sarcoidosis in a patient with adult-onset Kawasaki disease. J Cardiol Cases 12:68–71. https://doi.org/10.1016/j.jccase.2015.05.003
van Swieten JC, Koudstaal PJ, Visser MC, Schouten HJ, van Gijn J (1988) Interobserver agreement for the assessment of handicap in stroke patients. Stroke 19:604–607. https://doi.org/10.1161/01.str.19.5.604
Stern BJ (2004) Neurological complications of sarcoidosis. Curr Opin Neurol 17:311–316. https://doi.org/10.1097/00019052-200406000-00013
Lacomis D (2011) Neurosarcoidosis. Curr Neuropharmacol 9:429–436. https://doi.org/10.2174/157015911796557975
Ferriby D, de Seze J, Stojkovic T, Hachulla E, Wallaert B, Destée A, Hatron PY, Vermersch P (2001) Long-term follow-up of neurosarcoidosis. Neurology 57:927–929. https://doi.org/10.1212/wnl.57.5.927
Challenor YB, Felton CP, Brust JC (1984) Peripheral nerve involvement in sarcoidosis: an electrodiagnostic study. J Neurol Neurosurg Psychiatry 47:1219–1222. https://doi.org/10.1136/jnnp.47.11.1219
Petereit HF, Reske D, Tumani H, Jarius S, Markus Leweke F, Woitalla D, Pfister HW, Rubbert A (2010) Soluble CSF interleukin 2 receptor as indicator of neurosarcoidosis. J Neurol 257:1855–1863. https://doi.org/10.1007/s00415-010-5623-3
Otto C, Wengert O, Unterwalder N, Meisel C, Ruprecht K (2020) Analysis of soluble interleukin-2 receptor as CSF biomarker for neurosarcoidosis. Neurol Neuroimmunol Neuroinflamm 7:e725. https://doi.org/10.1212/NXI.0000000000000725
Eurelings LEM, Miedema JR, Dalm VASH, van Daele PLA, van Hagen PM, van Laar JAM, Dik WA (2019) Sensitivity and specificity of serum soluble interleukin-2 receptor for diagnosing sarcoidosis in a population of patients suspected of sarcoidosis. PLoS One 14:e0223897. https://doi.org/10.1371/journal.pone.0223897
Grutters JC, Fellrath JM, Mulder L, Janssen R, van den Bosch JM, van Velzen-Blad H (2003) Serum soluble interleukin-2 receptor measurement in patients with sarcoidosis: a clinical evaluation. Chest 124:186–195. https://doi.org/10.1378/chest.124.1.186
Acknowledgements
We are grateful to all the members of the Department of Neurology and Clinical Neuroscience at Yamaguchi University Graduate School of Medicine.
Funding
Project of Finding-Out and Crystallization of Subliminals (FOCS) in Yamaguchi University.
Author information
Authors and Affiliations
Contributions
This study was conceived by MF and MK; MF performed all data acquisition and analysis; MF an MK wrote the draft, which was revised by all authors; all authors contributed to clinical supports, data interpretation and approving the final version of the manuscript. TK provided supervision of the study.
Corresponding author
Ethics declarations
Conflicts of interest
All authors report no disclosures.
Ethics approval
The study was approved by the ethics committee of the Yamaguchi University Medical Faculty in accordance with the Declaration of Helsinki.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Fujisawa, M., Koga, M., Sato, R. et al. Spinal cord sarcoidosis in Japan: utility of cerebrospinal fluid examination and nerve conduction study for diagnosis and prognosis prediction. J Neurol 269, 4783–4790 (2022). https://doi.org/10.1007/s00415-022-11113-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-022-11113-y