
Abstract
Neurosarcoidosis is uncommon with an incidence of approximately 5 to 15%. Central nervous system involvement can be divided into brain and spinal cord neurosarcoidosis. Spinal cord sarcoidosis is extremely rare, occurring in less than 1% of all sarcoidosis cases. Its manifestations may include cauda equina syndrome, radiculopathy, syringomyelia, cord atrophy, arachnoiditis, and myelopathy or transverse myelitis. We highlight two cases of spinal cord sarcoidosis, each presenting with longitudinally extensive transverse myelitis, that demonstrate the dilemmas that physicians face with regard to diagnosis and treatment. Given its rarity and the diversity of possible manifestations, establishing the diagnosis of spinal cord sarcoidosis is often very difficult. Extensive evaluation must be conducted to rule out primary neurologic, primary rheumatologic, infectious, and neoplastic diseases. MRI often demonstrates hyperintensity on T2-weighted images and enhancement following gadolinium administration. CSF analysis most consistently shows a lymphocytic pleocytosis and elevated proteins. While these less invasive investigations may be helpful, the gold standard for diagnosis is biopsy of neurologic or non-neurologic tissue confirming the presence of non-caseating granulomas. Evidence-based guidelines for the treatment of transverse myelitis secondary to sarcoidosis are lacking due to its rarity; therefore, therapy is based on expert and anecdotal experience and usually consists of high doses of steroids in combination with various immunosuppressive agents. The use of infliximab in particular appears promising, but there is a need for further investigation into the ideal treatment regimen.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Sarcoidosis is a multisystem disorder, characterized by non-caseating granulomatous inflammation. The lungs and intrathoracic lymph nodes are the sites most commonly affected [1]. Nervous system involvement in sarcoidosis, both central and peripheral, is uncommon with an incidence of approximately 5 to 15%, and a great heterogeneity in presentation often makes the diagnosis very challenging [2,3,4]. Neurologic deficits are the initial presentation of sarcoidosis in 70–80% of these patients [2,3,4,5,6]. However, on further investigation, the majority are found to have systemic involvement that is often asymptomatic with bilateral hilar lymphadenopathy being the most common finding [4,5,6].
Central nervous system involvement can be divided into brain and spinal cord neurosarcoidosis [1]. Manifestations in the brain include neuroendocrine dysfunction, intraparenchymal lesions, cranial neuropathies, with cranial nerves II, VII, and VIII being the most frequently affected, encephalopathy/vasculopathy, and meningeal infiltration [2, 5, 7]. Spinal cord neurosarcoidosis is very rare, occurring in less than 1% of all sarcoidosis patients [3, 4]. Inflammation may involve the intramedullary, extramedullary, or extradural portions of the spinal cord, leading to a variety of manifestations including cauda equina syndrome, radiculopathy, syringomyelia, cord atrophy, arachnoiditis, and myelopathy or transverse myelitis [1, 5, 7]. The clinical presentation may be acute, subacute, or chronic [3, 4, 6]. Our report describes two patients with longitudinally extensive transverse myelitis secondary to sarcoidosis and highlights the diagnostic dilemmas and lack of consensus treatment guidelines that physicians face when caring for these patients.
Case 1:
A 47-year-old, previously healthy male, with a remote history of seizures (off all medications), presented to the emergency department with 3 weeks of lower back pain, 1 week of urinary retention, progressive bilateral lower extremity weakness, and numbness/tingling to the T6/7 level. His exam was concerning for myelopathy, demonstrated by moderate weakness in bilateral lower extremities (both proximal and distal muscle groups) with inability to ambulate, brisk reflexes, and sensory loss to the level of T6.
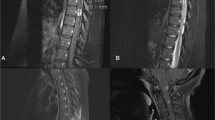
MRI showed a central cord lesion extending from the cervical medullary junction through the conus medullaris. The lesion was hyperintense on T2-weighted imaging and STIR and hypointense on T1-weighted imaging and demonstrated heterogeneous patchy enhancement most prominent posteriorly (Fig. 1). These findings were most consistent with transverse myelitis.

MRI findings from case 1 demonstrating hyperintensity on T2-weighted images of the cervical cord, sagittal image (a) and of the thoracic cord, sagittal image (b)
Routine labs including electrolytes, renal function, liver function, complete blood count, and urinalysis were all unremarkable. Inflammatory markers were mildly elevated with an ESR of 23 mm/h (normal 0–15) and CRP of 12.5 mg/L (normal 0–7.48). A lumbar puncture during this admission was significant for lymphocytic pleocytosis and elevated protein. He underwent extensive evaluation for infectious and autoimmune etiologies (Tables 1 and 2). Of note, the Lyme ELISA using the C6 assay returned positive, and the confirmatory Western Blot revealed nine positive IgG bands, no positive IgM bands.
Testing for paraneoplastic syndromes, neuromyelitis optica, multiple sclerosis, and various metabolic and nutritional myelopathies was negative (Table 3).
The patient was treated with 3 days of IV methylprednisolone 1 g daily and started on a 21-day course of IV ceftriaxone 2 g daily for possible neurologic Lyme disease. He improved clinically such that he was able to walk the hallway with assistance and was discharged to a rehabilitation facility 10 days after initial admission.
Five days into his rehabilitation stay, he developed worsening weakness in the lower extremities and more profound urinary retention requiring scheduled catheterizations. He rapidly progressed to complete paraplegia and was re-admitted to the hospital.
Additional infectious work-up was felt to be indicated (Table 4).
Findings on repeat MRI were unchanged from prior. The possibility of an autoimmune process vs a primary neurologic disease such as neuromyelitis optica was revisited. Ophthalmology exam was negative for optic neuritis. The decision was made to empirically start plasma exchange for a potential autoimmune process of unclear etiology. He received a total of five sessions.
Given lack of improvement in symptoms and largely negative work-up, CT chest/abdomen/pelvis was done which revealed bilateral hilar and mediastinal lymphadenopathy. Mediastinal lymph node biopsy, done via mediastinoscopy, demonstrated non-caseating granulomas, with no evidence of malignancy or infection. The patient was diagnosed with neurosarcoidosis now 7 weeks after initial symptom onset. He was started on a second round of IV methylprednisolone, 1 g daily for 5 days, followed by prednisone 80 mg daily.
At the time of discharge, his strength had not improved, and he remained paraplegic with continued numbness up to the T6 level. He was started on infliximab (3 mg/kg and up-titrated to 5 mg/kg) as an outpatient. The patient remained paraplegic 6 months after initial infusion, though repeat MRI has shown decrease in extent of enhancement.
Case 2:
A 56-year-old male with a history of HTN, DM, and bipolar disorder presented to the emergency department with right-sided hemiparesis. Two weeks prior to admission, he began experiencing right lower extremity followed by right upper extremity weakness and numbness, eventually affecting ambulation. Over the same time course, he noticed associated neck pain. There were no changes in level of consciousness, vision, or speech, and there was no history of recent trauma. He recalled having an upper respiratory infection about 4 weeks prior to onset of current symptoms. Exam revealed mild-to-moderate right upper and lower extremity weakness in both proximal and distal muscle groups, and decreased sensation in the right arm and leg (C5–6 level). He was also noted to be hyper-reflexic on the right.
Cervical spine MRI showed extensive T2 hyperintensity in the spinal cord extending from C2-C7, with patchy areas of enhancement, and evidence of cord expansion, consistent with transverse myelitis with associated cord edema (Fig. 2).

MRI findings from case 2 demonstrating hyperintensity on T2-weighted image of the cervical cord, sagittal image (a)
Complete blood count was significant for mild leukocytosis of 11.4 K/uL (normal 4.0–11.0), with 89% neutrophils. Remaining routine labs, including electrolytes, renal function, liver function, and urinalysis were unremarkable. Inflammatory markers were within normal range: ESR 8 mm/h (normal 0–20) and CRP 1.60 mg/L (normal 0.00–7.48). Lumbar puncture showed lymphocytic pleocytosis and elevated protein. Extensive evaluation for infectious and autoimmune etiologies was unrevealing (Tables 5 and 6).
Testing for metabolic/nutritional myelopathies, as well as multifocal neurologic diseases including multiple sclerosis and neuromyelitis optica, was also negative (Table 7).
The work-up was expanded to include CT chest/abdomen/pelvis to assess for underlying malignancy. These revealed multiple prominent mediastinal lymph nodes, with at least one that was pathologically enlarged, measuring up to 1.3 cm. Oncology had low suspicion for malignancy and short-term follow-up was recommended.
Neuro-ophthalmology found cupping of bilateral optic nerves, thought to be physiologic, but no evidence of optic neuritis.
At that point, the patient was started on IV methylprednisolone 1 g daily for 5 days, for a working diagnosis of post-infectious myelitis. This resulted in partial improvement in strength, such that he was able to ambulate without assistance. He was eventually discharged home with physical therapy, 7 days after admission.
However, 1 week later, he was readmitted with worsening right-sided hemiparesis, now requiring a walker. Symptoms progressed to involve the left side, with shoulder pain and hand numbness. In addition, he developed urinary retention, requiring catheterization.
Cervical spine MRI was repeated and again demonstrated extensive T2 hyperintensity within the spinal cord from C2-C7, with patchy areas of enhancement and cord expansion, slightly progressed from the prior study. Lumbar puncture showed continued lymphocytic pleocytosis (cell count 66/mcL, 88% lymphocytes) and elevated protein (67 mg/dl).
A mediastinal lymph node biopsy performed via mediastinoscopy revealed non-caseating granulomas. The diagnosis of neurosarcoidosis was made, now 6 weeks after initial symptom onset. He was started on a second round of pulse steroids with IV methylprednisolone 1 g daily for 5 days, to which he responded well with notable improvement in strength of right upper and lower extremities. Sensation to light touch remained slightly decreased along right arm and left fingertips. He was discharged to a rehabilitation facility on prednisone 60 mg daily.
Over the course of 3 months, the prednisone was tapered down to 20 mg daily; however, at that time his right-sided weakness worsened. He received a third round of pulse steroids with IV methylprednisolone 1 g daily for 3 days followed by prednisone 60 mg daily. Given the relapsing course of his disease, he was started on infliximab (3 mg/kg, up-titrated to 7 mg/kg), with moderate improvement in right-sided strength. Nine months after the initial infusion, he had only minimal deficits in proximal strength of the right lower extremity.
Discussion
We report two cases of transverse myelitis as the initial manifestation of neurosarcoidosis, each with longitudinally extensive lesions and presenting with varying degrees of weakness and sensory deficits, followed by catastrophic and rapid progression. Given the diversity of the possible signs and symptoms of neurosarcoidosis, its rarity, the multiple clinical mimics, and the absence of a specific non-invasive confirmatory test, the diagnosis is often delayed. In cases such as ours in which there is no preceding diagnosis of sarcoidosis multiple inflammatory, infectious and malignant etiologies must be carefully considered before the diagnosis of neurosarcoidosis can be established. Depending on the presentation, diagnostic considerations might include primary neurologic disorders such as multiple sclerosis and neuromyelitis optica; primary rheumatologic disorders including granulomatosis with polyangiitis, Behcet’s, systemic lupus erythematosus, and Sjogren’s syndrome; infectious diseases including Lyme disease, tuberculosis, herpes virus, and West Nile virus; and neoplastic diseases such as meningeal carcinomatosis and lymphomatosis [8]. Zajicek et al. published criteria for the diagnostic certainty of neurosarcoidosis in 1999 (Table 8).
Less invasive investigations looking for neurosarcoidosis may aid in the diagnosis, but biopsy demonstrating non-caseating granulomas is still gold standard. MRI often demonstrates hyperintensity on T2-weighted images and enhancement following gadolinium administration [1,2,3,4, 6]. In transverse myelitis, the thoracic and cervical spinal cord are most commonly affected, and inflammation typically spans more than three segments [1,2,3,4,5,6]. CSF analysis most consistently shows a lymphocytic pleocytosis and elevated proteins [3, 4, 6]. CSF and serum ACE levels are neither sensitive nor specific [2,3,4,5,6]. In the neurosarcoidosisseries of Zajicek et al. CSF ACE was assessed in 18 cases and was outside the normal laboratory range in only 6 cases (33%). Serum ACE was only elevated in 12/51 cases (23.5%).
Our first case was especially difficult to diagnose given the presence of positive serologic testing for Lyme disease with nine positive IgG bands on the Western Blot (reference positive: ≥ 5 of 10 IgG bands). However, while transverse myelitis has been described in the Lyme disease literature, this occurs almost exclusively in Europe where the neurogenic strain B. garinii is endemic and only very rarely in the USA [9, 10]. Our patient denied travel to Europe. In addition, nervous system involvement in Lyme disease is responsive to appropriate antimicrobial therapy, which contrasts with progression of our patient’s symptoms [10, 11]. However, there are no high-level studies looking specifically at the treatment of transverse myelitis due to Lyme disease. Finally, given that New England is an endemic area for Lyme disease, the baseline incidence of Lyme seropositivity is high thereby increasing the chance of positive Lyme titers that are coincidental and unrelated. After taking these factors into account along with the strong supporting evidence for sarcoidosis provided by the pathologic findings, we concluded that late Lyme disease was highly unlikely to be responsible for his neurologic manifestations.
There are no evidence-based guidelines for the treatment of transverse myelitis secondary to sarcoidosis due to its rarity and therefore treatment is largely based on experience. A consistent feature across the board is the initial use of high-dose steroids with several case reports and case series describing subsequent initiation of various immunosuppressive agents [1, 5]. If symptoms are severe, a short course of IV pulse steroids (e.g., 1 g methylprednisolone/day for 3–5 days) is usually administered. Thereafter, or if signs and symptoms are less severe, oral steroids (e.g., 40–80 mg prednisone/day) should be given for at least 1–3 months, and then slowly tapered to the lowest effective dose [1].
The prognosis in spinal cord sarcoidosis is guarded with mixed response to therapy. Wang and Li described a group of seven patients with transverse myelitis due to sarcoidosis affecting six or more contiguous spinal segments. Initial treatment in these cases involved the use of corticosteroids (methylprednisolone, prednisone, or dexamethasone). Other immunosuppressive agents used included infliximab (n = 6), methotrexate (n = 4), azathioprine (n = 3), leflunomide (n = 2), cyclophosphamide (n = 2), and adalimumab (n = 1). All patients improved following immunosuppressive treatment; the mean onset of improvement occurred at 1 month (range: 3 days to 3 months). However, all patients had some degree of residual neurological deficits at the study conclusion.
Durel et al. published a multicenter observational study of 20 biopsy-proven patients with spinal cord sarcoidosis manifesting with myelopathy. Only two cases involved direct sampling of neurologic tissue. The other pathology specimens were retrieved from minor salivary glands, peripheral lymph nodes, bronchial mucosa, mediastinal lymph nodes, skin, or liver. Longitudinal involvement (spanning more than three vertebral segments) was found in 15 patients. All patients received steroids initially, the majority of whom were administered pulse IV doses for 3–6 days. Concomitant immunosuppressive treatment was administered to ten patients (50%) because of disease severity: methotrexate (n = 4), mycophenolate mofetil (n = 3), cyclophosphamide (n = 2), or azathioprine (n = 1). Seven patients (35%) subsequently received anti-TNF-a therapy (mostly infliximab) during follow-up, four of whom had experienced progression of disease with the initial treatment regimens. Five of these seven patients were still receiving infliximab at the last follow-up visit with improvement or stabilization of the neurologic manifestations and no relapse of active disease. In the two other patients, the treatment was stopped because of suspicion of infection in one patient and lack of efficacy in the other. After a mean follow-up of 52.1 months, 5% of patients had complete symptom resolution, 90% had various degrees of partial recovery, and 5% showed no improvement.
Regardless of treatment, an important prognostic factor in transverse myelitis due to neurosarcoidosis is the severity of initial neurologic manifestations [4]. This is evident in the case of our first patient who ultimately developed complete paraplegia within the first month of symptom onset, and has thus far shown no significant clinical improvement, despite aggressive therapy with high doses of steroids and infliximab. However, our second patient has done relatively well on steroids and infliximab with partial improvement in his initial symptoms of right-sided hemiparesis and urinary retention. The literature is conflicting with regard to the relationship between the time from onset of neurologic manifestations to treatment and outcome. In Cohen-Aubart’s study, the time between the onset of neurological symptoms and initiation of steroid therapy was not found to correlate with outcome. In contrast, other studies have suggested that prompt corticosteroid and immunosuppressive therapy may be important in spinal cord sarcoidosis to obtain more favorable outcomes.
Conclusion
In summary, we report two cases of neurosarcoidosis manifesting with longitudinally extensive transverse myelitis. Establishing this diagnosis is often challenging as most patients who present with a clinical syndrome compatible with spinal cord sarcoidosis do not carry a preceding diagnosis of sarcoidosis necessitating a large differential diagnosis. When spinal cord sarcoidosis is considered, the work-up should include MRI, CSF analysis, and clinical as well as histological evaluation for sarcoidosis in non-neurologic tissues as these patients are often found to have systemic involvement, though other sites may be less clinically apparent. Current treatments for transverse myelitis secondary to sarcoidosis are guided by anecdotal experience and expert opinion leaving a great need for guidance from clinical trials in the future. However, based on existing reports and our current experience, high-dose corticosteroids in combination with infliximab appear promising.
References
Tana C, Wegener S, Borys E, Pambuccian S, Tchernev G, Tana M, Giamberardino MA, Silingardi M (2015) Challenges in the diagnosis and treatment of neurosarcoidosis. Ann Med 47(7):576–591
Cação G, Branco A, Meireles M, Alves JE, Mateus A, Silva AM, Santos E (2017) Neurosarcoidosis according to Zajicek and scolding criteria: 15 probable and definite cases, their treatment and outcomes. J Neurol Sci 379:84–88
Wang L, Li Y (2015) Longitudinal ultra-extensive transverse myelitis as a manifestation of neurosarcoidosis. J Neurol Sci 355(1–2):64–67
Durel CA, Marignier R, Maucort-Boulch D, Iwaz J, Berthoux E, Ruivard M, André M, le Guenno G, Pérard L, Dufour JF, Turcu A, Antoine JC, Camdessanche JP, Delboy T, Sève P (2016) Clinical features and prognostic factors of spinal cord sarcoidosis: a multicenter observational study of 20 biopsy-proven patients. J Neurol 263(5):981–990
Ungprasert P, Matteson EL (2017) Neurosarcoidosis. Rheum Dis Clin N Am 43(4):593–606
Cohen-Aubart F, Galanaud D, Grabli D, Haroche J, Amoura Z, Chapelon-Abric C, Lyon-Caen O, Valeyre D, Piette JC (2010) Spinal cord sarcoidosis: clinical and laboratory profile and outcome of 31 patients in a case-control study. Medicine (Baltimore) 89(2):133–140
Fried ED, Landau AJ, Sher JH, Rao C (1993) Spinal cord sarcoidosis: a case report and review of the literature. J Assoc Acad Minor Phys 4(4):132–137
Zajicek JP, Scolding NJ, Foster O, Rovaris M, Evanson J, Moseley IF, Scadding JW, Thompson EJ, Chamoun V, Miller DH, McDonald W, Mitchell D (1999) Central nervous system sarcoidosis—diagnosis and management. QJM 92(2):103–117
Cerar T, Strle F, Stupica D, Ruzic-Sabljic E, McHugh G, Steere AC, Strle K (2016) Differences in genotype, clinical features, and inflammatory potential of Borrelia burgdorferi sensu stricto strains from Europe and the United States. Emerg Infect Dis 22(5):818–827
Halperin JJ (2018) Diagnosis and management of Lyme neuroborreliosis. Expert Rev Anti-Infect Ther 16(1):5–11
Halperin JJ (2011) Neurologic manifestations of Lyme disease. Curr Infect Dis Rep 13(4):360–366
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical standards
The manuscript does not contain clinical studies or patient data.
Disclosures
None.
Rights and permissions
About this article

Cite this article
Scott, A.M., Yinh, J., McAlindon, T. et al. Two cases of sarcoidosis presenting as longitudinally extensive transverse myelitis. Clin Rheumatol 37, 2899–2905 (2018). https://doi.org/10.1007/s10067-018-4144-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10067-018-4144-9