
Abstract
Patients with Wernicke’s encephalopathy (WE) often have unusual patterns of vertical nystagmus. Initially there is often a spontaneous upbeating nystagmus that may change to downbeat nystagmus with a change in the direction of gaze, convergence or with vestibular stimuli. Patients also often show a profound loss of the horizontal but not the vertical vestibulo-ocular reflex (VOR). Furthermore, the acute upbeat nystagmus may change to a chronic downbeat nystagmus. We present hypotheses for these features based on (1) the location of vertical gaze-holding networks near the area postrema of the dorsomedial medulla where the blood–brain barrier is located, which we suggest becomes compromised in WE, (2) the location of the vestibular nuclei in the brainstem, medially for the horizontal VOR, and laterally for the vertical VOR, (3) neuronal circuits differ in susceptibility to and in the ability to recover from thiamine deficiency, and (4) impaired processing of otolith information in WE, normally used to modulate translational vestibulo-ocular reflexes, leads to some of the characteristics of the spontaneous vertical nystagmus including the peculiar reversal in its direction with a change in gaze or convergence.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Wernicke’s encephalopathy (WE) is caused by thiamine (B1) deficiency and classically is characterized by confusion, ophthalmoplegia and ataxia. Patients with WE may also have spontaneous vertical nystagmus with two curious features [1]. First, the direction of their nystagmus in straight-ahead gaze changes from upbeat (UBN) in the acute stage to an enduring downbeat (DBN) in the chronic stage. Second, the vertical nystagmus in straight-ahead gaze may be markedly attenuated or reverse direction on convergence, on looking to the side, or in response to vestibular stimuli such as head shaking, applying a vibrator to the skull, or changing the position of the head relative to gravity. Furthermore, patients with acute WE commonly have a profoundly impaired horizontal vestibulo-ocular reflex (VOR) with relative sparing of the vertical VOR [2, 3]. We have previously presented hypotheses to account for these findings and related the pathophysiology to the location and anatomical relationships of the circuitry within the cerebellum and brainstem that ensure that gaze is held steady during fixation and rotation of the head [1]. Here, we will (1) present two other patients (one recent from the literature) whose patterns of nystagmus are compatible with these ideas, (2) restate and refine our hypotheses to explain the patterns of nystagmus and VOR dysfunction observed in WE, and (3) expand on the idea that spontaneous vertical nystagmus may arise not only from imbalance in semicircular canal-ocular pathways that mediate the rotational VOR but also in otolith-ocular pathways that mediate the translational VOR.
Patient reports
From two patients in our original publication [1] we had information from several examinations relevant to the issues raised above (patients 1 and 2, Table 1). Here we describe another patient seen by one of us (patient 3, Table 1) and also identified another report in the literature [4] (patient 4, Table 1). Even though the precise transition from UBN, to permanent DBN is not recorded in any of these four patients together they illustrate its variable timeline using the pattern of nystagmus during the switch of UBN to DBN with a change in horizontal or vertical gaze as a possible marker. We will briefly describe the two new cases (Table 1).
Patient 3
A 46-year old man was admitted for the evaluation of confusion. He had been on a self-imposed, severely restrictive diet consisting primarily of juices for 12 weeks but did not use alcohol. On examination, he was awake but not speaking. His neuroimaging showed findings of WE (Fig. 1). He also had a mildly elevated lactic acid and low serum folate; thiamine levels were not ordered. He was treated with 100 mg of oral thiamine and eventually referred for neuro-ophthalmology consultation. Three months after the initial presentation he had UBN in straight-ahead gaze (Video), the nystagmus changed to DBN in right, left and down gaze. Ten weeks later, he showed a subtle UBN (noted best with ophthalmoscopy and slit lamp examination) in straight-ahead gaze, and obvious DBN in lateral gaze; the patient was unavailable for follow-up appointments.

Axial DWI MRI (a) the arrow points to an area of increased signal in the left medial thalamus, adjacent to the third ventricle, symmetric signal changes are also present in the medial thalamus. b Axial FLAIR MRI of the pontomedullary junction. The arrow points to the medial vestibular and prepostus hypoglossi nuclei, this region includes the nucleus intercalatus and the nucleus pararaphales (a caudal nucleus of the paramedian tract (PMT) neurons). c Coronal T1 weighed MRI post contrast. The arrow point to an area of contrast enhancement in the left medial thalamus and provides evidence of a disruption of the blood brain barrier during the acute phase of WE
Patient 4 [4]
A 42-year old woman was evaluated for dizziness and gait disturbance. Nine months earlier she had been diagnosed with acute WE, which at that time improved when given thiamine. Her examination with her recurrence of WE showed truncal ataxia and UBN on straight-ahead and down gaze and DBN in right, up and left gaze. The MRI was normal and a low serum thiamine level (61.5 nmol/L, normal 66.5–220 nmol/L) compatible with a diagnosis of recurrent WE. No follow-up examination was reported.
As a group, all four patients with UBN/DBN (two with permanent transition from UBN to DBN and two with UBN on straight-ahead gaze and DBN on lateral gaze) received thiamine when WE was diagnosed. In addition, the first two patients were dependent on alcohol, the third was on a voluntary, self-imposed nutritional deprivation diet and the fourth had vomiting. We followed patients 1 and 2 with a permanent switch from UBN to DBN for 7 and 8 years, respectively, without change in their nystagmus. Patient 3 is the closest to knowing when the permanent switch of UBN to DBN occurs; his recent exam showed a subtle UBN in straight-ahead gaze and a prominent DBN on lateral gaze. These findings are opposite from those noted in Patient 1 in the acute stage of WE (Table 1). Patient 4, reported by Yoon [4], had a second episode of WE nine months following an initial attack. On this presentation she had UBN in straight-ahead gaze and down gaze, and DBN in right, left and up gaze [4].
Discussion
Key concepts underlying the pattern of vertical nystagmus and vertical VOR in WE
Here we review the three key ideas to explain the characteristics of vertical nystagmus and relative sparing of the vertical VOR in WE. (1) Neurons within the circuits that generate the VOR and hold gaze steady during fixation show both a selective vulnerability to thiamine deficiency, and a selective pattern of recovery of function upon treatment [1, 5, 6]. (2) The structures most susceptible to WE are close to the areas where the blood–brain barrier (BBB) is most porous or susceptible to damage, and in our case, especially the area postrema in the medial medulla on the floor of the fourth ventricle [7,8,9,10] (Fig. 1). (3) Some of the characteristics of the spontaneous vertical nystagmus in WE reflect abnormal processing of the information normally used to ensure the translational VOR (t-VOR) generates compensatory eye movements that are appropriate for the point of regard [11].
Pathogenesis of the initial upbeat nystagmus
We attribute the initial upbeat nystagmus to the involvement of midline structures associated with vertical gaze and most striking the nucleus intercalatus and/or the nucleus of Roller, each part of the perihypoglossal complex (Fig. 2). These structures when damaged, can lead to a downward slow-phase bias, and an UBN [1]. Nearby are the nuclei of the paramedian tracts including the nucleus pararaphales [5, 6] which when damaged alone leads to an upward slow-phase bias, and DBN. We suggest that early in the course of thiamine deficiency, the damage to the perihypoglossal nuclei predominates over the damage to the nuclei of the paramedian tracts, producing an initial downward slow-phase bias and an UBN. We next hypothesize that recovery of neurons within the nucleus intercalatus and the nucleus of Roller is more complete than that of neurons in the paramedian tract nuclei, leading to an enduring upward slow-phase bias and a permanent DBN. The DBN emerges because the damaged paramedian tract nuclei no longer provide an excitatory drive to the cerebellar flocculus, leading to a functionally lesioned flocculus. When the flocculus is lesioned or its function is impaired an upward slow-phase bias and a DBN appears [12] because the flocculus no longer inhibits the upward slow-phase pathways of the VOR within the brainstem (Fig. 2).

Hypothesis for pattern of vertical nystagmus with lesions of brainstem and cerebellum in WE. We suggest that neurons in the perihypoglossal complex (PHC, nucleus intercalatus and nucleus of Roller) and in the paramedian tract nuclei (PTN) are initially both affected, but with a net imbalance producing a downward bias and upbeat nystagmus (UBN). When the PHC nuclei recover, but the paramedian tract nuclei do not, an upward bias predominates, and a permanent downbeat nystagmus (DBN) evolves. Lacking excitation from the PTN neurons, the flocculus remains functionally lesioned, leading to disinhibition of the vestibular nuclei producing upward slow phases and DBN. Dashed arrow is inhibition and solid arrow is excitation
Why is the horizontal VOR more affected than the vertical VOR?
Next, we asked why is the horizontal VOR more involved than the vertical VOR. One possibility is related to the dichotomy that the inhibitory transmitter for the vertical VOR is gamma amino butyric acid (GABA) and that for the horizontal VOR is glycine. Perhaps thiamine deficiency affects the function of these neurotransmitters differently, leaving the GABA-mediated inhibitory effects in the vertical VOR relatively intact. An alternative explanation relates to the difference in the location of the horizontal and vertical VOR pathways in the medulla. We suggest that involvement of the NPH/MVN complex, which is located medially in the medulla and just under the area postrema, causes the bilateral, symmetric loss of the horizontal VOR and a commonly associated horizontal gaze-evoked nystagmus [2, 3]. On the other hand, the more lateral structures (superior and lateral vestibular nuclei) that mediate the vertical VOR are spared, leaving the vertical VOR relatively intact. Key to this hypothesis is a hypothesized central role for breakdown of the BBB in WE, especially near the area postrema, leaving the underlying structures susceptible to metabolites or toxins coming from the blood stream [7].
Why does upbeat nystagmus switch to downbeat nystagmus with change in gaze, convergence or vestibular stimulation?
How can both UBN and DBN be present at the same time in the same patient, with the direction of nystagmus reversing depending on the point of regard or the response to provocative vestibular stimuli? We propose these changes in the pattern of nystagmus reflect disturbances in the circuits that process information about linear acceleration from the otoliths, which is necessary to generate the correct ocular motor response for translational motion of the head [11]. Recall that the direction of the response to head translation depends on the point of regard both across the visual field and in depth. In other words, the translational VOR (t-VOR) must be modulated by orbital position and the angle of vergence to generate compensatory eye movements. When one is translating forward or backward and looking at a target to the right or left, a horizontal eye movement is called for; if looking up or down, a vertical eye movement is called for, and if looking straight ahead, a convergence or divergence eye movement is called for.
Areas in the brainstem that receive information from the otoliths in the labyrinth, and may be important for the calculations necessary for the t-VOR, are also located medially in the medulla, including the medial portions of the medial and inferior vestibular nuclei [13, 14]. These nuclei also project to the cerebellar nodulus, a structure involved with calculating the translational components of motion of the head [15]. Disruption of these projections potentially explains the changes in the direction of the vertical nystagmus in WE patients. An important caveat is that abnormal processing of information in the circuits that mediate the velocity-storage mechanisms that modulate nystagmus relative to the gravity vector, which include the medial portions of the vestibular complex and the cerebellar nodulus, could also explain the change in direction of nystagmus in response to vestibular stimuli.
Can spontaneous vertical nystagmus arise from disorders in otolith-ocular pathways that mediate the t-VOR?
These speculations about the t-VOR in WE relate to the question of what kinds of imbalance can create a spontaneous vertical nystagmus. Most hypotheses for DBN suggest a tone imbalance in vertical semicircular canal pathways or an up-down asymmetry in the function of the neural “integrators” responsible for generating commands that hold vertical gaze steady. The cerebellum, and especially the flocculus/paraflocculus region is commonly invoked in the pathogenesis of spontaneous DBN because lesions there can produce it [12]. Furthermore, the cerebellar flocculus has inhibitory projections to anterior but not to posterior semicircular canal pathways in the brainstem so one can envision that a loss of these inhibitory projections could lead to an upward slow-phase bias and a DBN. An alternative explanation for DBN is that it relates not just to projections to canal pathways but also to other neurons in the brainstem that generate pursuit and gaze-holding commands [16]. But certain features of vertical nystagmus in WE also point to an imbalance in otolith-ocular pathways that mediate the t-VOR, including the curious effects of horizontal eye position, convergence and vestibular stimulation on the direction of a spontaneous vertical nystagmus in WE. And lesions in the posterior vermis including the cerebellar nodulus, which receives otolith projections, can also lead to a spontaneous DBN with abnormalities in the translational VOR [17, 18].
How can we distinguish an imbalance in the rotational VOR (r-VOR) versus the translational VOR (t-VOR) pathways?
Hypothetically, a tone imbalance in pathways that mediate the up–down (bob) t-VOR could also produce DBN, but its properties should be different from a spontaneous nystagmus arising from an imbalance in pathways that mediate the vertical r-VOR. The r-VOR functions to stabilize images on the entire retina and generates slow phases that are referenced to axes of rotation that are parallel to the orientation of the semicircular canals in the labyrinth; i.e., a head-fixed reference frame. The small exception to this rule is that compensation is also necessary for the inevitable small amount of translation of one or both orbits that accompanies any rotation of the head except for rotation around the interaural axis. In contrast, the t-VOR functions to stabilize images on the foveae of both eyes. Consider forward translation, as occurs during walking or running, the t-VOR must generate a different response for images that are located straight ahead (convergence), to the side (horizontal) or above or below eye level (vertical) [11]. In other words, the translational VOR must generate slow phases that are referenced to horizontal and vertical axes that move with the globe; i.e., an eye-fixed reference frame. Because of the inevitable increase in motion of images that are relatively close to the head when it is translating, the t-VOR must also modulate its output based on the distance of the orbits from the visual target.
With these considerations in mind, if DBN is in an eye-fixed reference, from a tone imbalance in t-VOR pathways, the nystagmus will appear vertical in all horizontal eye positions (with the observer remaining aligned with the patient’s line of sight) (Fig. 3b, Video). On the other hand, if DBN is in a head-fixed reference, from a tone imbalance in r-VOR pathways, the nystagmus should show an additional torsional component (with the observer remaining aligned with the patient’s line of sight) after the patient moves the eyes to a horizontal eccentric eye position (Fig. 3c). In other words, if there is a spontaneous vertical (or horizontal) nystagmus in the straight-ahead position, one can detect whether its reference frame is eye or head-fixed by examining the nystagmus when the patient looks in a direction orthogonal to the spontaneous nystagmus (Fig. 3b, c). Clearly this is sometimes difficult to see on simple bedside examination and quantitative recordings of the movements of the eyes around all three axes of rotation might be necessary to make this distinction.

Effect of direction of gaze on the waveform of nystagmus. a DBN in straight-ahead gaze. b Pattern of nystagmus with an imbalance in t-VOR or possibly pursuit pathways. c Pattern of nystagmus with an imbalance in r-VOR or possibly rotational OKN pathways. The text at the bottom of the figure defines the effect of eye position on an imbalance in the ocular motor integrators for holding gaze. See text for explanation of Listing’s law
Other potential causes of vertical nystagmus in WE
The analysis of DBN in WE is further complicated by the role of the brainstem and cerebellum in generating gaze-holding commands. The cerebellum projects to the brainstem neural integrator, which is a neural network located largely in the medulla for horizontal movements and in the midbrain for vertical movements [19]. The neural integrator takes velocity commands from the conjugate eye movement systems and creates a position command to hold the eyes steady after every movement. Lesions in the flocculus–paraflocculus complex, or the brainstem circuits that project to it, interfere with the function of this integrator. The integrator may become impaired or “leaky”, causing a gaze-evoked nystagmus with velocity-decreasing slow-phase waveforms, or become “unstable”, in which case the slow-phase waveforms are velocity increasing [19, 20]. These effects on the neural integrator can be seen by looking at how the spontaneous nystagmus is affected by changes in eye position along the same axis as the nystagmus, e.g., up and down for a vertical nystagmus (Fig. 3, bottom). If the integrator is impaired (“leaky”), slow-phase velocity increases as one looks in the direction of the quick phase. If the integrator is unstable, slow-phase velocity increases as one looks in the direction of the slow phase.
A pursuit imbalance has also been invoked to explain some of the properties of DBN, as the Purkinje cells of the flocculus and paraflocculus preferentially discharge during upward movements [16]. Hence their loss would lead to an upward slow-phase bias and a DBN. Hypothetically, an imbalance in pursuit could produce a nystagmus in eye coordinates (as the t-VOR) but there is also evidence that pursuit is developed on a head-coordinate frame [21]. Likewise, optokinetic nystagmus can be organized in rotational (head) or translational (eye) coordinates, and might be another source of a bias leading to a vertical nystagmus [22]. Another confounding factor is the change in torsion associated with Listing’s law. Listing’s law dictates how the torsional orientation of the globe must change as the eye rotates to any given eccentric eye position since the torsional orientation of the globe is fixed no matter from what direction the eye had come (Donders’ law) [23]. In other words, any torsion associated with a horizontal or vertical rotation of the globe that was driven by rotation or translation of the head would have to be distinguished from torsion superimposed because of Listing’s and Donders’ laws. Finally, inherent slow-phase biases in the vertical gaze holding networks have also been invoked to counteract the pervasive downward pull of gravity that constantly affects us as we move around in a natural environment [24]. One can see that spontaneous vertical nystagmus may have many factors contributing to its genesis. A first step in understanding its pathogenesis would be to analyze the slow phases of nystagmus of both eyes at different orbital positions and viewing distances, comparing their axis of eye rotation, slow-phase velocity and degree of conjugacy.
WE and the changing directions of vertical nystagmus
How can we relate these ideas about the changing patterns of vertical nystagmus to WE? Recall that at close viewing or on horizontal or vertical eccentric gaze, spontaneous vertical nystagmus may change direction in WE. Likewise, various vestibular stimuli including head shaking, positional testing and vibration, may lead to a change in the intensity or reverse the direction of a spontaneous vertical nystagmus. As WE is a complicated disorder, affecting many different circuits that could influence vertical gaze-holding, some of the unusual characteristics of its nystagmus could reflect bungled attempts by the brain to adjust otolith-ocular responses for head orientation, orbital position and viewing distance, based on the incorrect assumption the head is translating.
Conclusions
-
1.
We suggest that midline structures in the dorsal medulla are unusually susceptible to thiamine deficiency because of their proximity to the blood–brain barrier underneath the area postrema. In WE the blood–brain barrier, perhaps impaired, may be a conduit by which toxins reach these structures.
-
2.
The initial vertical nystagmus in WE could arise from involvement of midline structures in the dorsal medulla that are associated with vertical gaze, with greatest effect on the nucleus intercalatus and/or the nucleus of Roller, each part of the perihypoglossal complex. These structures when damaged lead to an upbeat nystagmus. We further suggest the perihypoglossal complex recovers but another involved midline structure, the paramedian tract nuclei that when lesioned leads to a downbeat nystagmus, does not, leaving a net bias causing enduring downbeat nystagmus. A caveat is that in WE there may be damage to other structures in the brainstem or cerebellum that could contribute to vertical gaze dysfunction.
-
3.
Patients also often show a profound loss of the horizontal but not the vertical VOR, probably because the vestibular nuclei that subserve the horizontal VOR are located medially underneath the area postrema, while the vestibular nuclei that subserve the vertical VOR are located laterally, away from the area postrema. A caveat is that different inhibitory transmitters (glycine and GABA, respectively) are used by these reflexes, and thiamine deficiency might selectively involve one more than the other.
-
4.
Patients with WE often show a horizontal gaze-evoked nystagmus which could reflect lesions of the medial vestibular nuclei and the adjacent nucleus prepositus hyoglossi.
-
5.
On a single visit the nystagmus may change from upbeat to downbeat nystagmus with a change in the direction of gaze or with vestibular stimuli. We suggest that impaired processing of otolith information, normally used to modulate translational vestibulo-ocular reflexes, is the cause, and in WE, leads to some of the characteristics of the spontaneous vertical nystagmus including the peculiar reversal in its direction with the direction of gaze and convergence.
References
Kattah JC, Tehrani AS, du Lac S, Newman-Toker DE, Zee DS (2018) Conversion of upbeat to downbeat nystagmus in Wernicke encephalopathy. Neurology 91:790–796
Choi KD, Oh SY, Kim HJ, Kim JS (2007) The vestibulo-ocular reflexes during head impulse in Wernicke's encephalopathy. J Neurol Neurosurg Psychiatry 78:1161–1162
Kattah JC, Guede C, Hassanzadeh B (2018) The medial vestibular nuclei, a vulnerable target in thiamine deficiency. J Neurol 265:213–215
Yoon W (2017) Changing vertical nystagmus in the opposite direction: is the transition from upbeat to downbeat nystagmus a diagnostic clue for Wernicke's encephalopathy? J Neurol Dis 5:370–374
Buttner-Ennever JA, Horn AK (1996) Pathways from cell groups of the paramedian tracts to the floccular region. Ann N Y Acad Sci 781:532–540
Nakamagoe K, Iwamoto Y, Yoshida K (2000) Evidence for brainstem structures participating in oculomotor integration. Science 288:857–859
McEntee WJ (1997) Wernicke's encephalopathy: an excitotoxicity hypothesis. Metab Brain Dis 12:183–192
Harata N, Iwasaki Y (1995) Evidence for early blood-brain barrier breakdown in experimental thiamine deficiency in the mouse. Metab Brain Dis 10:159–174
Nardone R, Holler Y, Storti M et al. (2013) Thiamine deficiency induced neurochemical, neuroanatomical, and neuropsychological alterations: a reappraisal. Sci World J 2013:309143. https://doi.org/10.1155/2013/309143
Jankowska-Kulawy A, Bielarczyk H, Pawelczyk T, Wroblewska M, Szutowicz A (2010) Acetyl-CoA and acetylcholine metabolism in nerve terminal compartment of thiamine deficient rat brain. J Neurochem 115:333–342
Patel VR, Zee DS (2015) The cerebellum in eye movement control: nystagmus, coordinate frames and disconjugacy. Eye (Lond) 29:191–195
Zee DS, Yamazaki A, Butler PH, Gucer G (1981) Effects of ablation of flocculus and paraflocculus of eye movements in primate. J Neurophysiol 46:878–899
Stein B, Carpente MB (1967) Central projections of portions of the vestibular ganglia innervating specific parts of the labyrinth in the rhesus monkey. Am J Anat 120:281–318
Buttner-Ennever JA (1999) A review of otolith pathways to brainstem and cerebellum. Ann N Y Acad Sci 871:51–64
Laurens J, Meng H, Angelaki DE (2013) Computation of linear acceleration through an internal model in the macaque cerebellum. Nat Neurosci 16:1701–1708
Marti S, Straumann D, Buttner U, Glasauer S (2008) A model-based theory on the origin of downbeat nystagmus. Exp Brain Res 188:613–631
Walker MF, Tian J, Shan X, Tamargo RJ, Ying H, Zee DS (2008) Lesions of the cerebellar nodulus and uvula impair downward pursuit. J Neurophysiol 100:1813–1823
Walker MF, Tian J, Shan X, Ying H, Tamargo RJ, Zee DS (2009) Enhancement of the bias component of downbeat nystagmus after lesions of the nodulus and uvula. Ann N Y Acad Sci 1164:482–485
Leigh J, Zee, DS (2015) The neurology of eye movements. Oxford University Press, New York
Helmchen C, Glasauer S, Sprenger A (2013) Inverse eye position dependency of downbeat nystagmus in midline medullary lesion. J Neurol 260:2908–2910
FitzGibbon EJ, Calvert PC, Dieterich M, Brandt T, Zee DS (1996) Torsional nystagmus during vertical pursuit. J Neurophthalmol 16:79–90
Tian J, Zee DS, Walker MF (2007) Rotational and translational optokinetic nystagmus have different kinematics. Vis Res 47:1003–1010
Straumann D, Zee DS, Solomon D (2000) Three-dimensional kinematics of ocular drift in humans with cerebellar atrophy. J Neurophysiol 83:1125–1140
Pierrot-Deseilligny C (2005) Vertical nystagmus: clinical facts and hypotheses. Brain 128:1237–1246
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that they have no competing interests.
Ethical approval
All experiments followed the tenets of the Declaration of Helsinki and this study was approved by the Institutional Review Board.
Informed consent
The patient gave written consent to use this video for publication.
Additional information
This manuscript is part of a supplement sponsored by the German Federal Ministry of Education and Research within the funding initiative for integrated research and treatment centers.
Electronic supplementary material
Below is the link to the electronic supplementary material.
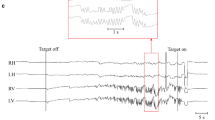
Baseline and Follow-up Video recording of case 3 (table 1). Baseline: The first video section was obtained three months after the diagnosis of WE, he has a robust UBN in straight-ahead gaze. In lateral gaze note DBN mixed with a horizontal component, right beat in right gaze and left beat in left gaze. Follow-up: The second Video section was obtained 10 weeks after the first recording: No nystagmus is present in straight-ahead gaze though with direct ophthalmoscopy and slit lamp examination there was a small-amplitude UBN. In lateral gaze he has a robust DBN (MP4 208212 kb)
Rights and permissions
About this article

Cite this article
Kattah, J.C., McClelland, C. & Zee, D.S. Vertical nystagmus in Wernicke’s encephalopathy: pathogenesis and role of central processing of information from the otoliths. J Neurol 266 (Suppl 1), 139–145 (2019). https://doi.org/10.1007/s00415-019-09326-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-019-09326-9