
Abstract
Objective
Neurological complications of systemic sarcoidosis are uncommon and the natural history and optimal treatments under-researched. With the advent of modern biological therapies, it is important to define the clinical characteristics and immunopathology of the disease.
Methods
Patients referred to and treated within the Centre for Neurosarcoidosis over a 15 year period who had biopsy-proven “highly probable” disease of the central nervous system were studied prospectively.
Results
166 patients were studied, of whom two-thirds had involvement of the brain and spinal cord and the remainder cranial neuropathies and radiculopathy. Imaging was abnormal in all those with meningeal and parenchymal diseases, and was normal in 37% of those with cranial neuropathy. Those with leptomeningeal disease had a more severe disorder, with hydrocephalus and tissue destruction, whereas those with pachymeningeal disease had more striking imaging features but less neurological impairment. The CSF was active in 70% of cases, even when imaging was normal. Disability correlated with CSF indices in those with a leptomeningitis. Oligoclonal bands were seen in 30% of cases and correlated with disability and the presence of hydrocephalus. Unmatched bands were seen only in isolated neurological disease.
Conclusions
This prospective study of neurosarcoidosis increases our understanding of the pathophysiology of the disease. A reclassification of the clinical and imaging features of the disease allows an understanding of its pathophysiology and correlation with CSF indices allows an early identification of those with a more destructive disease will help to define treatment and may thereby improve outcome.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Granulomatous inflammation develops when antigen presentation in a genetically susceptible host leads to activation of CD4+ lymphocytes which differentiate into TH-1 cells and cause amplification of the immune response, including the differentiation of macrophages into multinucleated giant cells. When these cells are surrounded by lymphocytes, they are termed epithelioid granulomas [1]. The severity of the inflammation, whether or not it accumulates and spreads, and whether or not it leads to deposition of fibrous tissue, varies from case to case, and may be related to the presence of SAA [1] and the efficiency of regulatory T-cell (Treg) function in suppressing TNFα and INFγ expression [2].
Sarcoidosis is a multisystem disease of uncertain aetiology in which granulomatous inflammation develops in numerous tissues within the body [3]. The respiratory system is involved in 95% of cases, with the skin, eye, lymph nodes, and liver next most commonly affected [3]. The nervous system is involved in 5% [4]. The ACCESS study [5] demonstrated that half had involvement of one tissue (usually the lung) at diagnosis, although many subsequently developed multisystem disease [6]. The disease is three times more common in people of African ancestry than Caucasians of northern European heritage3, with a much lower prevalence in people of Asian ancestry. Various HLA subtypes influence the susceptibility to the disease and its prognosis [1].
Our understanding of the pathophysiology of neurological involvement by the disorder comes from small case series and individual reports, and as a result, there are no conclusive data on which to base advice on investigation, correct diagnosis, and treatment. This is important bearing in mind the advent of modern biological therapies which appear to have significantly greater efficacy than corticosteroids and low-dose immunosuppression. This paper describes the clinical characteristics, imaging, and CSF abnormalities in 166 patients studied prospectively and treated at the Centre for Neurosarcoidosis over the past 15 years. A companion paper (Neurology, in press) correlates these and the systemic features with response to treatment and neurological prognosis.
Methods
The Centre for Neurosarcoidosis is a national referral centre for the investigation and treatment of the disease. The investigation results are reviewed (including the pathology when available) and further investigation made if necessary. Those in whom, after investigation, a diagnosis of systemic sarcoidosis with pathological support was agreed, in whom the neurological features and investigation results were considered to be in keeping with a neurological complication of the systemic disease, have been included in this prospective study. All patients, therefore, are considered to have a “highly probable” diagnosis of neurosarcoidosis according to the WASOG sarcoidosis organ assessment instrument [7]. Patients with an isolated form of the disease in whom the neuropathological features were considered to be typical for the disease were also included. An assessment of disablement was measured in each case before and after treatment using the modified Rankin score (MRS) [8].
The study was undertaken prospectively following permission from the local research ethics committee, and all patients gave informed consent to participate. Statistical analysis: group comparisons were made using Fisher’s exact test and the Mann–Whitney U test, correlations using Spearman’s rank correlation test. Analyses were performed with Minitab software, version 17, and multiple regression analysis using the Excel data analysis add-in.
Results
-
1.
Patient demographics and clinical features of the systemic disease
There were 166 patients; Table 1 summarises the patient demographic data and the clinical features of the systemic disease. Excluding those with isolated neurological disease, and counting mediastinal lymphadenopathy as a separate disorder to lung parenchymal involvement, 10 had single system disease in addition to neurological involvement, 55 showed involvement of two systems, 58 three, 30 four, four had five systems, and two and one had involvement of six and seven separate systems, respectively.
The serum angiotensin converting enzyme level was normal in 57 and elevated in 64 (53%). The median (range) ACE was 73 (12–283) iU/l (normal range 8–50). Twelve patients had hypercalcaemia, in each case at diagnosis; the median calcium in those was 2.6 (2.6–3.2) mmol/l. Four had leucopenia and two hypogammaglobulinaemia at diagnosis.
Fifty-eight patients underwent Gallium-67 scintigrams during their evaluation; none was normal, 16 showed abnormal uptake in one tissue, 16 in two, 30 in three, and 10 in four separate tissues. Thirty-three underwent F-18 FDG PET/CT scans; none was normal, two had uptake in one tissue, 13 in two, and nine each in three and four tissues.
-
2.
The clinical features of the neurological disease
The clinical syndromes are noted separately below and summarised with CSF and imaging results in Table 2. Six patients had isolated neurological disease, determined by a neuropathological examination; the remainder had clinically typical and histologically proven systemic sarcoidosis, whose characteristics are noted above. The systemic disease and the neurological syndrome coincided in 100 (60%) cases, preceded it in 55 (34%) cases, and developed after the neurological symptoms in 10 (6%) cases.
Of those with disorders of the brain, spinal cord, and cauda equina, abnormalities of MRI were seen in all cases. Complete data on spinal fluid examinations were available in 89 cases; the CSF protein was raised in 76% of samples, median 0.8 (0.19–8.35) g/l, and the cell count in 51%, median 5 (0–395). The blood/CSF glucose ratio was low in 81%, median 48 (20–81)%. The CSF ACE level, not considered to be a marker for sarcoidosis, correlated with CSF protein (ρ = 0.81) and was raised in 50% of the samples, median 1.2 (0.3–10.0) iU/l. Oligoclonal bands were negative in 73%, matched positive in 23% and positive with intrathecal synthesis alone in 4%, all of whom had isolated neurological disease.
The median MRS overall was 3 (1–5). A multiple regression analysis did not identify a link between MRS and age and any systemic feature (including number of tissues involved); F > 0.55.
Cranial neuropathy
-
1.
Facial neuropathy
There were 26 cases, 21 females. Twelve were bilateral and simultaneous, 14 unilateral. Five occurred at the onset of the systemic symptoms of the disease. Imaging when carried out at the time of the facial neuropathy was normal in 11 cases; in two, there was enhancement of the nerve only, and in four, there was evidence for more widespread meningeal enhancement.
A spinal fluid examination carried out at the time of the facial neuropathy was abnormal in 13 of 16 cases; the median protein was 0.68 g/l and the median cell count was 1 (Table 2).
In seven cases, the facial neuropathy arose before the onset of a more widespread neurological disorder; in one, it arose 2 years after a brain stem lesion. In the remaining cases, therefore, the facial neuropathy was the only neurological manifestation of the disease.
-
2.
“Polyneuritis cranialis”
The fifth and seventh nerves were affected together in two cases, in which imaging and CSF constituents were normal; the fifth, sixth, and seventh in two cases, in which a basal meningeal enhancement was seen in one case with normal imaging in the other, and both showed normal CSF indices; two cases had fifth, seventh and unilateral hearing loss, in which the imaging was abnormal in each case and one showed an active CSF.
There were six cases in which dysphonia and dysphagia were seen. All were female, imaging was normal in four with a mild basal meningeal enhancement in the other two, and only minor CSF changes (with a raised protein and no cells) were seen.
-
3.
Optic neuropathy
There were 27 cases, most of whom were included in a separate paper [9]. Optic neuritis arose in 17, perineuritis in two, and a compressive optic neuropathy related to an adjacent pachymeningitis of the orbital apex in eight cases.
Involvement of the brain by pachymeningitis
A predominate inflammation of the dura was seen in 23 cases, 15 of whom were female, mean age 44 (27–66) years. Headache occurred in 19 cases and seizures in six. Hydrocephalus did not occur (Table 2).
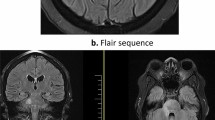
The imaging features were of a dural-based enhancing mass in 16 cases, with evidence for a more widespread but contiguous dural meningeal enhancement in the other seven. Twelve of those with a mass had single lesions, whilst the remaining four showed an adjacent dural spread. Seizures occurred only in those with mass lesions, which arose in the frontal and parietal regions in two and three cases respectively, each of which was associated with headache (Fig. 1a). The remaining cases involved the basal structures; seven at the orbital apex, each of which was associated with optic neuropathy (Fig. 1b), three at the cavernous sinus, each of which was associated with ophthalmoparesis and diminished trigeminal function (Fig. 1c), one extending into the hypothalamus and three into the brainstem causing ataxia. Three cases showed widespread pachymeningeal thickening with enhancement; one was associated with severe bilateral optic neuropathies (Fig. 1d), whilst the other two had few neurological signs.

Imaging features of pachymeningitis of the brain; a parietal mass, b lesion at the orbital apex extending through the middle and posterior cranial fossae, c lesion of the cavernous sinus, and d widespread pachymeningitis affecting both sides
A spinal fluid examination was undertaken in 12 cases; the median (range) CSF protein was 1.1 (0.3–3.6) g/l and cell count 4 (0–150) (Fig. 2 a–c). There was correlation between CSF protein and cell count; ρ = 0.86 (p < 0.0001). Matched positive oligoclonal bands were present in three cases. Neither CSF protein nor cell count correlated with MRS at diagnosis (p > 0.6).

Median CSF protein, cell count, and CSF/blood glucose ratio in pachymeningitis and leptomeningitis of the brain, with interquartile range
Involvement of the brain by leptomeningitis
The identification with MRI of enhancement of the pia and arachnoid mater associated with inflammation of the subjacent cortex and white matter occurred in 67 cases. 36 were females and the mean age was 43 (25–69) years. None had isolated meningitis without associated neurological signs.
The clinical syndromes often overlapped, but can reasonably be divided into two, in which an encephalopathy with headache, drowsiness, and cognitive slowing occurred in 42 cases, of whom 17 had seizures and eight hydrocephalus, and a brainstem syndrome with ataxia of gait, diplopia, vertigo, and other cranial neuropathies. There were 25 such cases, of whom 12 had headache; none had seizures and six had hydrocephalus. Overall, the median MRS was 3 (1–5) and was similar in each subgroup (Table 2).
Leptomeningeal enhancement was seen predominately in the basal regions in 52 cases, and in the remainder, the pattern of enhancement was adjacent to the skull vault (Fig. 3a). In those with predominately middle cranial fossa enhancement (42 cases), the hypothalamus was involved in 10 cases and the mesial temporal lobes in three. Hydrocephalus was seen in seven of these cases. Those with more posteriorly placed enhancement showed lesions within the brainstem and the cerebellum (25 cases) (Fig. 3b), and hydrocephalus occurred in eight of these cases (Fig. 3c). There was considerable overlap, and many cases had striking and widespread enhancement throughout the basal regions (Fig. 3d).

Imaging features of leptomeningitis of the brain; a involving the convexity on one side, b cerebellum and adjacent parietal cortex, c brainstem leading to an asymmetric hydrocephalus (notice that there are nodules of inflammation within the ependyma of the ventricles), and d widespread basal leptomeningitis
The median CSF protein was 0.72 (0.27–4.99) g/l and the CSF cell count 5 (0–395). The median CSF/blood glucose was 48 (23–72) % (Fig. 2a–c). 76% of the glucose levels recorded were below 60%. CSF/ blood glucose ratio was significantly associated with both CSF protein and cell count (p < 0.02). 13 had positive oligoclonal bands (11 matched and 2 unmatched) and 23 had none (Table 2). There was a correlation between CSF protein and MRS, ρ = 0.34 (p < 0.005), but not between cell count and MRS (ρ = 0.05). The median CSF protein was significantly higher in those with oligoclonal bands (0.96 g/l) than those without (0.62 g/l) (p < 0.04); so too was the median CSF cell count (11 vs 2) (p = 0.04).
The presence of hydrocephalus was associated with a significantly higher CSF protein (1.6 vs 0.62 g/l, p < 0.01) compared with those without, and with a lower median CSF/blood glucose ratio (31 vs 49%, p = 0.02), but the cell count was not associated (13 vs 4, p > 0.1).
Involvement of the spinal cord and cauda equina
Involvement of the cord and/or cauda equina was seen in 37 patients; 13 were female and the mean (range) age was 43 (25–60) years. Thirteen had evidence for disease involvement in other sites; one had an optic neuropathy, three involvement of the pituitary, five the basal meninges, and four the brain stem. The median MRS was 3 (2–5) at diagnosis. Lesions in the spinal cord were seen in 28 cases; four showed the appearances of a pachymeningitis, 21 a leptomeningitis, and three showed focal intrinsic lesions of the cord with a ring pattern of enhancement.
Of those with a dural-based lesion, two were cervical and two dorsal (Fig. 4a,b). In each case, the associated neurological symptoms were mild; each had sensory symptoms only although one had mild hyperreflexia in addition on examination. None had sphincter dysfunction or weakness.

Imaging features of cord and cauda equina involvement; pachymeningitis of the a cervical, b dorsal cord, and c the cauda equina; d ring enhancing lesion of the cervical cord and e thoracic lesion in spinal leptomeningitis; f leptomeningeal involvement of the cauda equine
Those with a ring pattern of enhancement presented with a subacute increasingly severe cord lesion with weakness and spasticity, sensory symptoms, and sphincter dysfunction. Each had prominent signs on examination and two were unable to walk. The MRI appearances were striking (Fig. 4d).
The remaining 21 showed leptomeningeal enhancement; the severity of the physical signs varied (Fig. 4e). All had sphincter dysfunction. Fifteen presented subacutely and six had a progressive disease course. Imaging showed lesions in the cervical region in seven cases and the dorsal region in eight, of whom five showed spread of enhancement into the cauda equina. Enhancement was seen in five of the cervical lesions, which extended for 2–6 segments, and in all dorsal lesions (4–12 segments) except for one; this patient was evaluated 10 years after he had developed weakness of he left leg and was left with wasting and spasticity. The dorsal cord exhibited atrophy with neither high signal nor enhancement. Of the patients with progressive disease only one showed leptomeningeal enhancement; high signal was seen in the cervical region (2–4 segments) in three and three had dorsal lesions (4–6 segments).
Ten patients (of whom five also had enhancement of the surface of the lower dorsal cord on imaging) had lesions of the cauda equina. There were two subtypes; a subacute severe distal weakness of L5 and S1 innervated muscles occurred in three. It was painful and asymmetric in one, in whom a response to steroids was observed, and in two painless and severe and did not recover. Imaging was normal in each case. The remaining seven presented with a painless sensory loss in the feet extending up subacutely, without weakness but with prominent sphincter disturbance. The neurological signs were of sensory loss and a reduction in or loss of the ankle reflexes. In each case, a prominent enhancement of the cauda equina was seen (Fig. 4c,f).
The median MRS was significantly higher in those with a leptomeningeal disorder [4 (2–5)] when compared with those with a pachymeningeal disorder [2 (2–3)], p < 0.02, and with those with a cauda equina disorder with sensory symptoms (2), p < 0.05.
Patients with a cauda equina syndrome had a greater median CSF protein [1.87 (0.7–8.35) g/l] than those with a leptomeningeal cord disorder [0.89 (0.4–3.33) g/l], p < 0.02, and those with a leptomeningeal cord disorder had a lower median CSF protein and cell count [7.5 (0–36)] than those with a pachymeningeal cord disorder [median protein 2.12 (0.4–3.99) g/l; median cell count 26.5 (0–150)], p < 0.02. Those with a progressive disease course had a significantly lower median CSF protein (0.55 (0.33–0.84 g/l) compared with those with a subacute leptomeningeal disorder, p < 0.02. The CSF white cell counts [median 5 (1–38), 5.5 (1–44) and 7 (0–53)] were not different when compared, p > 0.05.
Thoracic radiculopathy: eight patients, all women, have presented, at the onset of the systemic disease, with a burning numbness of the chest wall, which responds to steroids and, in four cases, has been relapsing and steroid—dependent for a time. One case was associated with a facial neuropathy and two had symptoms suggesting a more widespread sensory neuropathy, although nerve conduction studies were normal. The mean age was 44 (24–57) years. Imaging of the dorsal spinal cord was, in each case, normal. The spinal fluid, however, was active in all but one case; median CSF protein 0.65 (0.2–1.12) g/l, median CSF white cell count 3 (0–180). One had matched positive oligoclonal bands and the others were negative.
Isolated neurosarcoidosis
Six patients had biopsy-proven granulomatous disease whose histological appearances and clinical characteristics were in keeping with sarcoidosis; four showed a leptomeningeal infiltration, one of whom had a lesion of the dorsal spinal cord, and two had a pachymeningitis. No evidence for systemic disease was found using CT PET scans and blood investigations. The clinical features and MRI appearances were the same as those with an associated systemic disorder in each case. The median MRS was 4 (1–4) in those with isolated disease and 3 (1–5) in those with systemic disease, p = 0.15.
When compared with the patients with systemic disease and brain or spinal cord involvement, the CSF cell count was higher [median 35 (3–95) vs 4 (0–395), p < 0.02]; protein (median 1.05 vs 0.77 g/l, p = 0.27) and glucose (58 vs 47%, p = 0.28) levels were insignificantly different. The prevalence of oligoclonal bands was greater at 50% than in those with systemic disease at 23% (p = 0.001). Importantly, oligoclonal bands isolated to the CSF were seen only in those whose disease was also isolated to the nervous system; all those with systemic disease who had oligoclonal bands had matched bands.
Discussion
Accounts of neurological complications by sarcoidosis began to be published as soon as an understanding that it was a multisystem disease became recognised. In 1948, a review of all 118 cases published to that time noted that the facial and optic nerves were most commonly affected, but that all parts of the nervous system had been seen to be involved [10]. Later, hospital-based studies of cohorts with systemic sarcoidosis revealed a prevalence of neurological involvement in 3.5–7% of 285–807 patients [11,12,13,14]. In these series, cranial neuropathies were seen most commonly, in 47–64%, involvement of the central nervous system in 14–50%, the peripheral nervous system in 3–14%, and muscle in 0–21%.
More recently, a number of case series have been published, along with a multitude of single case reports. The largest series [13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29] have defined a prevalence of cranial neuropathy of around 50%, with peripheral nerve and muscle involvement in 10% and 5% respectively. Lesions of the brain, brain stem, hypothalamus, and spinal cord have been noted, accounting for 16–70% of cases. These reports are important but have not characterised the brain disorders and in particular have not been able to define the relationship of the neurological disorder with the systemic disease.
In this series, the prevalence of brain and spinal cord involvement was high owing undoubtedly to a referral bias. Neurological involvement most commonly arises at the time of the development of the systemic disease, and rarely, there is no evidence for other organ involvement at the time of the neurological presentation. Patients with cranial neuropathy when isolated without signs of more widespread parenchymal disease show normal imaging or only mild meningeal enhancement. The CSF although active in most cases shows modest changes only. Most do not go on to develop a more widespread form of neurosarcoidosis. Of 90 cases in which sarcoidosis has affected, the brain 75% had a leptomeningitis with inflammation of the subjacent cortex and white matter, of whom two-thirds presented with an encephalopathy and diencephalic dysfunction, in which seizures and hydrocephalus were common, and one-third had a predominate brain stem disorder, also associated with hydrocephalus. These cases showed a rapid disease onset which progressively deteriorated. 25% had a pachymeningitis in which there was less neurological impairment, although mass lesions were associated with seizures. The clinical course evolved more slowly than in the leptomeningeal subgroup.
The CSF was active in the majority of cases, raised CSF protein, and reduced CSF/blood glucose ratio correlated with disease severity, in particular hydrocephalus, but cell count did not. The presence of oligoclonal bands correlated with CSF protein, implying a more severe (or more long established) disease. Patients with systemic sarcoidosis had matched CSF/serum oligoclonal bands, and only those with isolated neurological granulomatous disease had unmatched bands.
Involvement of the spinal cord and cauda equina due to leptomeningitis was seen in 33 cases and to pachymeningitis in four, in whom the neurological impairments were seen to be less severe. Most presented with a subacute spasticity with sensory and sphincter disturbance [30,31,32,33]. There was a preponderance of males [30, 33]. Four patients presented with a progressive paraparesis in whom enhancement was not seen and atrophy of the cord noted. Half of those with lower dorsal lesions also had signs and MRI evidence for a cauda equina lesion, and some presented only with involvement of that region [34]. In these patients, sensory symptoms and a prominent sphincter dysfunction were seen. Clinical evidence for motor involvement was uncommon. A small subgroup presented with sensorimotor symptoms related to a spinal root lesion, in each case L5 or S1.
The MRI lesions were usually single and longitudinally extensive both within the cervical and dorsal cords [30,31,32,33, 35]. In these cases, enhancement was less intense than in corresponding brain lesions, and often only mild. The pattern of enhancement differs from other causes of a longitudinally extensive transverse myelitis or neuromyelitis optica spectrum disorder, in whom leptomeningeal enhancement is not seen [35].
Thoracic radiculopathy has been noted in the early reports; the CSF being active despite the normalcy of the imaging suggests that the disorder is a meningitis of the dorsal spinal cord, picking off the roots as they leave to form intercostal nerves. Other cases [36, 37] have shown prolongation of F-wave responses, compatible with this assumption.
Isolated granulomatous disease of the central nervous system
It has long been known that granulomatous disease of the nervous system may exist without evidence for systemic sarcoidosis; many appear to have a disease process indistinguishable from sarcoidosis. Some develop systemic features of the disease during investigation and follow-up [38]. Such cases need to be evaluated very carefully and this includes confirmation and a careful exclusion of alternative pathologies by a histological examination of affected neurological tissue; as noted above, other inflammatory, infectious, and neoplastic disorders may show striking similarities both on clinical and on imaging grounds. A series from two centres in the US 40 found a prevalence of 10% of their 91 cases, in which there was no difference in the clinical syndromes, the patient demographics or the CSF results. They noted a higher prevalence of meningeal involvement, owing to the concentration of brain disease in the isolated group [39].
This large series increases our understanding of the clinical features of the disease and, in particular, the relationship between neurological involvement and the systemic disease. The correlation between the imaging features and CSF findings will help those evaluating these patients, the majority of whom present during the first features of the systemic disease. A clear understanding of the systemic as well as the neurological manifestations will allow a correct diagnosis to be early and accurately, leading to more effective treatments and outcomes.
The differential diagnosis of sarcoidosis and its neurological complications is, however, complex, and it is extremely important to evaluate the disorder carefully in particular when the disease appears to be isolated to the nervous system, since there are many inflammatory, infective, and neoplastic disorders which affect the patients in the same way.
The data described herein provide important new information on the pathophysiology of neurosarcoidosis, in particular, the clinical and imaging characteristics of brain involvement, and their correlation with CSF constituents. This will allow a more rapid identification of those with more severe and destructive forms of the disease, enabling a more aggressive treatment pathway (Neurology, in press) and an improvement in outcome.
References
Chen ES, Moller DR (2011) Sarcoidosis—scientific progress and clinical challenges. Nat Rev Rheumatol 7:457–467
Rappl G, Pabst S, Riemann D et al (2011) Regulatory T cells with reduced repressor capacities are extensively amplified in pulmonary sarcoid lesions and sustain granuloma formation. Clin Immunol 140:71–83
Iannuzzi MC, Rybicki BA, Tierstein AS. Sarcoidosis (2007) N Engl J Med 357:2153–2165
Kidd DP, Beynon HLC (2003) Neurological complications of systemic sarcoidosis (review). Sarc Vasc Diffuse Lung Dis 20:85–94
Baughman RP, Tierstein AS, Judson MA et al (2001) Clinical characteristics of patients in a case control study of sarcoidosis. Am J Respir Crit Care Med 164:1885–1889
Judson MA, Baughman RP, Thompson BW et al (2003) Two year prognosis of sarcoidosis: the ACCESS experience. Sarc Vasc Diffuse Lung Dis 20:204–211
Judson MA, Costabel U, Drent M et al (2014) The WASOG sarcoidosis organ assessment instrument: an update of a previous clinical tool. Sarc Vasc Diffuse Lung Dis 31:19–27
Bonita R, Beaglehole R (1988) Modification of the Rankin scale: recovery of motor function after stroke. Stroke 19:1497–1500
Kidd DP, Burton BJ, Graham EM, Plant GT (2016) Optic neuropathy associated with systemic sarcoidosis. Neurol Neuroimmunol Neuroinflam 3:e270
Colover J (1948) Sarcoidosis with central nervous system involvement. Brain 71:451–475
Wiederholt WC, Siekert RG (1965) Neurological manifestations of sarcoidosis. Neurology 15:1147–1154
James DG, Sharma OP (1967) Neurological complications of sarcoidosis. Proc R Soc Med 60:1169–1170
Stern BJ, Krumholz A, Johns CJ, Scott P, Nissim J (1985) Sarcoidosis and its neurological manifestations. Arch Neurol 42:909–917
Chen RCY, McLeod JG (1987) Neurological complications of sarcoidosis. Clin Exp Neurol 26:99–112
Delaney P (1977) Neurologic manifestations in sarcoidosis: review of the literature, with a report of 23 cases. Ann Int Med 87:336–345
Pentland BJ, Mitchell D, Cull RE, Ford MJ (1985) Central nervous system sarcoidosis. Q J Med 56:457–465
Oksanen V (1986) Neurosarcoidosis: clinical presentations and course in 50 patients. Acta Neurol Scand 73:283–290
Chapelon C, Ziza JM, Piette JC et al (1990) Neurosarcoidosis: signs, course and treatment in 35 confirmed cases. Medicine 69:261–276
Sharma OP (1997) Neurosarcoidosis: a personal perspective based on the study of 37 patients. Chest 112:220–228
Pavese P, Brion JP, Chabre O, Fauconnier J, Pasquier B (1999) Les atteintes neurologiques de la sarcoïdose. Presse Med 28:168–172
Zajicek JP, Scolding NJ, Foster O et al (1999) Central nervous system sarcoidosis—diagnosis and management. QJM 92:103–117
Ferriby D, de Seze J, Stojkovic T et al (2001) Long-term follow-up of neurosarcoidosis. Neurology 57:927–929
Allen RK, Sellars RE, Sandstrom PA (2003) A prospective study of 32 patients with neurosarcoidosis. Sarc Vasc Diffuse Lung Dis 20:118–125
Kellinghaus C, Schilling M, Lüdemann P (2004) Neurosarcoidosis: clinical experience and diagnostic pitfalls. Eur Neurol 51:84–88
Spencer TS, Campellone JV, Maldonado I, Huang N, Usmani Q, Reginato AJ (2005) Clinical and magnetic resonance imaging manifestations of neurosarcoidosis. Semin Arthritis Rheum 34:649–661
Joseph FG, Scolding NJ (2009) Neurosarcoidosis: a study of 30 new cases. J Neurol Neurosurg Psychiatry 80:297–304
Pawate S, Moses H, Sriram S (2009) Presentations and outcomes of neurosarcoidosis: a study of 54 cases. Q J Med 102:449–460
Gascon-Bayarri J, Mana J, Martinez-Yelamos S, Murillo O, René R, Rubio F (2011) Neurosarcoidosis: report of 30 cases and a literature survey. Eur J Int Med 22:e125 – e132
Leonhard SE, Fritz D, Eftimov F, van der Kooi AJ, van de Beek D, Brouwer MC (2016) Neurosarcoidosis in a tertiary referral center: a cross-sectional cohort study. Medicine 95:e3277
Cohen-Aubart F, Galanaud D, Grabli D et al (2010) Spinal cord sarcoidosis: clinical and laboratory profile and outcome of 31 patients in a case–control study. Medicine (Baltimore) 89:133–140
Sakushima K, Yabe I, Nakano F et al (2011) Clinical features of spinal cord sarcoidosis: analysis of 17 neurosarcoidosis patients. J Neurol 258:2163–2167
Sohn M, Culver DA, Judson MA, Scott TF, Tavee J, Nozaki K (2014) Spinal cord neurosarcoidosis. Am J Med Sci 347:195–198
Durel CA, Marignier R, Maucort-Boulch D et al (2016) Clinical features and prognostic factors of spinal cord sarcoidosis: a multicenter observational study of 20 BIOPSY-PROVEN patients. J Neurol 263:981–990
Kaiboriboon K, Olsen TJ, Hayat GR (2005) Cauda equina and conus medullaris syndrome in sarcoidosis. Neurologist 11:179–183
Flanagan EP, Kaufmann TJ, Krecke KN et al (2016) Discriminating long myelitis of neuromyelitis optica from sarcoidosis. Ann Neurol 79:437–447
Miura S, Kusumoto M, Noda K et al (2007) Bell-shaped sensory impairments of all modalities in a neurosarcoidosis patient. Clin Neurol Neurosurg 109:794–798
Uzawa A, Kojima S, Yonezu T, Kanesaka T (2009) Truncal polyradiculopathy due to sarcoidosis. J Neurol Sci 281:108–109
Wegener S, Linnebank M, Martin R, Valavanis A, Weller M (2015) Clinically isolated neurosarcoidosis: a recommended diagnostic path. Eur Neurol 73:71–77
Nozaki K, Scott TF, Sohn M, Judson MA (2012) Isolated neurosarcoidosis: case series in 2 sarcoidosis centers. Neurologist 18:373–377
Author information
Authors and Affiliations
Contributions
Dr. Kidd was responsible for the study concept and design, acquisition of data, and analysis and interpretation. He was responsible for a critical revision of the manuscript for important intellectual content. He was the study supervisor.
Corresponding author
Ethics declarations
Conflicts of interest
Dr. Kidd receives royalties from Elsevier and Springer-Verlag. He reports no other disclosures.
Ethical standards
All patients consented to their clinical details being reported and the work has been conducted in compliance with the declaration of Helsinki.
Rights and permissions
About this article

Cite this article
Kidd, D.P. Sarcoidosis of the central nervous system: clinical features, imaging, and CSF results. J Neurol 265, 1906–1915 (2018). https://doi.org/10.1007/s00415-018-8928-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-018-8928-2