
Abstract
We report atypical opsoclonus in a patient with multiple system atrophy and propose a mechanism based on the patterns of modulation by visual, vestibular, and saccadic and vergence stimulation. Firstly, the 6-Hz opsoclonus mostly in the vertical plane occurred only after the development of downbeat nystagmus in darkness without visual fixation. Even after a substantial build-up, visual suppression of the opsoclonus was immediate and complete. Furthermore, the latency for re-emergence of opsoclonus in darkness was greater when the duration of preceding visual fixation was longer. Secondly, the effect of preceding downbeat nystagmus on the development of opsoclonus was evaluated by changing the head position. The opsoclonus did not occur in the supine position when the downbeat nystagmus was absent. After horizontal head shaking, the opsoclonus in the vertical plane gradually evolved into horizontal plane and resumed its vertical direction again after vertical head shaking. Thirdly, any opsoclonus was not triggered by imaginary saccades in the supine position. Lastly, combined vergence and saccadic eye movements during the Müller paradigm did not induce opsoclonus. From these findings of modulation, we suggest that the opsoclonus observed in our patient was invoked by vestibular signals. When the function of the omnipause neurons and saccadic system was impaired, the centrally mediated vestibular eye velocity signals may activate the saccadic system to generate opsoclonus. These atypical patterns of opsoclonus, distinct from the classic opsoclonus frequently observed in para-neoplastic or para-infectious disorders, may be an unrevealing sign of degenerative brainstem or cerebellar disorders.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Saccadic oscillations without an intersaccadic interval comprise ocular flutter and opsoclonus [1, 2]. When these oscillations are confined to the horizontal plane, they are called ocular flutter. In contrast, opsoclonus refers to the saccadic oscillations occurring in multiple planes [1, 2]. The typical opsoclonus and ocular flutter characterized by irregular short bursts of saccades and following quiescence usually inform potentially life-threatening underlying conditions such as para-infectious and para-neoplastic disorders [3, 4]. These eye movements may be ascribed to membrane dysfunction of the burst neurons for saccades and inherently unstable positive feedback neural circuit [5,6,7,8]. According to this explanation, the role of brainstem structures is more important for the generation of ocular flutter and opsoclonus. Otherwise, the cerebellar structures, especially the fastigial nuclei that are responsible for saccadic initiation and accuracy, may be involved in the generation of ocular flutter and opsoclonus [9, 10]. Even though these saccadic oscillations may be seen without any provocation, milder forms may be observed only when the activity of the omnipause neurons is suppressed, like during voluntary saccades. In addition, ocular flutter and opsoclonus may be provoked by non-saccadic stimuli including smooth pursuit [11], vergence [12], eye blinking [13], and positional change [14, 15].
The formulation of this study was based on our anecdotal observation of atypical opsoclonus in a patient with multiple system atrophy, developing only in association with downbeat nystagmus in darkness and changing its main direction from vertical into horizontal after horizontal head shaking. In this study, we evaluated modulation of opsoclonus with regard to visual, vestibular, and saccadic and vergence stimulation and propose the mechanism opsoclonus in this patient. In addition, we addressed the clinical significance of this type of opsoclonus distinct from the typical form of opsoclonus.
Method
Patient
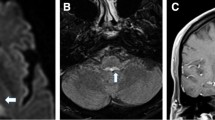
A 63-year-old man presented with dizziness, dysarthria, and unsteadiness that had worsened over the preceding 2 years. The patient had a diagnosis of rapid eye movement sleep disorder and orthostatic hypotension 4 months before. Examination showed no spontaneous or gaze-evoked nystagmus during visual fixation. In darkness without visual fixation, the patient showed spontaneous downbeat nystagmus that increased while lying down and straight head hanging. He also showed paroxysmal vertical oscillation of the eyes without visual fixation, which will be detailed further in the following section. Nevertheless, the patient denied worsening of dizziness with development of paroxysmal vertical oscillation. Smooth pursuit was impaired in both horizontal directions, and saccades were bilaterally hypermetric, but more to the right. Bedsides, horizontal head impulse test was normal in both directions. Visual suppression of the vestibulo-ocular reflex (VOR) was also impaired during en bloc eye-head motion. There was no palatal tremor. The deep tendon reflexes were increased, and coordination was impaired in both the upper and lower extremities. He could maintain balance while standing, but the gait was unstable and wide-based, especially initially, with a gradual improvement during continued gait. Brain MRIs 2 years before showed only mild atrophy of the superior and middle cerebellar peduncles, but follow-up MRIs disclosed prominent and diffuse atrophy in the whole brainstem and cerebellum with a pontine hot cross bun appearance (Fig. 1a, b). Evaluation for cerebellar dysfunction due to para-neoplastic, para-infectious, or other metabolic disorders was unremarkable. The patient was diagnosed with multiple system atrophy (MSA).

MRIs and saccadic oscillations of the patient. The initial (a) and follow-up MRIs (b) with a 2-year interval show progressive atrophy involving the brainstem and cerebellum with an interval development of the hot cross bun appearance. Vertical ocular oscillations (c) follow the downbeat nystagmus only in darkness. The amplitude and velocity profile of the ocular oscillations (d) fit into the amplitude-velocity relationship (main sequence) of normal saccades. RH/LH, right/left eye horizontal; RV/LV, right/left eye vertical
Atypical Opsoclonus
The patient showed paroxysmal involuntary vertical ocular oscillation that always followed spontaneous downbeat nystagmus in darkness (Video 1). These oscillations were also accompanied by minor but synchronized horizontal oscillations at 6 Hz with an intermittent pause of 500 ms (Fig. 1c). In each cluster of ocular oscillations, the amplitude of eye motion gradually increased from about 5° to 25° with the maximal peak velocity of oscillations at 360°/s. These eye movements immediately disappeared with visual fixation. The relationship between the amplitude and velocity of ocular oscillation well fit into that of normal saccades (the main sequence, Fig. 1d). Thus, the ocular oscillations were most consistent with opsoclonus (refer to the “Discussion” section for additional explanation). Repeated examinations 1 and 3 months later showed a persistence of the opsoclonus without an interval change. Single or combined administration of baclofen, betahistidine, clonazepam, and 3,4-diaminopyridine did not improve the symptoms or ocular motor signs.
Quantified Ocular Motor Paradigms
We noted two interesting features of ocular flutter in this patient. One was that it developed only in darkness and ceased with visual fixation. The other was that it mostly occurred in the vertical plane and only after downbeat nystagmus. Ocular flutter and opsoclonus may occur spontaneously but may also be triggered when the omnipause neurons are suppressed, likewise during voluntary saccades, eye blinking, saccadic-vergence responses in response to the Müller paradigm, or positional changes. However, the effects of visual fixation and preceding nystagmus on the development of opsoclonus have not been investigated. Therefore, we evaluated modulation of opsoclonus during visual fixation and preceding nystagmus using the following protocol. First, we determined the effect of visual fixation on opsoclonus by applying a visual target (paradigm 1). For this, we measured the latency for emergence of opsoclonus after elimination of the visual target. Because the initial saccadic oscillations were weak and coarse, we defined the latency as the time required for the eye velocity to reach 200°/s. The patient fixated a visual target for 3 min with the head upright while sitting. The target was then eliminated and the latency for development of ocular flutter was measured. In addition, we evaluated the post-suppression effect of visual fixation on opsoclonus as follows. After the development of substantial opsoclonus in darkness, the target for visual fixation was presented for three different durations (5, 10, and 15 s). Then, we measured the latency for re-emergence of opsoclonus according to the duration of visual fixation. Second, we determined the effect of preceding downbeat nystagmus on the development of opsoclonus. For this, we measured opsoclonus in supine and head-bending position (paradigm 2). As suggested in a previous study [16], the downbeat nystagmus was absent in the supine (− 90° in pitch axis) but present in the head bending (30° in pitch axis) position in our patient. In each position, we observed the development of opsoclonus with recording of eye motion for more than three times the latency measured in darkness while sitting. Third, we further investigated the effects of mastoid vibration, head impulses, and horizontal and vertical head shaking (paradigm 3), visually mediated and imaginary saccades, and saccade-vergence responses using the Müller paradigm (paradigm 4). As described elsewhere [17, 18], the horizontal and vertical head shakings were delivered for 15 s with an approximate amplitude of 15° and frequency at 2 Hz. For visually mediated saccades, the target jumped horizontally and vertically at the amplitudes of 15° and 30° with a 2-s fixation period. For imaginary saccades, the patient was asked to generate saccades to the imaginary target at the positions adopted in the previous visually mediated saccade trials. In the Müller experiments [19], the far and near targets were aligned to the patient’s right eye with distances of 1.2 m and 30 cm. During the gaze shifts between the far and near targets, the change in vergence angle was measured at about 10°. Eye movements were recorded using three-dimensional video-oculography with a sampling rate of 120 Hz (Easy-Eye, SLMED, Korea). The study protocols were approved by the Institutional Review Board of Seoul National University Bundang Hospital (B-1109/135-106), and written informed consent was obtained from the patient.
Results
The latency of opsoclonus in darkness was about 24.7 s (Fig. 2a). After the opsoclonus built up substantially, a visual target was presented at 1.2 m for the duration of 5, 10, and 15 s. For each condition, the opsoclonus was suppressed completely and re-emerged with a latency of 0.5, 1.7, and 4.7 s after the removal of the visual target (paradigm 1, Fig. 2b).

Effect of visual fixation and downbeat nystagmus on ocular flutter. The latency defined as the time required for the peak velocity of saccades first reaches at 200°/s is 24.7 s after an elimination of visual fixation (a). The flutter ceased immediately with visual fixation, and the effect of post-visual suppression is proportional to the duration of visual fixation (b). While supine (− 90° pitch head position) when downbeat nystagmus was absent, vertical flutter did not develop. When the head was bended forward (30° pitch head position), the ocular flutter followed downbeat nystagmus (c). VV, vertical velocity; RV/LV, right/left eye vertical; VHV, vertical head velocity
When the supine position was assumed slowly so that no downbeat nystagmus was present, opsoclonus did not develop for an observation period of 80 s. In contrast, when the head was bended to reach 30° pitch position and downbeat nystagmus appeared, the opsoclonus developed with a latency of 50 s (paradigm 2, Fig. 2c).
Opsoclonus did not change the direction after mastoid vibration or single head impulses. After horizontal head shaking, the main direction of opsoclonus slowly changed from vertical to oblique, and then to horizontal (Video 2, Fig. 3a). After the direction of opsoclonus was mostly aligned horizontally, vertical head shaking reversed the main direction of opsoclonus from horizontal into oblique and then to vertical (paradigm 3, Video 3, Fig. 3b).

Effects of head shaking and imaginary saccades on ocular flutter. After horizontal head shaking, the vertical flutter evolves into horizontal flutter (a). After vertical head shaking, the flutter resumes its vertical direction (b). No flutter was induced by imaginary horizontal (c) and vertical saccades (d) while supine (− 90° pitch position) without visual fixation when downbeat nystagmus was absent. VV, vertical velocity; RV/LV, right/left eye vertical; VHV, vertical head velocity
Finally, either the visually mediated or imaginary saccades did not induce opsoclonus. Müller experiments did not induce opsoclonus either (paradigm 4, Fig. 3c, d).
Discussion
Opsoclonus and ocular flutter have been considered signs of dysfunction of the brainstem and cerebellum [1, 2] where the saccadic neural connections create positive and negative feedback loops among the ocular motor nuclei, excitatory and inhibitory premotor burst neurons, and fastigial nuclei [5,6,7,8]. The typical opsoclonus and ocular flutter occur primarily in para-infectious or para-neoplastic disorders, but may be rarely observed in drug toxicity, metabolic disorders [1,2,3,4] and degenerative disorders including Friedreich’s ataxia and Krabbe’s disease, and MSA [14, 20, 21]. Given that the opsoclonus in our patient was observed only after downbeat nystagmus in darkness with a substantial latency, ocular flutter and opsoclonus might have been underrecognized in patients with cerebellar degeneration.
With regard to opsoclonus of this patient, there are several key observations to be discussed. First is the effect of visual fixation on the opsoclonus. In our patient, the opsoclonus was only seen in darkness without visual fixation. There was no ocular flutter during visual fixation and during visually mediated saccades and smooth pursuit. As is well known, the release of omnipause suppression is essential for the development of ocular flutter and opsoclonus [1, 2]. The omnipause neurons are involved in visual fixation by receiving crossed projections from the rostral pole of superior colliculus, and their steady firing rate decreases in darkness [22, 23]. It appears that hypoactive omnipause neuron in darkness would lead to vulnerable condition for developing saccadic intrusions as shown for square wave jerks in healthy subjects, which increased their frequency and amplitude in darkness without fixation [24, 25]. Therefore, we may assume that the omnipause neurons, when partially damaged, manage to suppress ocular oscillations during visual fixation but not in darkness. It should be noted that, in our study, the visual suppression of ocular flutter was immediate and strong, but the latency for re-emergence of opsoclonus in darkness depended on the duration of preceding visual fixation. Thus, it seemed to take more time for the opsoclonus to emerge when the duration of preceding visual fixation was longer. The conductance-based model of burst neurons for ocular flutter and opsoclonus stated that glycinergic inhibition by the omnipause neurons induce hyperpolarization and resultantly increase in the depolarizing threshold of the burst neuron membrane [8, 26]. Therefore, the immediate visual suppression of opsoclonus in our patient may reflect the instantaneous effect of glycinergic inhibition by increasing the depolarization threshold. On the other hand, the post-suppression effect suggests a gradual recovery of the hyperpolarization shift in the resting potential, which depends on the duration of glycinergic suppression.
Second is the relationship between the vestibular eye motion and opsoclonus. This relationship was initially inferred from downbeat nystagmus always preceding the opsoclonus in darkness. In addition, the opsoclonus did not occur in the static head position when downbeat nystagmus was not present. Furthermore, the direction of opsoclonus was changed into horizontal after horizontal head shaking and returned to vertical after vertical head shaking. Based on these findings, we assumed that the opsoclonus may have been evoked by vestibular eye motion. Since nystagmus contains both slow vestibular and fast saccadic components, then we further defined the trigger of opsoclonus by inducing saccades. Because opsoclonus was not induced during imaginary horizontal and vertical saccades in the supine position when downbeat nystagmus was absent, we could get to the conclusion that the slow components of vestibular eye motion were involved in the generation of opsoclonus in this patient.
Then, how could the vestibular signals induce opsoclonus in this patient. In a previous report, the opsoclonus induced during positioning was explained by a lesion-induced ephaptic transmission between the vestibular pathways and saccadic burst neurons [15]. However, given the strict alignment in the direction of opsoclonus and vestibular stimulation in our patient, we may propose another explanation (Fig. 4). The vestibular and saccadic systems are interconnected within the brainstem to function for the eye-head movements and quick phases of vestibular or optokinetic nystagmus [27]. Though it still remains uncertain in humans, anatomical and physiological evidence explains the mechanism of quick phases of vestibular nystagmus as follows [28]: during vestibular stimulation, the vestibular-only neurons in the ipsilateral vestibular nucleus project to the burst driver neurons in the region of contralateral nucleus prepositus hypoglossi [29]. The burst driver neurons then send excitatory projections to several neurons including the contralateral long lead burst neurons which turn off the omnipause neurons and the contralateral excitatory and inhibitory burst neurons that generate ipsilateral saccades and prevent contralateral eye motion [29]. In addition to this multi-synaptic pathway, vestibular nucleus neurons are known to project directly to the nucleus raphe interpositus where omnipause neurons lie [30]. Through these direct and indirect neural connections, the slow and quick phases of vestibular nystagmus can occur harmoniously during vestibular stimulation. In our patient, the continuous velocity bias from downbeat nystagmus may have led to inhibition of the omnipause neurons and excitation of the burst neurons in the rostral interstitial nucleus of medial longitudinal fasciculus via the multi-synaptic pathway. The velocity bias in the vestibular nucleus may also have reduced the neuronal activity of omnipause neurons directly. If the omnipause neurons are damaged partially as we postulated, the excited burst neurons can oscillate within their unstable neural circuit and finally generate opsoclonus in darkness where the activity of damaged omnipause neurons diminishes further. This can also explain the modulation of opsoclonus direction after horizontal and vertical head shaking.

Hypothetical mechanism of opsoclonus by adopting the interaction between the vestibular and saccadic systems. A suggested mechanism for the fast component of vestibular nystagmus in cats (a). In darkness, the velocity signals in the vestibular nucleus are integrated into position signals in the nucleus prepositus hypoglossi or interstitial nucleus of Cajal. At a certain level of threshold, which may be related to the customary ocular motor range, the position signals excite the long-lead burst neurons, which in turn inhibit the omnipause neurons suppressing the bust neurons. With this decrease of omnipause suppression, the burst neurons generate the fast component of vestibular nystagmus. In our patient, visual fixation in the light may have prevented any kind of eye movements by suppressing the vestibular eye motion and enhancing the omnipause activity despite the pathologically unstable positive and negative feedback loops (b). In contrast, with an elimination of visual fixation, vestibular eye velocity signals may activate the pathologically unstable saccade system, leading to opsoclonus (c). VN, vestibular nucleus; NPH, nucleus prepositus hypoglossi; INC, interstitial nucleus of Cajal; LLBN, long-lead burst neuron; OPN, omnipause neuron; EBN/IBN, excitatory/inhibitory burst neuron; FN, fastigial nucleus; OMN, ocular motor nucleus; EP, eye position
Another observation to note is the continuation of rhythmic opsoclonus at 6 Hz. These characteristics are distinct from those of typical opsoclonus showing irregular alteration of burst and pause and relatively higher frequency of 10 to 25 Hz [1, 2]. Accordingly, the ocular oscillations in our patient may be misinterpreted as a form of acquired pendular nystagmus that mimics opsoclonus. However, the peak velocity of the oscillations more than 200°/s (up to 360°/s) seems inconsistent with pendular nystagmus. The relationship between the amplitude and velocity of ocular oscillation fits with that of normal saccades. Thus, it is reasonable to diagnose opsoclonus. Additionally, the proposed conductance-based model of burst neurons may explain the sustained rhythmicity [2, 8]. The neuronal excitability of burst neurons is determined by mixed cation currents (Ih) activated by hyperpolarization and low-threshold calcium currents (It) [7]. The burst neurons show rhythmic and sustained oscillatory activity due to these cation currents when the membrane resting potential is below the certain level (i.e., − 58 mV). In contrast, they show tonic firing responses above that level [31]. These characteristics are important for post-inhibitory rebound activity of saccadic premotor burst neurons [5, 7, 8]. If the damage to omnipause neurons is partial but stable, the premotor burst neurons may oscillate by themselves and generate continuous and rhythmic saccadic oscillation, like those observed in the Müller paradigm [12, 19]. In contrast, typical opsoclonus may be attributed to lesions involving both the burst and omnipause neurons with an immune-mediated phasic irritation [32, 33]. In terms of frequency, opsoclonus of higher amplitude (up to 25°) may show a lower frequency. Indeed, higher frequency oscillations were mostly observed in ocular flutter or opsoclonus of smaller amplitude (usually less than 2°) [5,6,7,8]. In addition, the proposed mechanisms of ocular flutter and opsoclonus can also account for the low frequency opsoclonus in our patient. The brainstem mechanism of ocular flutter and opsoclonus assumes two conditions. One is the inherently unstable positive feedback loop that comprises the excitatory and inhibitory burst neurons within the brainstem. The other is the pathologically increased membrane excitability of the burst neurons [5,6,7,8]. A previous model simulation weighing on the brainstem mechanism indeed succeeded in generating eye oscillation (low-frequency and high-velocity) similar to that observed in our patient by changing the parameters related to the characteristics of burst neuron membrane [5]. Alternatively, the cerebellar mechanism may more easily explain the relatively lower frequency oscillation due to multi-synaptic connections. Our patient had saccadic hypermetria more to the right, suggesting asymmetric dysfunction of the fastigial nuclei. The fastigial nucleus is known to control the onset and termination of saccades [1, 2], and the cerebellar mechanism of opsoclonus involves a negative feedback loop that may contain the efference copy of eye position signals passing through the fastigial nucleus [9, 10]. Therefore, in our patient, asymmetric dysfunction of the fastigial nuclei and impaired omnipause neurons may have contributed to the generation of ocular flutter and opsoclonus at various frequencies along with unstable cerebellar feedback loop [9, 10].
The low frequency, sustained rhythmicity, developments only in darkness and modulation by the vestibular signals are the features distinct from those of classic opsoclonus. This saccadic abnormality may be an occult sign of degenerative brainstem and cerebellar dysfunction and needs to be evaluated further in more patients.
References
Leigh RJ and Zee DS. The neurology of eye movements. OUP USA; 2015.
Ramat S, Leigh RJ, Zee DS, Optican LM. What clinical disorders tell us about the neural control of saccadic eye movements. Brain. 2007;130:10–35. https://doi.org/10.1093/brain/awl309.
Armangue T, Sabater L, Torres-Vega E, Martinez-Hernandez E, Arino H, Petit-Pedrol M, et al. Clinical and immunological features of opsoclonus-myoclonus syndrome in the era of neuronal cell surface antibodies. JAMA Neurol. 2016;73:417–24. https://doi.org/10.1001/jamaneurol.2015.4607.
Oh SY, Kim JS, Dieterich M. Update on opsoclonus-myoclonus syndrome in adults. J Neurol. 2019;266:1541–8. https://doi.org/10.1007/s00415-018-9138-7.
Ramat S, Leigh RJ, Zee DS, Optican LM. Ocular oscillations generated by coupling of brainstem excitatory and inhibitory saccadic burst neurons. Exp Brain Res. 2005;160:89–106. https://doi.org/10.1007/s00221-004-1989-8.
Ramat S, Leigh RJ, Zee DS, Shaikh AG, Optican LM. Applying saccade models to account for oscillations. Prog Brain Res. 2008;171:123–30. https://doi.org/10.1016/S0079-6123(08)00616-X.
Shaikh AG, Ramat S, Optican LM, Miura K, Leigh RJ, Zee DS. Saccadic burst cell membrane dysfunction is responsible for saccadic oscillations. J Neuroophthalmol. 2008;28:329–36. https://doi.org/10.1097/WNO.0b013e31818eb3a5.
Shaikh AG, Zee DS, Optican LM, Miura K, Ramat S, Leigh RJ. The effects of ion channel blockers validate the conductance-based model of saccadic oscillations. Ann N Y Acad Sci. 2011;1233:58–63. https://doi.org/10.1111/j.1749-6632.2011.06130.x.
Zee DS, Robinson DA. A hypothetical explanation of saccadic oscillations. Ann Neurol. 1979;5:405–14. https://doi.org/10.1002/ana.410050502.
Wong AM, Musallam S, Tomlinson RD, Shannon P, Sharpe JA. Opsoclonus in three dimensions: oculographic, neuropathologic and modelling correlates. J Neurol Sci. 2001;189:71–81.
Scholz J, Vieregge P, Ruff C. Paraneoplastic opsoclonus-myoclonus syndrome in metastatic ovarian carcinoma. J Neurol Neurosurg Psychiatry. 1994;57:763–4. https://doi.org/10.1136/jnnp.57.6.763-a.
Bhidayasiri R, Somers JT, Kim JI, Ramat S, Nayak S, Bokil HS, et al. Ocular oscillations induced by shifts of the direction and depth of visual fixation. Ann Neurol. 2001;49:24–8. https://doi.org/10.1002/1531-8249(200101)49:1<24::aid-ana6>3.0.co;2-t.
Hain TC, Zee DS, Mordes M. Blink-induced saccadic oscillations. Ann Neurol. 1986;19:299–301. https://doi.org/10.1002/ana.410190315.
Brodsky MC, Hunter JS. Positional ocular flutter and thickened optic nerves as sentinel signs of Krabbe disease. J Am Assocr Pediat Ophth Strab. 2011;15:595–7.
Martins AI, Nunes C, Macario MC, Lemos J. Positional ocular flutter associated with middle cerebellar peduncle demyelination. J Neuroophthalmol. 2019;39:117–9. https://doi.org/10.1097/WNO.0000000000000720.
Marti S, Palla A, Straumann D. Gravity dependence of ocular drift in patients with cerebellar downbeat nystagmus. Ann Neurol. 2002;52:712–21. https://doi.org/10.1002/ana.10370.
Choi KD, Oh SY, Park SH, Kim JH, Koo JW, Kim JS. Head-shaking nystagmus in lateral medullary infarction: patterns and possible mechanisms. Neurology. 2007;68:1337–44. https://doi.org/10.1212/01.wnl.0000260224.60943.c2.
Yang Y, Kim JS, Kim S, Kim YK, Kwak YT, Han IW. Cerebellar hypoperfusion during transient global amnesia: an MRI and oculographic study. J Clin Neurol. 2009;5:74–80. https://doi.org/10.3988/jcn.2009.5.2.74.
Ramat S, Das VE, Somers JT, Leigh RJ. Tests of two hypotheses to account for different-sized saccades during disjunctive gaze shifts. Exp Brain Res. 1999;129:500–10. https://doi.org/10.1007/s002210050920.
Fahey MC, Cremer PD, Aw ST, Millist L, Todd MJ, White OB, et al. Vestibular, saccadic and fixation abnormalities in genetically confirmed Friedreich ataxia. Brain. 2008;131:1035–45. https://doi.org/10.1093/brain/awm323.
Shindo K, Onohara A, Hata T, Kobayashi F, Nagasaka K, Nagasaka T, et al. Opsoclonus-myoclonus syndrome associated with multiple system atrophy. Cerebellum Ataxias. 2014;1:15. https://doi.org/10.1186/s40673-014-0015-6.
Evinger C, Kaneko CR, Fuchs AF. Activity of omnipause neurons in alert cats during saccadic eye movements and visual stimuli. J Neurophysiol. 1982;47:827–44. https://doi.org/10.1152/jn.1982.47.5.827.
Munoz DP, Wurtz RH. Fixation cells in monkey superior colliculus. II. Reversible activation and deactivation. J Neurophysiol. 1993;70:576–89. https://doi.org/10.1152/jn.1993.70.2.576.
Shallo-Hoffmann J, Sendler B, Muhlendyck H. Normal square wave jerks in differing age groups. Invest Ophthalmol Vis Sci. 1990;31:1649–52.
Ohtsuka K, Mukuno K, Ukai K, Ishikawa S. The origin of square wave jerks: conditions of fixation and microsaccades. Jpn J Ophthalmol. 1986;30:209–15.
Shaikh AG, Miura K, Optican LM, Ramat S, Leigh RJ, Zee DS. A new familial disease of saccadic oscillations and limb tremor provides clues to mechanisms of common tremor disorders. Brain. 2007;130:3020–31. https://doi.org/10.1093/brain/awm240.
Guitton D, Mandl G. A comparison between saccades and quick phases of vestibular nystagmus in the cat. Vis Res. 1980;20:865–73.
Curthoys IS. Generation of the quick phase of horizontal vestibular nystagmus. Exp Brain Res. 2002;143:397–405. https://doi.org/10.1007/s00221-002-1022-z.
Ohki Y, Shimazu H, Suzuki I. Excitatory input to burst neurons from the labyrinth and its mediating pathway in the cat: location and functional characteristics of burster-driving neurons. Exp Brain Res. 1988;72:457–72.
Langer TP, Kaneko CR. Brainstem afferents to the oculomotor omnipause neurons in monkey. J Comp Neurol. 1990;295:413–27. https://doi.org/10.1002/cne.902950306.
McCormick DA, Pape HC. Properties of a hyperpolarization-activated cation current and its role in rhythmic oscillation in thalamic relay neurons. J Physiol. 1990;431:291–318. https://doi.org/10.1113/jphysiol.1990.sp018331.
Shaikh AG, Hain TC, Zee DS. Oculomotor disorders in adult-onset Still’s disease. J Neurol. 2010;257:136–8. https://doi.org/10.1007/s00415-009-5308-y.
Markakis I, Alexiou E, Xifaras M, Gekas G, Rombos A. Opsoclonus-myoclonus-ataxia syndrome with autoantibodies to glutamic acid decarboxylase. Clin Neurol Neurosurg. 2008;110:619–21. https://doi.org/10.1016/j.clineuro.2008.03.005.
Funding
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (NRF-2017R1C1B1008582).
Author information
Authors and Affiliations
Contributions
Dr. J.Y. Lee analyzed and interpreted the data and wrote the manuscript. Drs. H.J. Kim and H.J. Oh analyzed and interpreted the data and revised the manuscript. Dr. J.Y. Choi designed and conceptualized the study, interpreted the data, and revised the manuscript. Dr. J. S Kim interpreted the data and critically revised the manuscript.
Corresponding authors
Ethics declarations
Conflict of Interest
Dr. J.S. Kim serves as an associate editor of Frontiers in Neuro-otology and on the editorial boards of the Journal of Korean Society of Clinical Neurophysiology, the Journal of Clinical Neurology, Frontiers in Neuro-ophthalmology, the Journal of Neuro-ophthalmology, the Journal of Vestibular Research, and the Journal of Neurology, and Medicine. Others have no conflicts of interest to disclose.
Ethical Standard
This study followed the tenets of the Declaration of Helsinki and was performed according to the guidelines of the Institutional Review Board of Seoul National University Bundang Hospital (B-1109/135-106).
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
(MP4 61,462 kb)
(MP4 107,753 kb)
(MP4 100,614 kb)
Rights and permissions
About this article

Cite this article
Lee, JY., Kwon, E., Kim, HJ. et al. Opsoclonus Following Downbeat Nystagmus in Absence of Visual Fixation in Multiple System Atrophy: Modulation and Mechanisms. Cerebellum 20, 724–733 (2021). https://doi.org/10.1007/s12311-019-01090-w
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12311-019-01090-w