
Abstract
The pathomechanism of downbeat nystagmus (DBN), an ocular motor sign typical for vestibulo-cerebellar lesions, remains unclear. Previous hypotheses conjectured various deficits such as an imbalance of central vertical vestibular or smooth pursuit pathways to be causative for the generation of spontaneous upward drift. However, none of the previous theories explains the full range of ocular motor deficits associated with DBN, i.e., impaired vertical smooth pursuit (SP), gaze evoked nystagmus, and gravity dependence of the upward drift. We propose a new hypothesis, which explains the ocular motor signs of DBN by damage of the inhibitory vertical gaze-velocity sensitive Purkinje cells (PCs) in the cerebellar flocculus (FL). These PCs show spontaneous activity and a physiological asymmetry in that most of them exhibit downward on-directions. Accordingly, a loss of vertical floccular PCs will lead to disinhibition of their brainstem target neurons and, consequently, to spontaneous upward drift, i.e., DBN. Since the FL is involved in generation and control of SP and gaze holding, a single lesion, e.g., damage to vertical floccular PCs, may also explain the associated ocular motor deficits. To test our hypothesis, we developed a computational model of vertical eye movements based on known ocular motor anatomy and physiology, which illustrates how cortical, cerebellar, and brainstem regions interact to generate the range of vertical eye movements seen in healthy subjects. Model simulation of the effect of extensive loss of floccular PCs resulted in ocular motor features typically associated with cerebellar DBN: (1) spontaneous upward drift due to decreased spontaneous PC activity, (2) gaze evoked nystagmus corresponding to failure of the cerebellar loop supporting neural integrator function, (3) asymmetric vertical SP deficit due to low gain and asymmetric attenuation of PC firing, and (4) gravity-dependence of DBN caused by an interaction of otolith-ocular pathways with impaired neural integrator function.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Downbeat nystagmus (DBN) is an acquired persisting nystagmus which, if present during attempted fixation, indicates CNS dysfunction (Leigh and Zee 2006). The retinal slippage caused by the upward ocular drift of DBN leads to disabling oscillopsia and blurred vision. Most frequently, DBN occurs with cerebellar degeneration (about 20% of all cases, Wagner et al. 2007), especially if it affects the vestibulo-cerebellum. However, approximately 40% of the patients present with idiopathic DBN, i.e., an underlying pathology cannot be found (Wagner et al. 2007). Typically, DBN depends on gaze direction obeying Alexander’s law, i.e., vertical drift velocity increases with gaze in direction of the fast phase, which here is downward. Other ocular motor signs typically associated with DBN include impairment of smooth pursuit (SP) eye movements and combined eye- and head-tracking [deficient visual cancellation of the vestibulo-ocular reflex (VOR)].
The upward directed drift in patients with cerebellar DBN consists of two components (Straumann et al. 2000; Glasauer et al. 2003): vertical gaze-evoked drift attributed to leakiness of the vertical gaze-holding mechanism, and (II) an upward directed spontaneous or bias drift present with gaze straight ahead. The pathomechanism of this spontaneous upward drift is still debated. Most hypotheses assume a tone imbalance within vertical VOR pathways (Baloh and Spooner 1981; Chambers et al. 1983; Gresty et al. 1986; Halmagyi et al. 1983; Böhmer and Straumann 1998; Pierrot-Deseilligny and Milea 2005). Other theories conjecture an asymmetric impairment of vertical SP pathways (Zee et al. 1974) or a dissociation between internal coordinate systems for vertical saccade generation and gaze holding (Glasauer et al. 2003). However, none of these previous hypotheses on the origin of DBN (1) covers all associated ocular motor signs without assuming at least two different mechanisms, and (2) can plausibly explain why the drift is directed upward.Footnote 1 The purpose of the present paper is to resolve these open questions and to present a unifying theory on the mechanism of DBN, based on known anatomy and physiology of the primate ocular motor system as well as on our own experimental findings in patients.
Floccular Purkinje cells (PCs), the sole output of the cerebellar FL,Footnote 2 play the leading part in our hypothesis. These cells send inhibitory projections to the vestibular nuclei (VN) in the brainstem. Floccular PCs modulate their activity with eye movements, predominantly gaze velocity, and exhibit distinct, either roughly horizontal or roughly vertical directional sensitivities (Miles et al. 1980; Chubb and Fuchs 1982; Partsalis et al. 1995). Remarkably, 90% of vertical floccular PCs are tuned for downward on-directions, i.e., they increase their firing rate to eye movements evoked by downward visual motion. Thus, downward pursuit is caused by a net increase of the population firing rate above resting level, while upward pursuit requires a decrease in activity below resting level. There are, however, virtually no PCs with upward on-directions (Partsalis et al. 1995; Krauzlis and Lisberger 1996). This substantial asymmetry in the distribution of on-directions of vertical gaze-velocity PCs constitutes the core of our hypothesis: The spontaneous upward drift in cerebellar disease may result from the inherent asymmetric distribution of on-directions of vertical PCs. We further propose that the ocular motor features commonly associated with DBN, e.g., gaze and gravity dependence of vertical ocular drift and asymmetrically impaired vertical SP eye movements, may also be explained on the basis of an extensive loss of vertical floccular PCs.
Based on established anatomical and physiological facts, including the asymmetric distribution on-directions of vertical floccular PCs, we developed a computational model of vertical eye movements to formalize and test our hypothesis. Our principal demand to the model was that (1) it could simulate various types of eye movements under intact conditions, and (2) it includes structures and pathways to an extent necessary for simulation of lesions. The theoretical framework and the different components of the model are detailed in the “Methods” section. We present examples of patient data illustrating the various ocular motor deficits typically found with DBN. Computer simulations of intact eye movements and in the presence of cerebellar lesions are compared with the experimental data of DBN patients. For simulation of cerebellar disease, the following assumptions were made: An extensive loss of vertical floccular PCs, as it occurs with cerebellar degeneration, will lead to a lowering of the total resting discharge rate, as well as a decrease in overall gain and modulation amplitude of these neurons. We finally discuss the relevance of the model for the pathomechanism of DBN in the context of previous theories. A preliminary version of the computer model was published earlier in a proceeding (Marti et al. 2005b).
Methods
To test our hypothesis that a lesion to floccular PCs results in spontaneous upward drift and, at the same time, causes the various ocular motor signs typically associated with DBN, we developed a computational model of normal vertical eye movements. Using this model, we then simulated cerebellar lesions. The proposed model (Fig. 1) for vertical eye movements is based on a previous proposal of how the cerebellum contributes to gaze holding (Glasauer 2003). The core assumption of this model is that the cerebellar FL is an integral part of vertical (and horizontal) gaze holding (as found experimentally, e.g., Zee et al. 1981), which in our model is achieved by an eye-velocity feedback loop including an internal model of the eye plant to minimize errors between desired eye velocity (zero in case of fixation) and currently estimated eye velocity. To sufficiently explain ocular motor findings in cerebellar DBN, the model has to include structures and pathways (1) subserving gaze holding, (2) of the angular VOR, (3) mediating otolith signals, (4) for vertical smooth pursuit eye movements, and (5) a saccadic burst generator for vertical saccades. Figure 1 shows the complete model structure together with simulated responses (insets) at the level of various model elements for VOR cancellation (head and target move together).

Model outline. Boxes show responses of the respective model elements to vestibulo-ocular reflex (VOR) cancellation during sinusoidal pitch head rotation (target rotates with the head, leftmost boxes). Note that even though the eye remains stable within the head (rightmost box), floccular Purkinje cells are strongly modulated (central box), since the VOR is suppressed by pursuit commands. Model structure: via a negative feedback loop (bold connections), the cerebellar flocculus (‘cerebellum’) augments the time constant of the inherently leaky brainstem integrator (‘brainstem integrator’, time constant 5 s). The cerebellum receives an efference copy of the outgoing motor command via the paramedian tract neurons (PMT) and/or Y-group (‘motor command efference copy (via PMT)’) and estimates the actual eye velocity on behalf of an internal model of the eye plant (see below). Head position (‘head position in space’) is processed by the otoliths (‘otoliths’) and by the semicircular canals (‘SCC’) yielding angular head velocity. Target position (‘target position in space’) is converted to retinal coordinates and processed by the visual system (‘retina, V1, MT’) to yield retinal target position and retinal slip velocity. From retinal slip, a target velocity estimate (‘estimate of target velocity in space’) is supposed to be computed in middle superior temporal area (MST) using an efference copy feedback about gaze velocity (‘estimated gaze velocity’) conveyed from floccular Purkinje cells (‘Purkinje cells to deep nuclei’) via thalamic relay neurons (‘delayed gaze velocity via thalamus’). Canal afferents are sent to the cerebellum, to the brainstem velocity-position integrator (interstitial nucleus of Cajal, ‘INC’), and via the direct pathway (‘direct pathway (VOR + pursuit)’) to the ocular motor neurons (‘OMN’) and the eye plant (‘eye plant’). The cerebellum contains an internal model of the eye plant (‘forward model of eye plant’) in order to compute an estimate of eye velocity from the motor command, which is compared to the desired eye velocity composed of target (‘estimate of target velocity in space’) and head velocity estimates. This error signal (‘eye velocity error’) is supposed to be sent back to the brainstem via the floccular Purkinje cells (‘FL-Purkinje cells to vestibular nuclei’). The transfer function of this population of Purkinje cells was changed for simulation of downbeat nystagmus (DBN). It conveys pursuit commands and augments the time constant of the neural integrator. Saccades and nystagmus quick phases are generated via a simplified burst generator (‘saccadic burst generator’) that is driven by retinal error (‘retinal error for saccadic eye movements’). For model equations, see Appendix
The various subsystems of the model and their interaction are based on the concept of internal estimation and predictive models (see, e.g., review by Davidson and Wolpert 2005). For example, the model proposes that the cerebellum uses an internal forward model of the eye plant to estimate current eye velocity, and that this estimate is combined with vestibular input to yield an estimate of gaze velocity. The latter is supposed to be used by cortical visual areas to estimate target velocity in space, which, in turn, serves as drive for the smooth pursuit system.
Model simulations were performed using Matlab/Simulink version 7.1 (The MathWorks, Natick, MA, USA), using the same set of parameters for all simulations except for the change to the FL PC population described below (see Appendix for mathematical formulation). The Simulink model code is available online (Supplementary material) and on request from one of the authors (SG). The differential equations governing the model and all parameters are given in the Appendix.
Visual processing
The visual processing by the retina, the primary visual cortex, and higher regions (Box ‘Retina, V1, MT’ in Fig. 1) is simplified to generate only two signals from target position in eye coordinates, a retinal error signal for saccade generation, and a retinal slip signal as input to the pursuit system.
Gaze holding
For all types of eye movements, the final common pathway includes the so-called velocity-to-position integrator in the brainstem, which helps to generate appropriate motor commands for slow eye movements and to hold the eyes steady in an eccentric gaze position (Robinson et al. 1984; Cannon and Robinson 1987). Lesions of the neural integrator typically cause gaze-evoked nystagmus. The interstitial nucleus of Cajal (INC) (Crawford et al. 1991) in the midbrain and cell groups of the paramedian tract (PMT) (Büttner-Ennever et al. 1989; Büttner-Ennever and Horn 1996), which project to the FL, are involved in vertical gaze-holding.
The performance of the brainstem integrator is supported by feedback pathways through the FL, which augment the time constant of the inherently leakyFootnote 3 brainstem integrator (Zee et al. 1981). In our model, these pathways consist in a negative velocity feedback loop (bold pathway in Fig. 1) that minimizes errors between desired and currently estimated eye velocity. In previous models, the important role of the FL for the gaze-holding mechanism was explained by a positive position feedback from the cerebellum to the brainstem integrator (Zee et al. 1980). However, since floccular PC output is solely inhibitory, the assumption of a negative feedback may be more plausible (Glasauer 2003). Furthermore, while positive feedback loops tend to become unstable and require exact tuning of the feedback gain, the negative velocity feedback loop proposed here is inherently stable (see Appendix). In the present model, the feedback gain implemented at the level of the PCs (labeled ‘FL Purkinje cells’ in Fig. 1) controls the effective time constant of the integrator. The cerebellum, receiving an efference copy of the outgoing motor command (see also Hirata and Highstein 2001; Green et al. 2007) via the PMT and/or Y, estimates the current eye velocity on behalf of an internal forward model of the eye plant, similar to the proposal of Porrill et al. (2004). By comparing this central estimate of current eye velocity with desired eye velocity, the latter being given by the SCCs for VOR and by middle superior temporal (MST) for pursuit, the cerebellum obtains an error signal that is amplified and then supplied back to the brainstem integrator via the VN. During pure gaze holding, the desired eye velocity is zero, and the model reduces to a negative velocity feedback loop around the brainstem neural integrator, which, in this scheme, is more appropriately conceived as a neural controller minimizing velocity errors. Depending on the gain of this negative feedback loop implemented at the level of the lumped PC population (gain ten for simulation of healthy subjects), the overall time constant of gaze holding will be augmented (for a gain of ten from a brainstem time constant of 5 s to about 50 s).
Smooth pursuit
Smooth pursuit (SP) eye movements stabilize the image of a slowly moving target on the retina. The classic pursuit pathways (Fig. 2a) connect temporal (middle temporal, MT; middle superior temporal, MST) and frontal cortical areas (frontal eye fields, FEF) with pursuit related cerebellar regions via the pontine nuclei (for review: Krauzlis 2004; Fukushima 2003).

a Vertical smooth pursuit (SP) pathways. Solid bold arrow lines cortico-ponto-cerebellar pathway from middle temporal (MT) and middle superior temporal area (MST) to the dorsolateral pontine nuclei (DLPN) and the floccular lobe (FL). Motor commands for pursuit eye movements reach the ocular motor neurons via projections of the FL to the VN and Y-group. Gray thin arrow lines parallel cortico-ponto-cerebellar pathway from the frontal eye field (FEF) via the nucleus reticularis tegmentum pontis (NRTP) to the cerebellar dorsal vermis (DV), which projects to the fastigial nucleus (FN) and from there to the VN. A feedback pathway via deep cerebellar nuclei (DCN) and the thalamus (thalamic pathway) connects the FL with area MST (gray dashed lines). The VN-PMT-FL loop and the brainstem integrator are shown in b for clarity. b Vertical vestibulo-ocular reflec (VOR) pathways. Direct pathway (solid lines): a three-neuron arc runs from the vertical semicircular canals (SCC) to the vestibular nuclei (VN) and from there to the vertical ocular motor neurons (OMN). Indirect pathways: a polysynaptic pathway (dashed-dotted lines) connects the SCC, the VN and the brainstem integrator for vertical gaze holding (Nucleus interstitialis Cajal, INC) with the OMN. Parallel circuitries (dashed lines), which are important for VOR adaptation and gaze holding, link the VN, the paramedian tract neurons (PMT) and the floccular lobe (FL)
Smooth pursuit is a negative feedback loop in itself with the central nervous system acting as controller to minimize retinal slip. As recognized early on (Yasui and Young 1975; Robinson et al. 1986), in such a system, several problems arise: a simple servoing mechanism would need a high feedback gain to achieve near-zero tracking errors (or a pursuit gain close to unity). However, a high feedback gain together with the inevitable neural delays in the loop (the retina alone is assumed to delay the visual signal by 20–40 ms) would cause the system to become instable. As suggested by Yasui and Young (1975), instead of using highly amplified retinal slip as drive for pursuit, target velocity may be internally reconstructed by using an efference copy of the motor output, and utilize this estimate of target velocity as drive. Robinson et al. (1986) showed that such a scheme also allows to overcome the delay problem.
In our framework, the estimated target velocity (in Fig. 1: labeled output of MST) is used as desired gaze velocity, and is exactly what is required as input to the cerebellum. Consequently, we assume that the FL receives a cortical estimate of the actual target velocity in space, i.e., desired pursuit gaze velocity, from area MST via the pontine nuclei (Fukushima 2003; Krauzlis 2004; Thier and Ilg 2005). This signal is computed from retinal slip supplied by MT and an efference copy of gaze velocity relayed through the deep cerebellar nuclei and the thalamus (Tanaka 2005). In our model, as outlined above, the cerebellum computes an estimate of current eye velocity and, by comparing it with desired pursuit velocity, generates an error signal. During pursuit, this error signal represents the motor command needed to drive SP eye movements. The motor command is then passed to the OMN via cerebellar projections to target neurons in the VN and Y. Thus, the FL provides the main SP drive, thereby continuously adjusting outgoing motor commands to improve smooth pursuit performance. The model proposes two pursuit-related cerebellar output signals serving different purposes, basically equivalent to the hypothesis of Fukushima (2003): one, related to eye velocity and eye position in the orbit, projecting to the VN, and the other, related to gaze velocity (eye in space), projecting as estimate of gaze velocity in space via the thalamus to the cortex. To help solving the problem of the substantial delay of the retinal slip signal (about 100 ms, included in the model), the gaze velocity feedback signal projecting to MST was equivalently delayed to yield an accurate estimate of target velocity in space when fused with retinal slip in MST, thus effectively canceling the delay within the inner feedback loop. Our model approach to SP thus differs slightly from previous models, which propose a combination of retinal slip velocity and acceleration as input to the cerebellum (Krauzlis and Lisberger 1994), but incorporates an internal feedback loop similar to others (cf. Tabata et al. 2002 for review), except that feedback from the brainstem is conveyed via the cerebellum and thalamus to area MST.
Saccades
Modeling of saccades, even though not affected in DBN patients, was necessary to simulate nystagmus quick phases, saccadic gaze shifts, and the interaction of the saccadic burst command with the other ocular motor subsystems. Since the cortical processing of retinal error and the spatiotemporal transformation from the superior colliculus to the brainstem burst generator was not the focus of the current model, we used a simplified version of saccade generation (see Appendix for algorithm). A retinal error signal encoding target position in retinotopic coordinates is transmitted from the visual cortex to the saccadic burst generator in the brainstem, which transforms retinal position error into a saccadic burst command via an internal feedback loop (e.g., Jürgens et al. 1981). Saccadic burst neurons project directly to the OMN and to the neural integrator. Additionally, the burst, constituting desired eye velocity during saccades, is subtracted from the cerebellar estimate of current eye velocity (not shown in Fig. 1, but see Appendix), thus accounting for the fact that FL PCs show no consistent modulation during saccades (Noda and Suzuki 1979).
VOR pathways
The VOR maintains stable gaze during head movements by evoking compensatory eye movements of equal velocity, but in the opposite direction. Both direct and indirect neural pathways complement each other to achieve a sufficiently compensatory VOR (Fig. 2b).
The direct VOR pathway, a three-neuron arc, consists of the projections that run from the vertical SCC, modeled as high-pass filter (time constant 5 s), to the VN and from there directly to the OMN. The indirect VOR pathway runs through the integrator for vertical gaze holding, the INC (Crawford et al. 1991), to the OMN. Projections from the SCC to the cerebellum serve two purposes: they provide the desired eye velocity signal to help augmenting the time constant of the integrator during VOR as mentioned above, but also supply head movement information to construct the gaze velocity signal sent to MST. VOR and pursuit pathways converge at the brainstem level by additive combination, as has been confirmed experimentally and suggested previously (e.g., Schweigart et al. 1999; Hirata and Highstein 2001; Fukushima 2003).
Otolith pathways for static eye position
Similar central projections as for semicircular canal signals exist for otolith signals from the utricles. In our model, analogous to the recently proposed pathway for static torsional counterroll (Crawford et al. 2003; Glasauer et al. 2001), otolith signals from the utricle are conveyed to the vertical OMN via a pathway through the brainstem integrator. The cerebellum is only indirectly involved in this pathway, due to the feedback loop involving the PMT and the inhibitory projection back to the VN.
Apart from an influence on static eye position, otolith input also causes small but detectable gravity-dependent positional nystagmus in darkness (Bisdorff et al. 2000). Gravity-dependent modulation of nystagmus becomes evident in cerebellar disease, probably due to overacting otolith-ocular reflexes (Marti et al. 2002).
Floccular PCs
The cerebellar flocculus (FL) is of major importance for both the gaze-holding mechanism and the pursuit system. Accordingly, lesions of the FL severely impair smooth pursuit eye movements and lead to gaze-evoked nystagmus, whereas the angular VOR response is not critically affected (Zee et al. 1981; Waespe et al. 1983; Rambold et al. 2002). Downbeat nystagmus (DBN) is a distinct ocular motor phenomenon in patients with floccular lesions. Floccular PCs are mainly sensitive to gaze-velocity and exhibit either roughly horizontal or vertical on-directions (Lisberger and Fuchs 1978; Miles et al. 1980; Krauzlis and Lisberger 1996). According to the literature, about 90% of floccular PCs showing vertical gaze-velocity sensitivity have downward on-directions, i.e., increase their firing rate in response to downward gaze movements during pursuit or visual VOR cancellation (Miles et al. 1980; Stone and Lisberger 1990). Stimulation of these floccular PCs causes a slow downward eye movement (Sato et al. 1984). Most of the floccular PCs do not modulate during normal vertical VOR in the dark (Stone and Lisberger 1990). Fukushima and coauthors described some modulation of floccular PCs in response to vertical VOR, but in contrast to other studies, these results were obtained in response to rotation in the upright position, thus including otolith signals (Fukushima et al. 1999).
Floccular target neurons (FTNs) in the superior vestibular nucleus (SVN) and the adjacent group y (Y) receive inhibitory input from floccular PCs and have predominantly upward on-directions (Chubb and Fuchs 1982; Zhang et al. 1995a; McCrea et al. 1987). FTNs are part of the vertical smooth pursuit system (De Zeeuw et al. 1994; Zhang et al. 1995a, b). Most of these cells also modulate during vertical VOR in the dark, but not during visual cancellation of vertical VOR (Zhang et al. 1995a); only a minority of these upward FTNs behave like gaze-velocity units, i.e., show modulation during visual VOR cancellation, but not during VOR in the dark.
We modeled healthy floccular PCs as population using a firing-rate model (e.g., Dayan and Abbott 2001, chap. 7) with a log-sigmoid activation function (see Appendix and Fig. 3a), a time constant of 10 ms, and a high resting discharge rate that is balanced by the resting discharge of their brainstem target neurons (FTNs). The on-direction of the PCs is downward, whereas FTNs show an upward on-direction, as was observed experimentally and mentioned above. Sigmoid activation functions can be conceived as describing the summation of many neurons with different resting discharges, input–output slopes, and saturations. A partial lesion to the FL was simulated by scaling the activation function of the PC population resulting in a lower resting rate (see Appendix and Fig. 3a) and lower slope at zero input. The slow phase velocity of nystagmus with gaze straight ahead is determined by the scaling factor, or saturation, of the non-linearity (see Appendix and Fig. 3b).

a Assumed log-sigmoid input–output relationship of vertical Purkinje cells (PCs). The non-linearity for intact healthy PCs (gray) with saturation at 1 and the relation for the damaged PC population (black) with saturation set to 0.6 are shown together with their respective resting discharges (filled circles) at zero input. b Dependence of nystagmus slow phase velocity with gaze straight ahead (here, positive values correspond to DBN) on the saturation of PC non-linearity. Note that, using the activation function shown in a, the saturation determines the scaling of the whole function, i.e., the resting discharge and the slope. The two cases shown in a (1 and 0.6) are marked by filled circles. Complete loss of the flocculus corresponds to saturation zero (a flat line at zero in a), and would thus cause more than 25° per s DBN with gaze straight-ahead
Results
Patient data
This section presents data recorded in typical patients with idiopathic DBN or DBN due to cerebellar atrophy to better illustrate some of the main characteristics of DBN. The presented data are intended as representative examples for comparison with model simulations. For details of the experimental settings and procedures and quantitative results, please refer to the respective publications (see below). In all examples, eye movement recordings were performed with dual search coils; details of calibration procedure and computation of three-dimensional eye position are given elsewhere (Straumann et al. 1995; Glasauer et al. 2003).
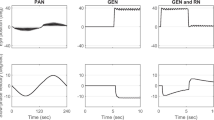
Figure 4a and b demonstrates the gaze-position dependence of vertical ocular drift velocity in a typical patient with idiopathic DBN. The visual target was visible throughout the whole experiment (for detailed “Methods”, see Glasauer et al. 2003). Vertical ocular drift velocity clearly increases with downward gaze position and almost decreases to zero with upward gaze position. The patient is not able to visually suppress the nystagmus.

Typical examples of gaze-position dependence and impaired vertical smooth pursuit (SP) in DBN patients. a Vertical target (gray dashed line) and eye position (black solid line) in degrees (downward: negative). The target, a laser dot projected on a screen, was continuously visible. b Corresponding vertical eye velocity (black solid line, in ° per s) together with target position (gray dashed line, not to scale). Vertical slow phase eye velocity clearly decreases with upward gaze almost to zero, and substantially increases with downward gaze position as expected from Alexander’s law. c Target (gray solid line) and eye position (black solid line) during vertical SP. d Corresponding vertical target (gray solid line) and eye velocity (black solid line). Downward SP is saccadic with a staircase-like eye position trace (c) showing many catch-up saccades (d), but still shows a clear modulation with target velocity, while upward SP is comparatively preserved
As depicted in Fig. 4c and d, vertical SP eye movements in response to a sinusoidally moving laser dot are asymmetrically impaired in patients with DBN (representative patient with idiopathic DBN; for detailed “Methods”, see Glasauer et al. 2005a): While downward SP is profoundly saccadic, upward SP eye movements are largely preserved (Fig. 4c). The asymmetry of the modulation of slow phase eye velocity can be clearly seen in Fig. 4d.
Figure 5 shows that slow phase velocity of DBN modulates as a function of whole-body pitch position in a cerebellar patient (for detailed “Methods” see Marti et al. 2002): Vertical ocular drift velocity increases with prone and decreases with supine body positions.

Gravity-dependent modulation of vertical ocular drift velocity in a patient with DBN. Average slow phase velocity (black circles connected with dashed line) plotted over whole-body position in the pitch plane (cartoons show head position; 0°: upright; 90°: nose up or supine position; 270°: nose down or prone). Vertical slow phase velocity increases with prone and slighty decreases with supine positions and shows a roughly sinusoidal modulation
Model simulations
Model validation
To validate the model and to show that the model generates realistic vertical eye movements, various classes of eye movements were simulated in response to head rotation or target movement. The simulations of normal eye movements is shown in Fig. 6a for vertical saccades together with gaze holding, smooth pursuit eye movements and VOR. In response to target steps, the model generated accurate vertical saccades (Fig. 6a, top row). Sinusoidal target motion led, after an initial catch-up saccade to smooth pursuit eye movements (Fig. 6a, middle row). Simulated pitch head rotation in darkness (in the upright position) generated appropriate nystagmic VOR responses with slow phase velocity modulated oppositely to head velocity (Fig. 6a, bottom row). Note that the Figure shows only slow phase velocity; the fast phases of simulated saccades have been omitted for clarity. Visual VOR cancellation is shown in the insets in Fig. 1. Other combined eye movements, such as VOR in light, superposition of VOR and pursuit with different velocities, superposition of saccades and VOR, etc., can also be simulated but are not shown. All simulations are in accordance with experimental results.

Model simulations of vertical saccades (upper row), smooth pursuit (SP, middle row), and vestibulo-ocular reflex (VOR) in the dark (lower row) for a control subject (part a, ‘control’) and a patient with downbeat nystagmus (part b, ‘downbeat’). First and third columns (‘position’): target position (dashed), eye position (solid), and, for VOR, head position (dotted). All position values are given in degrees (y-axis). Second and fourth column (‘velocity’): corresponding slow phase eye velocity (° per s). Fast phases of saccades are not shown for clarity. Abscissa: time (in seconds). Compare left panels to patient data in Fig. 4
Simulation of cerebellar downbeat nystagmus
To simulate DBN, it was assumed that damage to the FL, e.g., in cerebellar atrophy, reduces the number of floccular PCs. Although the individual discharge characteristics of the remaining floccular PCs may not be altered, a decreased number of floccular PCs will (1) lower the total resting discharge rate and (2) impair their total modulation amplitude. In our model, we represent these changes at the level of a lumped floccular PC by lowering the saturation of the non-linear sigmoid input–output relationship (see Appendix). We deliberately did not try to fit the specific form or parameters of the non-linearity simulating the damage to any data. Rather, the simulations shown are meant to demonstrate that all aspects of eye movement disorders related to DBN can be explained by a single lesion.
Spontaneous drift
As expected, the simulated damage leads to spontaneous upward drift (Fig. 6b, top row) due to the decrease of the resting discharge rate of the PC population. Notably, spontaneous drift is not compensated for by visual input via the smooth pursuit pathway, i.e., the spontaneous drift cannot be visually suppressed. Note that the simulated velocity traces show only slow phase velocity; the saccades have been omitted for clarity.
Eye position dependence of vertical drift
Simulations for vertical saccades are shown in Fig. 6b (top row). Accuracy of vertical saccades is not altered. Vertical ocular drift in DBN, however, is clearly eye-position dependent, i.e., increasing in gaze down and decreasing in gaze up (Fig. 6b, top row, left column). This eye-position dependence of ocular drift is known as Alexander’s law, which states that nystagmus is stronger with gaze in the direction of the fast phase (Robinson et al. 1984). Since the intact cerebellum augments the time constant of the inherently leaky brainstem integrator, cerebellar disease results in shortening of the time constant of the integrator, and therefore, in gaze-evoked drift. This vertical gaze-evoked drift component adds to the spontaneous upward bias drift, leading to the observed modulation of vertical drift as a function of vertical eye position. Comparison with patient data in Fig. 4 shows the close agreement of simulation and experiment.
Asymmetrically impaired vertical smooth pursuit
Due to the modification of floccular PCs with a non-linear sigmoid in–out-put relationship, saturation limits the increase of the resting rate in response to downward eye movements, i.e., in PCs on-direction, leading to saccadic downward pursuit (Fig. 6b, middle row; in the velocity traces, saccadic fast phases are omitted for clarity). The response to upward eye movements, however, is not impaired for low target velocities, since the PCs are still able to decrease their resting rate to zero. Note that slow phase velocity for downward pursuit is still modulated. This modulation pattern explains the finding of asymmetrically impaired smooth pursuit in patients with cerebellar DBN (see Fig. 4 for comparison).
VOR responses are not affected in DBN
Since floccular PCs are not sensitive to pure head velocity signals, they modulate only slightly during normal VOR in darkness. Accordingly, in DBN, the VOR response is not affected except for an offset in eye velocity, which corresponds to the spontaneous drift in gaze straight-ahead: in other words, the upward drift is simply superimposed on the VOR response (Fig. 6b, bottom row).
Gravity dependence
Vertical ocular drift in DBN is strongly modulated by gravity (Fig. 7, see also Fig. 5 for corresponding patient data). This increased modulation is a direct consequence of the decreased time constant of the neural integrator, and occurs without any additional assumption (e.g., we do not assume direct otolith pathways to the FL). Since otolith signals are supposed to pass exclusively through the brainstem integrator, the drift elicited by the otolith signals depends on the overall integrator time constant. With intact floccular PCs and a long time constant, the drift is very slow (Fig. 7 left), but reduced cerebellar inhibition in DBN results in increased modulation of vertical drift as function of head position (Fig. 7 right), which can be interpreted as overacting of these otolith-ocular reflexes. Note also that our model assumes that FL PC discharge modulates with head position (Fig. 7 lower row), which, to our knowledge, has not been tested so far.

Model simulations of gravity dependence of vertical drift in downbeat nystagmus (DBN). Modulation of vertical slow phase eye velocity (upper row) and corresponding modulation of Purkinje cell (PC) discharge rate (lower row) as a function of head position in the pitch plane (‘head position’, abcissa) in a control subject (‘control’, left column) and a patient with DBN (‘downbeat’, right column). Simulation was performed with a quasi-static change in pitch position (angular velocity 1° per s). In the left upper panel, the patient data (Fig. 5) are inserted (gray dots). Patient data show a similar shape, but are shifted by about 45° to the left, which may be due to individual initial head pitch or the pitch of the otoliths within the head. Note that PCs in the simulated patient show a slightly lower average discharge rate
Choice of lesion site and parameters
The specific choice of the parameters affecting the non-linear input–output relationshipFootnote 4 of the PC population and the site of the lesion determine the oculomotor disorders simulated by the model. The dependence of DBN slow phase velocity in gaze straight ahead upon the saturation of the PC population was already shown in Fig. 3. To further demonstrate the effects of different parameters or of additional lesions, we also simulated (1) additional damage of the PCs projecting back to the cortex (Fig. 8a), and (2) the effect of complete loss of the FL (Fig. 8b). Damaging the PCs projecting back to MST caused stronger DBN with lower gaze-direction dependence and a SP deficit which was asymmetric only due to an upward shift of the SPV (Fig. 8a). The complete loss of the PCs projecting to the VN was simulated using a lower resting discharge of Y neurons to avoid excessive DBN, which would reach about 28° per s in our model. Pursuit was abolished, the remaining modulation (Fig. 8b) is an effect of the low integrator time constant of 5 s. See Appendix for exact choice of parameters for these simulations.

Simulation of gaze holding and smooth pursuit (SP) responses for a combined partial damage to both Purkinje cell (PC) populations and b of complete loss of the PCs projecting to the VN. See legend to Fig. 6 for explanation of the figure outline
Discussion
We present a new hypothesis on the pathomechanism of cerebellar downbeat nystagmus (DBN). Our hypothesis is based on the inherently asymmetric distribution of on-directions of vertical gaze-velocity sensitive Purkinje cells (PC) in the cerebellar FL. This asymmetry has been found in monkeys, but recent fMRI studies suggest that it also exists in humans (Stephan et al. 2005; Kalla et al. 2006). We formalized our hypothesis in a computational model of vertical eye movements. Damage of the model element corresponding to the floccular PCs demonstrates that simulated vertical eye movements show all ocular motor deficits associated with DBN. Our hypothesis thus provides a unifying theory for the origin of DBN. In the following sections, we discuss our hypothesis and its potential implications in the context of the relevant literature.
Recasting the vestibular hypothesis on DBN
Most authors, who conjectured a directional imbalance of vertical vestibular pathways to be the main cause for the upward drift in patients with DBN (Leigh and Zee 2006; Pierrot-Deseilligny and Milea 2005), refer to an early study in rabbits by Ito and coworkers (Ito et al. 1977). These authors demonstrated that electrical stimulation of FL inhibits upward eye movements evoked by concurrent electrical stimulation of the anterior SCC nerve, but not downward eye movements evoked by stimulation of the posterior SCC nerve. Therefore, with lesions of the FL, a relative predominance of anterior over posterior SCC signals may occur due to the missing floccular inhibition of the anterior canals. Such predominance then could result in spontaneous upward drift, i.e., DBN (Baloh and Spooner 1981; Halmagyi et al. 1983; Walker and Zee 2005a, b).
Our hypothesis on the pathomechanism of cerebellar DBN offers a different explanation of the findings of Ito and colleagues. Vertical floccular gaze-velocity sensitive PCs have predominantly downward on-directions and project inhibitory to target neurons in the SVN and Y, whose activity is correlated with upward eye movements (Miles et al. 1980; Chubb and Fuchs 1982; Partsalis et al. 1995; Fukushima et al. 1999). Stimulation of vertical floccular PCs enhances inhibition of SVN and Y neurons and reduces upward eye movements, which, for example, are evoked by the downward head impulses. Thus, floccular stimulation in fact does not inhibit anterior SCC signals, but decreases activation of brainstem target neurons with upward on-directions, effectively causing cancellation of upward VOR. Lesions—or inhibition—of the floccular PCs will have the reverse effect, i.e., facilitate activation of upward eye movements. Our hypothesis is thus completely compatible with Ito’s findings.
Recent studies also favor a crucial role of an imbalance of central vestibular pathways for the pathomechanism of spontaneous vertical nystagmus (Pierrot-Deseilligny et al. 2005; Pierrot-Deseilligny and Milea 2005). It was proposed that upbeat nystagmus (UBN) and DBN may result from primary hypo- or hyperactivity, respectively, in the ventral tegmental tract (VTT), which originates in the SVN and transmits excitatory vestibular signals from the anterior SCC to the superior rectus und inferior oblique motoneurons of the third nucleus, probably of both sides. Our hypothesis indeed implies a hyperactivity of SVN and Y-group pathways to the OMNs in DBN, but proposes that this hyperactivity has its origin in a hypoactivity of the FL rather than in the SVN. For UBN, our hypothesis assumes a hypoactivity in the SVN or Y.
If we consider an imbalance of upward and downward vestibular signals (Böhmer and Straumann 1998) to be critical for the pathogenesis of DBN, we might expect asymmetric vertical VOR gains in these patients, specifically a higher gain for the upward VOR (elicited by downward head impulses) than for the downward VOR (elicited by upward head impulses). Glasauer and coworkers tested vertical VOR responses in patients with isolated DBN and found, on average, no significant impairment or asymmetry of the vertical VOR (Glasauer et al. 2004). Walker and Zee, on the other hand, measured, on average, higher gains for downward than for upward pitch head impulses in patients with cerebellar disease (Walker and Zee 2005a, b). Differences in the selected patient populations may account for the discrepancy between the two studies. Half of patients in the study by Glasauer et al. showed isolated DBN without any other signs of cerebellar disease, while Walker and Zee included only patients with cerebellar degeneration. In the latter condition it is more likely that, besides the FL, other vestibulo-cerebellar structures are lesioned, e.g., the nodulus, uvula, or even the VN (Migliaccio et al. 2004), in which case asymmetries of vertical VOR responses are highly plausible.
To summarize, we redraft the ‘vestibular’ hypothesis on the origin of DBN in the sense that DBN is primarily due to a lesion uncovering a cerebellar asymmetry, and thus causing vertical cerebellar nystagmus rather than a type of central vestibular nystagmus. As our model illustrates, a primary dysfunction of the vertical gaze-velocity sensitive floccular PCs, explains the upward direction of the spontaneous drift and all of the associated ocular motor deficits. A recent functional magnetic imaging study on patients with cerebellar DBN supports this view (Kalla et al. 2006).
With respect to our model, it is important to consider the functional role of the various SCC pathways: (1) the main VOR pathway runs through the brainstem, (2) in light, the floccular SCC input is used to first transform the estimate of eye velocity to an estimate of gaze velocity (estimated gaze velocity in Fig. 1) to be sent to MST, and then transform desired gaze velocity back to desired eye velocity (in the model, this latter step almost cancels out the SCC input), and (3) in darkness, the floccular SCC input is used as desired eye velocity signal to improve brainstem integration. The net effect of the two floccular SCC pathways is that of improved neural integration of the SCC signal. Therefore, we argue that dysfunction of FL-PCs as proposed here does not constitute a lesion to central vestibular pathways, but rather to smooth pursuit and/or integrator pathways.
Lesion sites for DBN
DBN has been described with various disorders, e.g., cerebellar degeneration, anomalies of the cranio-cervical junction, brainstem infarction, dolichoectasia of the vertebrobasilar artery, multiple sclerosis and syringobulbia. Our model is based on the hypothesis that, despite the variety of etiologies, the spontaneous upward ocular drift in DBN always originates from a primary dysfunction of the vertical gaze-velocity sensitive floccular PCs. However, focal brainstem lesions selectively affecting afferent floccular projections may mimic failure of floccular PCs and probably result in spontaneous upward drift as well. In cats, for example, experimental lesion of a subgroup of PMT neurons in the upper pons level led to DBN (Nakamagoe et al. 2000). These neurons receive afferents from the vestibular nuclei and project to the FL. A similar syndrome has, so far, not been described in humans, probably because specific lesion of this PMT subgroup without involvement of the adjacent MLF structures is unlikely to occur (Pierrot-Deseilligny and Milea 2005). Bertholon and coauthors described DBN in a patient with syringomyelia and focal bilateral cavities in the medulla (Bertholon et al. 1993). Probably, selective damage of afferent floccular projections may account for the DBN in this patient as well.
In summary, we hypothesize that the spontaneous upward drift in patients with DBN results from dysfunction of vertical floccular PCs with downward on-directions or selective damage of floccular afferents or efferents.
DBN and vertical gaze-holding
Vertical ocular drift in patients with DBN is composed of a spontaneous or bias upward drift and a vertical gaze-evoked drift (Straumann et al. 2000). The bias drift is already present with gaze straight-ahead and, as outlined above, primarily caused by failure of the floccular gaze-velocity sensitive PCs with downward on-directions, leading to a decreased inhibition of upward-on neurons in the SVN and Y.
The gaze-evoked drift component is only evoked with vertical gaze eccentricity, and indicates impairment of the vertical neural integrator. The important role of the FL for gaze-holding was previously explained by a positive feedback loop between cerebellum and the brainstem neural integrator (Zee et al. 1980). We modified this hypothesis by proposing a negative feedback loop (Glasauer 2003), which is compatible with the exclusively inhibitory cerebellar output. Simulations illustrate how upward eye velocity in DBN depends on vertical eye position (see Fig. 5).
Most DBN patients not only show a failure of the vertical neural integrator, but also deficient horizontal gaze-holding (Glasauer 2003). This is possibly due to the involvement of the FL in both vertical and horizontal gaze holding. Finally, Glasauer and coauthors reported one patient without failure of the (vertical) neural integrator (Glasauer 2003). This type of DBN seems to be the rare exception, but can be explained by specific loss of cerebellar PCs transmitting efference copy feedback signals via thalamic relays to the cortex while sparing those PC populations which are involved in gaze-holding and project back to the VN. Model simulations (not shown) confirm this hypothesis.
DBN and vertical smooth pursuit
Vertical smooth pursuit (SP) eye movements are asymmetrically impaired in cerebellar patients, i.e., upward are better preserved than downward smooth eye movements (Zee et al. 1974; Glasauer et al. 2005a). Notably, all DBN patients show impairment of vertical pursuit (Zee et al. 1974; Baloh and Spooner 1981; Glasauer et al. 2005a), which strongly suggests that the neural structures damaged in DBN are part of the vertical pursuit system. In support, Glasauer et al. (2005a) showed that the vertical pursuit deficit in DBN is not due to superposition of normal pursuit commands with upward drift as proposed earlier (Baloh and Spooner 1981), but constitutes a specific pursuit failure. The severe impairment of downward SP eye movements can be well explained by our model, since, due to failure of gaze-velocity sensitive floccular PCs with downward on-directions, the increase of the net firing rate in response to downward eye movements is limited. Decrease of the resting rate to zero is still possible, thus upward SP eye movements will be preserved, at least at lower target velocities. In animal experiments (Zee et al. 1981; Rambold et al. 2002), bilateral lesion of the FL led to severe pursuit deficits, but, for vertical pursuit, an asymmetry as seen in patients was not evident. Nevertheless, these observations are in agreement with simulations of our model: a simulated complete lesion of the FL causes DBN together with a symmetric pursuit deficit (Fig. 8).
Based on their experimental findings, Zee et al. (1974) already had proposed that DBN is due to an asymmetry in pursuit pathways. However, to also explain their finding of impaired gaze holding, they had to additionally assume failure of the neural integrator. Our present model unifies both assumptions: the underlying damage affects SP and neural integration at the same time. The same probably also applies to the findings of Büttner and Grundei (1995) who also found a closed correlation between gaze evoked nystagmus and SP deficit in the horizontal plane.
Additional support comes from a study by Marti et al. (2005a), who showed that repeated asymmetric SP stimulation in the vertical directions elicits DBN in healthy subjects. This experimental DBN was only seen in the dark and disappeared in the light, since healthy subjects were able to suppress the upward drift by activation of SP eye movements. However, failure of floccular PCs also entails loss of the ability to cancel the spontaneous upward drift by counteractive downward SP eye movements, and therefore DBN cannot be suppressed in cerebellar patients.
Apart from the FL, other cerebellar structures are involved in the generation and control of vertical smooth pursuit eye movements, specifically the dorsal vermis (see Fig. 2a) (Robinson and Fuchs 2001; Krauzlis 2004). Accordingly, complete flocculectomy does not completely abolish vertical smooth pursuit eye movements (Zee et al. 1981). It has been suggested that the pathway through the dorsal vermis is more critical for initial acceleration at pursuit onset, whereas the FL is primarily involved in the maintenance of smooth pursuit (Robinson and Fuchs 2001; Krauzlis 2004). An alternative model-based explanation involving smooth pursuit gain control, which is compatible with this distinction, is proposed in Nuding et al. (2008).
Gravity and DBN
Gravity influences vertical ocular drift both in healthy human subjects (Goltz et al. 1997; Bisdorff et al. 2000) and patients with DBN (Baloh and Spooner 1981; Chambers et al. 1983; Gresty et al. 1986; Marti et al. 2005b; Helmchen et al. 2004; Halmagyi and Leigh 2004), most likely via otolith-ocular reflexes. Spontaneous vertical drift in healthy humans and cerebellar patients with DBN consists of a gravity-independent component, already present in gaze straight ahead, and a gravity-dependent component being maximal in prone and minimal in supine position (Marti et al. 2002). Interestingly, the potassium-channel blocker 3,4-Diaminopyridine (3,4-DAP) mainly reduced the gravity-dependent and less the gravity-independent component in a patient with DBN (Helmchen et al. 2004).
The gravity-dependent modulation of ocular drift can be modeled by assuming static vertical otolith-ocular reflexes, which, similar to ocular counterroll for torsional eye position, shift the direction of primary gaze upward with downward pitching of the head and vice versa. This otolith mediated shift can be conceived as change in the zero position of the neural integrator, which becomes apparent as gravity-dependent drift in DBN patients with damaged integrator function, but remains small in healthy subjects with intact gaze holding. While being important in lateral-eyed animals without foveal vision like rabbits, otolith-ocular reflexes have lost their function in frontal-eyed, foveal animals. The proposed FL feedback loop may therefore exert an inhibitory influence on these otolith-ocular reflex pathways to minimize gravity-dependent drift modulation. Our findings support this hypothesis, since modulation of the gravity-dependent drift component is much larger in patients with DBN than in healthy subjects (Marti et al. 2002). With loss of floccular inhibition, these reflexes will become overacting, resulting in the observed patterns of gravity-dependent drift modulation. Theoretical support for this hypothesis comes from modeling of central positional nystagmus (Glasauer et al. 2001). However, at present, our hypothesis about otolith pathways needs further experimental confirmation. In this respect, three predictions for future experiments are made by the model: (1) floccular PCs should be modulated by static head pitch, (2) gaze position dependence and gravity dependence of DBN should be positively correlated, and (3) patients with idiopathic DBN should show similar head position dependence as those with cerebellar DBN. If these predictions turn out to be wrong, the observed gravity-dependent modulation in cerebellar DBN may be caused by damage to other structures such as the nodulus or uvula.
The well-documented influence of gravity on DBN may be related to the open question of why the distribution of on-directions of vertical FL PCs is asymmetric. Apart from this asymmetry, the symmetric push–pull principle is realized everywhere in the ocular motor and vestibular systems from SCC to extra-ocular eye muscles. However, gravity acts asymmetrically and, therefore, the floccular asymmetry may originate in the processing of visual or vestibular gravity-related signals.
Upbeat nystagmus
UBN usually occurs transiently with pontine or medullary brainstem lesions, but not with cerebellar lesions (Pierrot-Deseilligny and Milea 2005), which is plausible in the framework of our hypothesis, since only a minority of vertical floccular PCs has upward on-directions, and lesions affecting selectively these upward tuned PCs are unlikely to occur.
Given intact vestibulo-cerebellar function, activation of adaptive smooth pursuit eye movements will cancel the downward ocular drift and the UBN will gradually decline. This explains why UBN is only seen transiently with focal brainstem lesions, unlike DBN, were the adaptive cerebellar pathway is damaged making a gradual decline of nystagmus impossible.
A comparison of the effects of the potassium channel blocker 4AP in two patients with DBN and UBN also supports our hypothesis (Glasauer et al. 2005b): while DBN was reduced in light and in darkness due to 4AP, UBN was only reduced in light, but remained equally strong in darkness. The latter suggests that the reduction of UBN with 4AP was due to increased efficacy of smooth pursuit pathways leading to UBN cancellation.
Treatment options
Until recently, treatment options for DBN were poor. Fortunately, there is now evidence for substantial improvement of DBN under therapy with the potassium-channel-blockers 3,4-DAP and 4-AP (Strupp et al. 2003; Kalla et al. 2004, 2007). The exact mechanism by which aminopyridines reduce ocular drift in cerebellar DBN is not yet kown, but 4-Aminopyridine (4-AP) was shown to increase the excitability of cerebellar Purkinje cells (PC) in the guinea pig (Etzion and Grossman 2001). Such mechanism is highly compatible with our hypothesis, since increased excitability of the floccular PCs is likely to augment both their resting rate and their sensitivity, leading to decreased upward ocular drift and to improved integrator and smooth pursuit function (Glasauer et al. 2005b).
Limitations of the model
The increase of DBN with lateral gaze is a well-known clinical phenomenon, which has been documented in cerebellar DBN patients (Straumann et al. 2000), but also patients with DBN of unknown origin (Glasauer et al. 2003). The mechanism which leads to the observed increase of bias upward drift velocity with horizontal gaze eccentricity still remains speculative, and our model, confined to vertical eye movements, naturally does not provide an explanation. Probably, vertical floccular PCs may exhibit some horizontal eye-position-sensitivity (Leung et al. 2000), i.e., their resting rate may vary slightly as a function of horizontal eye position. With failure of these PCs in floccular disease, modulation of upward drift depending on horizontal eye position may be become more prominent, i.e., result in an increase of drift velocity with lateral gaze. Another hypothetical explanation of increased upward drift in lateral gaze supposes a downward shift of the ocular motor coordinate system as a result of the floccular lobe lesion. Relative to the shifted coordinate system, primary gaze direction would now correspond to an ocular elevation. As gaze direction moves rightward or leftward parallel to the earth-horizontal, the eye leaves the ‘line of latitude southward’, resulting in an increase of vertical deviation from the primary position. As a direct consequence, upward drift increases.
Our model explains the origin of DBN with dysfunction of the FL. The model does not account for other ocular motor phenomena seen with damage of the vestibulo-cerebellum. For example, lesions of the nodulus and uvula are likely to generate asymmetries of vertical velocity storage mechanisms, which are not included in detail in our model. Also, the model does not include some other pathways such as those mediating the translational VOR, and, since it is one-dimensional, does not capture the influence of vergence on DBN. However, the vergence-associated changes of Listing’s plane, i.e., of the intrinsic ocular motor coordinates, may provide a basis for these effects (see also Glasauer et al. 2003). The translational VOR, which is caused by otolith activation, is usually tested with interaural acceleration, but it also exists for vertical translations (Paige 1989). In our model, otolith pathways are included only to capture the effects of sustained otolith stimulation such as during tilt. In contrast, translational VOR is transient. Sustained nystagmus, such as seen in DBN or UBN, may theoretically be caused by central deficits in translational VOR pathways, if these pathways are directionally selective, but should not lead to the associated ocular motor deficits seen with DBN.
Our model necessarily does not capture all features and pathways of the SP system. The pursuit pathway originating in the FEF is not included. This pathway, thought to mediate signals important for smooth pursuit initiation and gain control (e.g., Tanaka and Lisberger 2001), was left out since the main focus of the model was on steady state pursuit and the role of the FL. Also, we made no attempt to include the well-known predictive and anticipatory aspects of pursuit (see, for example, Shibata et al. 2005 for a model) to keep our model as simple as possible without compromising its ability to simulate various classes of eye movements.
Finally, our model does currently not include cerebellar plasticity. Abnormal cerebellar plasticity in DBN may contribute to the observed oculomotor symptoms, but, as shown here, is not necessary to produce eye movement deficits that closely resemble those seen in patients.
Conclusions
We present a unifying theory on the pathomechanism of cerebellar DBN, which explains the origin of the upward directed drift and of the associated ocular motor deficits, i.e., impaired vertical smooth pursuit, deficient vertical gaze-holding, and gravity dependence of the upward directed drift. Our results suggest that DBN constitutes primarily a disorder of vertical ocular motor pathways mediating gaze holding and smooth pursuit, rather than a sign of vestibular tone imbalance. The inherent asymmetry in the distribution of on-directions of vertical floccular PCs seems to be crucial for the origin of a spontaneous upward drift in patients with floccular damage, e.g., in cerebellar atrophy, or dysfunction of floccular PCs due to other reasons, e.g., altered calcium-channel-kinematics in episodic ataxia.
Notes
While upbeat nystagmus also exists, it is a rare ocular motor sign, usually not caused by cerebellar disease, and it resolves within weeks or month even without treatment.
The floccular lobe consists of the flocculus proper, and the ventral and dorsal paraflocculus. Unless stated otherwise, we treat the floccular lobe as an entity, since the exact role of its different parts is not completely clear (Fukushima 2003).
A leaky integrator ‘forgets’ over time, i.e., the eye slowly drifts back to its resting position.
The input–output relationship determines how the overall firing rate of all neurons projecting to the population is translated to the PC firing rate. Its shape is shown in Fig. 3a. The log-sigmoid function we have chosen has two free parameters: output saturation and input gain factor.
Abbreviations
- 4-AP:
-
4-Aminopyridine
- 3,4-DAP:
-
3,4-Diaminopyridine
- DBN:
-
Downbeat nystagmus
- DCN:
-
Deep cerebellar nuclei
- DLPN:
-
Dorsolateral pontine nuclei
- DV:
-
Dorsal vermis
- FEF:
-
Frontal eye field
- FEFsem:
-
Smooth pursuit subregion of the FEF
- FL:
-
Floccular lobe
- FN:
-
Fastigial nucleus
- FTN:
-
Floccular target neurons
- INC:
-
Interstitial nucleus of Cajal
- MLF:
-
Medial longitudinal fasciculus
- MST:
-
Middle superior temporal area
- MT:
-
Middle temporal area
- NRTP:
-
Nucleus reticularis tegmentum pontis
- OMN:
-
Ocular motor neurons
- PC:
-
Purkinje cell
- PMT:
-
Paramedian tract
- SCC:
-
Semicircular canal
- SVN:
-
Superior vestibular nucleus
- SP:
-
Smooth pursuit
- UBN:
-
Upbeat nystagmus
- VOR:
-
Vestibulo-ocular reflex
- VTT:
-
Ventral tegmental tract
- Y:
-
Y-group
References
Baloh RW, Spooner JW (1981) Downbeat nystagmus: a type of central vestibular nystagmus. Neurology 31:304–310
Bertholon P, Convers P, Barral FG, Duthel R, Michel D (1993) Post-traumatic syringomyelobulbia and inferior vertical nystagmus. Rev Neurol (Paris) 149:355–358
Bisdorff A, Sancovic S, Debatisse D et al (2000) Positional nystagmus in the dark in normal subjects. Neurophthalmology 24:283–290
Böhmer A, Straumann D (1998) Pathomechanism of mammalian downbeat nystagmus due to cerebellar lesion: a simple hypothesis. [Review]. Neurosci Lett 250:127–130
Büttner-Ennever JA, Horn AK, Schmidtke K (1989) Cell groups of the medial longitudinal fasciculus and paramedian tracts. Rev Neurol (Paris) 145:533–539
Büttner-Ennever JA, Horn AK (1996) Pathways from cell groups of the paramedian tracts to the floccular region. Ann NY Acad Sci 781:532–540
Büttner U, Grundei T (1995) Gaze-evoked nystagmus and smooth pursuit deficits: their relationship studied in 52 patients. J Neurol 242:384–389
Cannon SC, Robinson DA (1987) Loss of the neural integrator of the oculomotor system from brain stem lesions in monkey. J Neurophysiol 57:1383–1409
Chambers BR, Ell JJ, Gresty MA (1983) Case of downbeat nystagmus influenced by otolith stimulation. Ann Neurol 13:204–207
Chubb MC, Fuchs AF (1982) Contribution of y group of vestibular nuclei and dentate nucleus of cerebellum to generation of vertical smooth eye movements. J Neurophysiol 48:75–99
Crawford JD, Cadera W, Vilis T (1991) Generation of torsional and vertical eye position signals by the interstitial nucleus of Cajal. Science 252:1551–1553
Crawford JD, Tweed DB, Vilis T (2003) Static ocular counterroll is implemented through the 3-D neural integrator. J Neurophysiol 90:2777–2784
Davidson PR, Wolpert DM (2005) Widespread access to predictive models in the motor system: a short review. J Neural Eng 2:S313–S319
Dayan P, Abbott LF (2001) Theoretical neuroscience: computational and mathematical modeling of neural systems. MIT Press, Cambridge
De Zeeuw CI, Wylie DR, DiGiorgi PL, Simpson JI (1994) Projections of individual Purkinje cells of identified zones in the flocculus to the vestibular and cerebellar nuclei in the rabbit. J Comp Neurol 349:428–447
Etzion Y, Grossman Y (2001) Highly 4-aminopyridine sensitive delayed rectifier current modulates the excitability of guinea pig cerebellar Purkinje cells. Exp Brain Res 139:419–425
Fukushima K, Fukushima J, Kaneko CR, Fuchs AF (1999) Vertical Purkinje cells of the monkey floccular lobe: simple-spike activity during pursuit and passive whole body rotation. J Neurophysiol 82:787–803
Fukushima K (2003) Roles of the cerebellum in pursuit-vestibular interactions. Cerebellum 2:223–232
Glasauer S, Dieterich M, Brandt T (2001) Central positional nystagmus simulated by a mathematical ocular motor model of otolith-dependent modification of Listing’s plane. J Neurophysiol 86:1546–1554
Glasauer S (2003) Cerebellar contribution to saccades and gaze holding: a modeling approach. Ann NY Acad Sci 1004:206–219
Glasauer S, Hoshi M, Kempermann U, Eggert T, Büttner U (2003) Three-dimensional eye position and slow phase velocity in humans with downbeat nystagmus. J Neurophysiol 89:338–354
Glasauer S, von Lindeiner H, Siebold C, Büttner U (2004) Vertical vestibular responses to head impulses are symmetric in downbeat nystagmus. Neurology 63:621–625
Glasauer S, Hoshi M, Büttner U (2005a) Smooth pursuit in patients with downbeat nystagmus. Ann NY Acad Sci 1039:532–535
Glasauer S, Strupp M, Kalla R, Büttner U, Brandt T (2005b) Effect of 4-aminopyridine on upbeat and downbeat nystagmus elucidates the mechanism of downbeat nystagmus. Ann NY Acad Sci 1039:528–531
Glasauer S (2007) Current models of the ocular motor system. Dev Ophthalmol 40:158–174
Goltz HC, Irving EL, Steinbach MJ, Eizenman M (1997) Vertical eye position control in darkness: orbital position and body orientation interact to modulate drift velocity. Vision Res 37:789–798
Green AM, Meng H, Angelaki DE (2007) A reevaluation of the inverse dynamic model for eye movements. J Neurosci 27:1346–1355
Gresty M, Barratt H, Rudge P, Page N (1986) Analysis of downbeat nystagmus. Otolithic vs semicircular canal influences. Arch Neurol 43:52–55
Halmagyi GM, Rudge P, Gresty MA, Sanders MD (1983) Downbeating nystagmus. A review of 62 cases. Arch Neurol 40:777–784
Halmagyi GM, Leigh RJ (2004) Upbeat about downbeat nystagmus. Neurology 63:606–607
Hirata Y, Highstein SM (2001) Acute adaptation of the vestibuloocular reflex: signal processing by floccular and ventral parafloccular Purkinje cells. J Neurophysiol 85:2267–2288
Helmchen C, Sprenger A, Rambold H, Sander T, Kömpf D, Straumann D (2004) Effect of 3, 4-diaminopyridine on the gravity dependence of ocular drift in downbeat nystagmus. Neurology 63:752–753
Ito M, Nisimaru N, Yamamoto M (1977) Specific patterns of neuronal connexions involved in the control of the rabbit’s vestibulo-ocular reflexes by the cerebellar flocculus. J Physiol 265:833–854
Jürgens R, Becker W, Kornhuber HH (1981) Natural and drug-induced variations of velocity and duration of human saccadic eye movements: evidence for a control of the neural pulse generator by local feedback. Biol Cybern 39:87–96
Kalla R, Glasauer S, Schautzer F, Lehnen N, Büttner U, Strupp M, Brandt T (2004) 4-Aminopyridine improves downbeat nystagmus, smooth pursuit, and VOR gain. Neurology 62:1228–1229
Kalla R, Deutschländer A, Hüfner K, Stephan T, Jahn K, Glasauer S, Brandt T, Strupp M (2006) Detection of floccular hypometabolism in downbeat nystagmus by fMRI. Neurology 66:281–283
Kalla R, Glasauer S, Büttner U, Brandt T, Strupp M (2007) 4-Aminopyridine restores vertical and horizontal neural integrator function in downbeat nystagmus. Brain 130:2441–2451
Krauzlis RJ (2004) Recasting the smooth pursuit eye movement system. J Neurophysiol 91:591–603
Krauzlis RJ, Lisberger SG (1994) A model of visually-guided smooth pursuit eye movements based on behavioral observations. J Comput Neurosci 1:265–283
Krauzlis RJ, Lisberger SG (1996) Directional organization of eye movement and visual signals in the floccular lobe of the monkey cerebellum. Exp Brain Res 109:289–302
Leigh RJ, Zee DS (2006) The neurology of eye movements, 4th edn. Oxford Press, New York
Leung HC, Suh M, Kettner RE (2000) Cerebellar flocculus and paraflocculus Purkinje cell activity during circular pursuit in monkey. J Neurophysiol 83:13–30
Lisberger SG, Fuchs AF (1978) Role of primate flocculus during rapid behavioral modification of vestibuloocular reflex. I. Purkinje cell activity during visually guided horizontal smooth-pursuit eye movements and passive head rotation. J Neurophysiol 41:733–763
Marti S, Palla A, Straumann D (2002) Gravity dependence of ocular drift in patients with cerebellar downbeat nystagmus. Ann Neurol 52:712–721
Marti S, Bockisch CJ, Straumann D (2005a) Prolonged asymmetric smooth-pursuit stimulation leads to downbeat nystagmus in healthy human subjects. Invest Ophthalmol Vis Sci 46:143–149
Marti S, Straumann D, Glasauer S (2005b) The origin of downbeat nystagmus: an asymmetry in the distribution of on-directions of vertical gaze-velocity purkinje cells. Ann NY Acad Sci 1039:548–553
McCrea RA, Strassman A, Highstein SM (1987) Anatomical and physiological characteristics of vestibular neurons mediating the vertical vestibulo-ocular reflexes of the squirrel monkey. J Comp Neurol 264:571–594
Migliaccio AA, Halmagyi GM, McGarvie LA, Cremer PD (2004) Cerebellar ataxia with bilateral vestibulopathy: description of a syndrome and its characteristic clinical sign. Brain 127:280–293
Miles FA, Fuller JH, Braitman DJ, Dow BM (1980) Long-term adaptive changes in primate vestibuloocular reflex. III. Electrophysiological observations in flocculus of normal monkeys. J Neurophysiol 43:1437–1476
Nakamagoe K, Iwamoto Y, Yoshida K (2000) Evidence for brainstem structures participating in oculomotor integration. Science 288:857–859
Noda H, Suzuki DA (1979) The role of the flocculus of the monkey in saccadic eye movements. J Physiol 294:317–334
Nuding U, Ono S, Mustari M, Büttner U, Glasauer S (2008) A theory of the dual pathways for smooth pursuit based on dynamic gain control. J Neurophysiol [Epub ahead of print]
Paige GD (1989) The influence of target distance on eye movement responses during vertical linear motion. Exp Brain Res 77:585–593
Partsalis AM, Zhang Y, Highstein SM (1995) Dorsal Y group in the squirrel monkey. II. Contribution of the cerebellar flocculus to neuronal responses in normal and adapted animals. J Neurophysiol 73:632–650
Pierrot-Deseilligny C, Milea D (2005) Vertical nystagmus: clinical facts and hypotheses. Brain 128:1237–1246
Pierrot-Deseilligny C, Milea D, Sirmai J, Papeix C, Rivaud-Pechoux S (2005) Upbeat nystagmus due to a small pontine lesion: evidence for the existence of a crossing ventral tegmental tract. Eur Neurol 54:186–190
Porrill J, Dean P, Stone JV (2004) Recurrent cerebellar architecture solves the motor-error problem. Proc Biol Sci 271:789–796
Rambold H, Churchland A, Selig Y, Jasmin L, Lisberger SG (2002) Partial ablations of the flocculus and ventral paraflocculus in monkeys cause linked deficits in smooth pursuit eye movements and adaptive modification of the VOR. J Neurophysiol 87:912–924
Robinson FR, Fuchs AF (2001) The role of the cerebellum in voluntary eye movements. Annu Rev Neurosci 24:981–1004
Robinson DA, Zee DS, Hain TC, Holmes A, Rosenberg LF (1984) Alexander’s law: its behavior and origin in the human vestibulo-ocular reflex. Ann Neurol 16:714–722
Robinson DA, Gordon JL, Gordon SE (1986) A model of the smooth pursuit eye movement system. Biol Cybern 55:43–57
Sato Y, Yamamoto F, Shojaku H, Kawasaki T (1984) Neuronal pathway from floccular caudal zone contributing to vertical eye movements in cats—role of group y nucleus of vestibular nuclei. Brain Res 294:375–380
Schweigart G, Mergner T, Barnes G (1999) Eye movements during combined pursuit, optokinetic and vestibular stimulation in macaque monkey. Exp Brain Res 127:54–66
Shibata T, Tabata H, Schaal S, Kawato M (2005) A model of smooth pursuit in primates based on learning the target dynamics. Neural Netw 18:213–224
Stephan T, Kalla R, Marti S, Straumann D, Glasauer S (2005) Asymmetric cerebellar flocculus activation for vertical smooth pursuit eye movements. In: Abstract for the 11th OHBM meeting, Toronto
Stone LS, Lisberger SG (1990) Visual responses of Purkinje cells in the cerebellar flocculus during smooth-pursuit eye movements in monkeys. I. Simple spikes. J Neurophysiol 63:1241–1261
Straumann D, Zee DS, Solomon D, Lasker AG, Roberts DC (1995) Transient torsion during and after saccades. Vision Res 35:3321–3334
Straumann D, Zee DS, Solomon D (2000) Three-dimensional kinematics of ocular drift in humans with cerebellar atrophy. J Neurophysiol 83:1125–1140
Strupp M, Schüler O, Krafczyk S, Jahn K, Schautzer F, Büttner U, Brandt T (2003) Treatment of downbeat nystagmus with 3,4-diaminopyridine: a placebo-controlled study. Neurology 61:165–170
Tabata H, Yamamoto K, Kawato M (2002) Computational study on monkey VOR adaptation and smooth pursuit based on the parallel control-pathway theory. J Neurophysiol 87:2176–2189
Tanaka M, Lisberger SG (2001) Regulation of the gain of visually guided smooth-pursuit eye movements by frontal cortex. Nature 409:191–194
Tanaka M (2005) Involvement of the central thalamus in the control of smooth pursuit eye movements. J Neurosci 25:5866–5876
Thier P, Ilg UJ (2005) The neural basis of smooth-pursuit eye movements. Curr Opin Neurobiol 15:645–652
Waespe W, Cohen B, Raphan T (1983) Role of the flocculus and paraflocculus in optokinetic nystagmus and visual-vestibular interactions: effects of lesions. Exp Brain Res 50:9–33
Wagner JN, Glaser M, Brandt T, Strupp M (2007) Aetiology of downbeat nystagmus: a retrospective study on 117 patients. In: Abstract for the 59th annual meeting of the AAN.2007.Boston.S07.005
Walker MF, Zee DS (2005a) Asymmetry of the pitch vestibulo-ocular reflex in patients with cerebellar disease. Ann NY Acad Sci 1039:349–358
Walker MF, Zee DS (2005b) Cerebellar disease alters the axis of the high-acceleration vestibuloocular reflex. J Neurophysiol 94:3417–3429
Yasui S, Young LR (1975) Perceived visual motion as effective stimulus to pursuit eye movement system. Science 190:906–908
Zee DS, Friendlich AR, Robinson DA (1974) The mechanism of downbeat nystagmus. Arch Neurol 30:227–237
Zee DS, Leigh RJ, Mathieu-Millaire F (1980) Cerebellar control of ocular gaze stability. Ann Neurol 7:37–40
Zee DS, Yamazaki A, Butler PH, Gücer G (1981) Effects of ablation of flocculus and paraflocculus of eye movements in primate. J Neurophysiol 46:878–899
Zhang Y, Partsalis AM, Highstein SM (1995a) Properties of superior vestibular nucleus flocculus target neurons in the squirrel monkey. I. General properties in comparison with flocculus projecting neurons. J Neurophysiol 73:2261–2278
Zhang Y, Partsalis AM, Highstein SM (1995b) Properties of superior vestibular nucleus flocculus target neurons in the squirrel monkey. II. Signal components revealed by reversible flocculus inactivation. J Neurophysiol 73:2279–2292
Zupan LH, Merfeld DM, Darlot C (2002) Using sensory weighting to model the influence of canal, otolith and visual cues on spatial orientation and eye movements. Biol Cybern 86:209–230
Acknowledgments
This work was supported by the Swiss National Science Foundation (3231-051938.97/31-63465.00/#3200BO-1054534), the Betty and David Koetser Foundation for Brain Research (Zurich, Switzerland), the Bonizzi-Theler Foundation (Zurich, Switzerland), the Deutsche Forschungsgemeinschaft (GL 342/1-3), and the BMBF (BCCN Munich, project 01GQ0440). We wish to thank two anonymous reviewers for their helpful comments.
Competing interest
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Appendix
Appendix
Model equations (healthy system)
The model equations are given in Laplace notation, with s denoting the complex frequency (see Fig. 9 for graphical illustration of the model structure and the equations). Thus, the temporal derivative of a variable is given by multiplying it with s, and the temporal integration is given by dividing by s. As an example, the first equation is given both in the Laplace notation and as differential equation.

The model output, vertical eye position e, is determined by the motor command m according to the eye plant equation
with τ e = 0.2 s being the dominant time constant of the eye plant, or, as differential equation, by
Thus, the eye plant is modeled as first-order low-pass filter (see Glasauer 2007 for a review on eye plant models).
Inputs to the system are head angular position α in the pitch plane and vertical target position t. The semicircular canal transfer function relates head angular position α to afferent canal output ω c by
with a dominant time constant τ c = 5 s (e.g., Zupan et al. 2002) and a secondary time constant τ d = 0.01 s.
Utricular afferent output u is proportional to the sine of the head pitch angle with a gain factor g u (see below):
Thus, for the upright position (u = 0) there is no otolith influence on eye movements.
Retinal slip r s, which is the input to the smooth pursuit system, depends on head angular position, eye position, and target position by
where \( e^{ - \Delta t \cdot s} \)models a time delay of Δt = 100 ms due to visual processing. Retinal error, used to drive saccades, is simply given as \( r_{\text{e}} = \left( {t - \alpha - e} \right) \cdot e^{ - \Delta t \cdot s} . \)
The brainstem integrator is modeled as leaky integrator (e.g., Zee et al. 1981) with a time constant τ b = 5 s. It receives the burst input b from the saccadic burst generator (see below), the canal afferent signal ω c, the utricular afferent signal u, and the Purkinje cell (PC) afferent discharge p. Its output e i can thus be written as
Note that the stead state gain is not unity, but slightly lower, to achieve an inverse model of the eye plant together with the direct pathway. The motor command m is composed of the weighted direct pathway carrying b, ω c, and p, and the integrator output:
with c ft being a constant bias term at the level of the FTN compensating for the resting discharge of the PC discharge p (see Eqns. 8 and 9 below). Note that the utricular signal is supposed to pass exclusively through the integrator (Eq. 5), as suggested by studies on static ocular counterroll (Glasauer et al. 2001, Crawford et al. 2003).
The floccular PC population is supposed to receive an efference copy of the motor command m processed by an internal model of the eye plant (Eq. 1) to yield an internal estimate of eye velocity v e (Glasauer 2003):
Additionally the PCs receive a copy of the canal signal, a copy of the burst command b, and visual input v from MST via the pontine nuclei. The output of the PCs is thus given by
with g being a gain parameter (see below), τ pc = 0.01 s the time constant of the PCs, and F(x) being the non-linear activation function which determines the resting discharge rate of the PCs and limits their discharge to positive values. F(x) was modeled as log-sigmoid function (e.g., Dayan and Abbott 2001):
with the saturation parameter g pc = 1 and the factor c = 4 yielding a resting discharge c ft = 0.5 and unity slope at zero input (Fig. 3a). In the range of normal gaze holding and smooth pursuit velocity, this non-linearity can safely be ignored, since the PC input remains in the linear range. For very high target velocity, pursuit would break down due to the non-linearity.
The visual input driving the smooth pursuit response is thought to originate in middle superior temporal area (MST) as internal estimate of target velocity in space. Therefore, it consists of retinal slip, the internal estimate of eye velocity, and the canal output as internal estimate of head angular velocity multiplied by a gain factor g v (see below):
Note that estimated gaze velocity in space (eye-in-head plus head-in-space) has to be delayed by the same amount of time Δt as the retinal slip (Eq. 4) to allow for reconstruction of target velocity. If light is off, e.g., during VOR in darkness, this signal is set to zero, i.e., desired gaze velocity in space then becomes zero.
Finally, the saccadic burst b is computed from the retinal error in a simplistic fashion via an internal feedback loop so that the saccade simply has constant velocity and reaches the target. Note that the exact implementation of the burst generator is not of importance for the overall performance of the model. Therefore, it is described separately below.
The ten equations above comprise the complete model for slow eye movements or saccades, if the burst command is regarded as additional model input. The equations can be reduced to a single overall transfer function for each input. Ignoring the time delay for smooth pursuit and the small time constant of the PCs, this transfer function is a leaky integrator with a time constant \( \tau = \tau _{\text{b}} \cdot \left( {1 + g} \right) \) (see also Eq. 12 below).
Free parameters of the model are the utricular gain factor g u , the PC gain g, the visual pathway gain g v, the PC saturation term g pc, and the bias term c ft. All of these parameters can be determined on the basis of the desired ocular motor responses. As mentioned above, g pc and c ft are closely related, since c ft compensated for the PC resting discharge, which is determined by g pc (see below). PC saturation has to be large enough to keep the PC response from reaching zero within the range of smooth pursuit eye velocity, and was set to g pc = 1, which results in c ft = 0.5. The gain factor g u has been chosen as g u = 1/τ b, which results in a slow ocular drift towards an eye position \( e = \sin \left( \alpha \right). \) That is, around the upright position, the static vertical eye position would perfectly compensate for head tilt. Accordingly, the model predicts positional nystagmus as found in normal subjects (see Fig. 7). However, whether the assumed gain is valid would necessitate a more detailed analysis of positional drifts. The PC input gain has been chosen to g = 10 to achieve an overall time constant τ for gaze holding of about 50 s. The visual pathway gain g v is set to g v = (1 + g)/g to result in an appropriate smooth pursuit response, i.e., eye velocity equals target velocity except for the delay and the effect of the leaky integration.
Damage to the PC population
In the patient simulations (Figs. 6, 7), the parameter g pc of the activation function of the PC population (Eq. 9) was changed to g pc = 0.6 to mimic, e.g., a loss of Purkinje cells such as in cerebellar atrophy. No other parameters were changed. The scaling of the non-linearity resulted in a lower resting discharge of the PC population and a lower sensitivity (see Fig. 3a). Simulations in Fig. 8a were done by applying the same change in g pc to second PC population. For the simulation in Fig. 8b, we set g pc = 0 to abolish floccular output completely, and set c ft = 0.25 to avoid having nystagmus SPV of more than 25° per s (cf. Fig. 3b). Note that changes in c ft do not always imply that FTN resting rate changed: due to the feedback loop, the same effect can be achieved by a horizontal shift of the activation function (see Eq. 11 below).
Due to the feedback loop, the fixed point of the PC population for gaze straight ahead is not equal to its resting discharge. The resting discharge, i.e., the discharge for zero input is 0.5·g pc. The discharge of the PC population for gaze straight ahead is determined by:
with x being the PC input. x/g is, in this case, equivalent to the slow phase eye velocity of DBN (see Fig. 3b). Equation 11 has no closed-form solution for x, but for small g pc its solution x converges to \( x = \left( {c_{{\text{ft}}} - g_{{\text{pc}}} } \right) \cdot g \cdot \left( {\tau _{\text{b}} - \tau _{\text{e}} } \right)/\tau _{\text{b}} . \) Thus, the fixed point is shifted away from the point of symmetry (x = 0) of the non-linearity towards saturation. Consequently, additional pursuit input will cause an asymmetric output due to the saturation.
The negative velocity feedback loop
The negative velocity feedback loop constituting the central element responsible for gaze holding and integrator function (ignoring the PC time constant and linearizing the non-linearity) basically consists of the following three equations (here for a velocity command ω):
which can be reduced to a standard negative feedback system of the form
where F r is the feedback transfer function (basically the internal model estimating eye velocity) and F f the feedforward controller (the brainstem integrator and direct pathway generating the motor command). Its solution is
For stability, the real part of every pole of the right-hand-side transfer function must be negative. For our special case
and, therefore, for g > −1 the system is stable.
So far, we have assumed that the internal eye plant model and the feed-forward inverse model are both perfectly tuned. It is therefore worthwhile to consider a more general case. Let the forward transfer function be F f = A/B and the feedback function F r = gs/C with A, B, and C being first-order polynomials of s, i.e., A, B, and C are of the form \( 1 + \tau _{\text{i}} s \) with positive τ i and g (additional gain factors can be ignored without loss of generality). The feedback function is thus a high-pass filter. Then the overall transfer function becomes \( F = \frac{{BC}}{{BC + Ags}}. \) By solving the denominator for its roots, one can show that the two resulting roots always have negative real parts. Thus, the negative velocity feedback loop is stable independently of the time constants of the inverse or forward models or the brainstem integrator. Note that overall instable integrator behavior (centrifugal drift) as seen in some DBN patients can be generated by the model, if the inverse model is no longer a highpass filter, but contains a negative position component (effectively leading to a positive position feedback).
Saccadic burst generator
The motor error necessary to trigger a saccade is derived by first reconstructing target position in head from retinal error r e (subject to a delay Δt = 100 ms, see above) and delayed eye position, and then subtracting actual eye position from this estimate of target position in head:
If the motor error was lower than a threshold, the burst generator was silent:
If the motor error exceeded this threshold, a constant saccadic burst in the appropriate direction was issued which continued until the motor error reached zero:
where sgn() is the sign function (sgn(x) = 0 for x = 0; sgn(x) = 1 for x > 0; sgn(x) = −1 for x < 0). Accordingly, the burst terminates if eye position equals target position in head.
Rights and permissions
About this article
Cite this article
Marti, S., Straumann, D., Büttner, U. et al. A model-based theory on the origin of downbeat nystagmus. Exp Brain Res 188, 613–631 (2008). https://doi.org/10.1007/s00221-008-1396-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00221-008-1396-7