
Abstract
Mutations in ganglioside-induced differentiation-associated-protein 1 (GDAP1) have been associated with both subtypes of Charcot–Marie–Tooth (CMT) disease, autosomal recessive (CMT4A and AR-CMT2K) and autosomal dominant (AD-CMT2K). Over 80 different mutations have been identified so far. With the use of Sanger sequencing and Next Generation Sequencing (NGS) technologies, we screened a cohort of 306 unrelated Chinese CMT patients and identified 8 variants in the GDAP1 gene in 4 families, 5 of which were novel (R41H, N201Kfs*5, Q38X, V215I and Q38R). Application of Bioinformatics tool and classification according to the American College of Medical Genetics (ACMG) predicted them of being likely pathogenic with the exception of one variant of uncertain significance (VUS). In addition, we detected the presence of a single heterozygous variant of uncertain significance H256R in one additional family from the CMT cohorts. We found a GDAP1 prevalence rate of 1.63% (5/306). Three families (families 1, 2 and 3) were found to have an autosomal recessive (AR) pattern of inheritance while family 4 displayed an autosomal dominant (AD) mode of inheritance. Electro-physiologic studies revealed an axonal type of neuropathy (AR-CMT2K and AD-CMT2K) in all affected individuals. Clinical characteristics and findings in our study demonstrated that the recessive form presented earlier in life and exhibited severe symptoms and rapid evolution compared to the dominant form. We observed a marked intra-familial variability within the AD family in terms of age at disease onset, symptom severity and clinical progression. Our study expands the mutational spectrum of GDAP1-related CMT disease with the identification of new and unreported GDAP1 variants and demonstrates the predominance of the axonal form of neuropathy in CMT disease associated with GDAP1. We highlight the clinical characteristics associated with these genotypes and describe the relative frequency of GDAP1 variants amongst the Chinese population.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Charcot–Marie–Tooth (CMT) disease is the most common inherited neuropathy and is a clinically and genetically heterogeneous group of disorders. The classical phenotype is characterized by an early age at onset with slowly progressive weakness and atrophy of the distal muscles predominantly affecting the lower limbs, foot deformities, reduced or absent deep tendon reflexes and mild sensory deficit [1], with an estimated prevalence of 1 in 2500 [2]. The mode of inheritance of CMT includes autosomal dominant (AD), autosomal recessive (AR), and X-linked inheritance. However, sporadic cases have also been largely reported in literatures. Neurophysiology differentiates CMT into 3 groups: CMT1 (demyelinating) with very slow motor nerve conduction velocity (MNCV) (< 38 m/s) at upper limbs with nerve biopsy demonstrating segmental de- and remyelination in association with onion bulb formation; CMT2 (axonal) with normal or a slight reduction of MNCV (> 38 m/s) at upper limbs with histopathology showing signs of axonal impairment characterized by axonal loss and regeneration clusters and ICMT (intermediate) with MNCV between 25 and 45 m/s at upper limbs and features of both demyelination and axonal degeneration [3, 4].
The ganglioside-induced differentiation-associated protein 1 (GDAP1) is an integral membrane protein of the outer mitochondrial membrane with a ubiquitous tissue distribution but predominantly expressed in neurons, particularly in the brain, spinal cord and dorsal root ganglion [5, 6]. It has a strong influence and impact on the maintenance and regulation of normal functioning, structural integrity and intracellular networking of the mitochondria [6]. It is one of the few genes linked to both autosomal recessive and autosomal dominant forms of CMT disease. The recessive forms include a demyelinating subtype CMT4A (OMIM 214400) [7], an axonal subtype AR-CMT2K (OMIM 607831) [8, 9] and an intermediate form RI-CMT (OMIM 608340) [10, 11]. The dominant form AD-CMT2K (OMIM 607831), accounts for a relatively few cases [12,13,14].
Population data and prevalence rate of GDAP1 mutations are extremely limited and highly variable within the general population. This is attributed to numerous factors such as the geographical distribution, ethnic background, areas of high consanguinity and the effects of founder mutations. The rarity of GDAP1-related CMTs in the Asian population can be acknowledged by the very few cases reported in literatures so far [15,16,17,18,19]. The relationship between the genotype and the phenotype is poorly understood because of the complex heterogeneity. In the present study, we performed both Sanger sequencing and targeted multi-gene panel analysis in a large cohort of Chinese patients with CMT to investigate genotype–phenotype correlation and determine the frequency of distribution.
Subjects and methods
Patients and evaluation
We analyzed 306 unrelated Chinese CMT patients/families, enrolled from the outpatient neurology clinic of The Third Xiangya Hospital and Xiangya Hospital from 2000 to 2016. All patients underwent a complete neurological examination by two neurologists and met with the CMT diagnosis criteria based on the clinical phenotype, mode of inheritance, electro-physiologic findings and molecular analyses [20]. Ethics approval was acquired at all institutions and written informed consent was obtained from all participants and/or their parents prior to their inclusion in this study. Disease severity was evaluated with the application of CMT neuropathy score (CMTNS) [21].
Molecular genetic analysis
Genomic DNA was isolated from peripheral blood obtained from index patients, their family members and healthy controls using standard procedures. All the demyelinating patients were found to be negative for PMP22 duplication/deletion as identified by multiplex ligation probe amplification (MLPA). From 2000 to 2014, we carried out GDAP1 mutation screening by direct sequencing. GDAP1 coding exons were amplified by Polymerase Chain Reaction (PCR) and the PCR products were analyzed on ABI 3730 XL DNA analyzer (Applied Biosystems, Waltham, MA, USA), according to the manufacturer’s protocol. From 2014, molecular diagnosis of CMT patients was established by a designed targeted gene panel, which covered 60 genes. The sample was captured by SureSelect Human All ExonV5 Kit (Agilent). Genomic DNA sequencing was performed on the IlluminaHiseq 2500 platform (San Diego, CA, USA).
Data analysis for the determination of pathogenic mutations
The co-segregation analysis of the candidate sequence variants was performed using PCR and Sanger sequencing. Sequence variants were filtered against the following database: dbSNP129 (http://www.ncbi.nlm.nih.gov/SNP), HapMap 8, Exome Aggregation Consortium (http://exac.broadinstitute.org/), and 1000 Genome project (http://www.1000genomes.org). All variants were screened in 200 healthy controls and numbered according to the Human Genome Variation Society (HGVS) nomenclature on the basis of standard reference sequences of mRNA (NM_018972) and protein (NP_061845). To assess their functional effects, in silico analysis was performed using PolyPhen2 (http://genetics.bwh.harvardPedu/pph2/), SIFT (http://sift.jcvi.org/), PROVEAN (http://provean.jcvi.org/) and Mutation Taster (http://www.mutationtaster.org/). ACMG standards and guidelines were applied to determine the pathogenicity of each variation.
Result
Genetic findings and analysis
We identified 7 pathogenic and/or likely pathogenic GDAP1 variants and 1 variant of uncertain significance (VUS) in 4 families, and 5 of them were novel. These included 3 missense (p.R41H, p.V215I and p.Q38R), 1 nonsense (p.Q38X) and 1 frame-shift variant (p.N201Kfs*5). Proband from family 1 harbored 2 missense pathogenic mutations N178S and H256R and have previously been reported [16]. Proband in family 2 carried a likely pathogenic missense variant R41H and a likely pathogenic frame-shift variant N201Kfs*5. Both were novel variants. Proband from family 3 carried a nonsense variant Q38X and a missense variant V215I. Both variants were novel and likely pathogenic. Proband from family 4 harbored a likely pathogenic missense variant V219G and a novel missense variant of uncertain significance Q38R. The former was found to be inherited from the mother and has been described previously in a Korean family [18], whereas the latter was inherited from the father. Eight other affected individuals in this family carried only the V219G variant. In addition, we detected the presence of a single heterozygous variant of uncertain significance H256R in one family (family 5) from the CMT cohorts. We summarize the pedigree and genotypes of families with GDAP1 variants in this study in Fig. 1 and the location and distribution of these variants in a 2D schematic representation of the GDAP1 protein in Fig. 2, respectively.

Pedigree and genotypes of families with GDAP1 variants in this study. Circles represent females; squares, males; shaded symbols, patients affected; arrows, probands

Schematic representation of GDAP1 with the location and distribution of all variants identified in the present series. Novel mutations are marked by a hash tag (#). Mutations inherited in an autosomal recessive manner are highlighted in black and those with autosomal dominant inheritance in red. GST glutathione-S transferase, HD hydrophobic domain, TMD trans-membrane domain, N-TER N terminal, C-TER C terminal
To establish the functional effects and determine the pathogenicity of the GDAP1 variants, we utilized the application of Bioinformatics tool and classified them according to the standard and guidelines of American College of Medical Genetics (ACMG). We illustrate the results of in silico analysis and predicted pathogenicity of these variants in Table 1.
Prevalence and frequency distribution
Of the 306 CMT patients, 120 patients (39%) presented with the axonal phenotype and 112 patients (37%) with the demyelinating phenotype. Of the remaining 74 patients, 57 (18.5%) presented with the intermediate type and the rest 17 patients (5.5%) remained unclassified.
We detected 5 families with GDAP1 variants amongst the CMT cohorts and they demonstrated a GDAP1 mutation frequency rate of 1.63% (5/306).
Clinical features
All affected individuals belonging to the AR families (families 1,2 and 3) initially presented with onset of symptoms in the first decade of life, with the mean age of onset at 5 years (range 1–7). The most common initial clinical symptom included walking difficulties. They shared a common phenotype characterized by a symmetric distal muscle weakness and atrophy of the lower limbs, foot deformities, walking difficulties, reduced or absent deep tendon reflexes and a mild sensory deficit localized primarily over the ankles. Walking abilities were limited or reduced in all but they maintained the ability to walk without assistance. None of the patients were wheelchair dependent. The disease severity was moderate as evidenced by the CMTNS (range 11–17).
Electro-physiologic studies classified all affected individuals as having an axonal type of neuropathy (AR-CMT2K). The average MNCV of the median nerve was 51 m/s (range 41.8–61.9) and the mean compound muscle action potential (CMAP) value was 4.9 mv (range 3.6–6.2).
The autosomal dominant family (family 4) included the proband and 8 other affected family members. The proband, 2-year-old female developed foot drop and frequent falls as the initial clinical symptoms observed at first year of life. She presented with limited and reduced dorsiflexion of both feet and clubfeet. Over the course, she failed to squat, walk and climb stairs unaided and this was evident when she was seen in our clinic at the age of 2 years.
Muscle atrophy and weakness were mainly restricted to the distal lower limbs along with absent reflexes in the lower extremities. The symptoms were predominantly motor. Electro-physiologic study revealed a median MNCV of 40.9 m/s with a marked reduction in CMAP value of 2.1 mv, demonstrating a strong association with an axonal type of neuropathy (AD-CMT2K). The disease severity was moderate with a CMTNS of 13. She presently uses an orthotic device for ambulation. Most of the other affected individuals could lead an independent life and developed mild symptoms such as mild distal leg atrophy and weakness, foot deformity, gait instability and preserved walking but with reduced pace. Nerve conduction studies were not performed due to their unavailability. The average age of onset among all affected members was 15.8 (range 1–22) years, higher than that of AR cases. The proband presented with an earlier disease onset and a more severe phenotype than other affected members in the family.
Discussion
We identified 8 GDAP1 variants (2 pathogenic, 5 likely pathogenic and 1 with uncertain significance) in 4 families, which included 5 novel variants, expanding the mutational spectrum of GDAP1 variants associated with CMT. Three previously reported pathogenic and likely pathogenic variants were also identified. Most variants were distributed in the glutathione S-transferase (GST) domains (3 variants each in GST-N and GST-C), 1 in the α4–α5 loop and 1 in the inter domain between α4–α5 loop and GST-C. Exon 5 was most commonly affected.
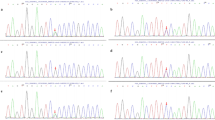
Additionally, a single heterozygous variant H256R was identified in one family (family 5) belonging to the CMT cohorts. The parents were found to be normal by history, clinical examination and Nerve Conduction Studies. The father carried a single heterozygous H256R variant in the GDAP1 gene while the genetic screening in the mother detected no abnormalities. The proband and her affected younger sister were symptomatic for CMT disease and carried the same H256R variant as their father. Further, we performed a sequencing analysis of this heterozygous variant in the family and we show the results of this analysis in Fig. 3.

Pedigree and chromatograms showing sequencing analysis of heterozygous c.767A>G; (p. H256R) of GDAP1 in Family 5
We examined all 6 exons of GDAP1 gene in the proband and did not find any variations except for H256R. Nonetheless, we cannot rule out two possibilities that may have existed. First is the presence of a pathogenic mutation in a different gene other than GDAP1 and the second is the existence of an intronic variant predicting altered splicing or a large deletion that was undetectable by the genetic screening methods employed in our study.
In the absence of a standard database, we could not identify the control frequency for this variant in the Chinese population. However, we found out that in the East Asian population, its allele count is 4 with an allele frequency of 0.0002120 (http://gnomad.broadinstitute.org/variant/8-75276292-A-G).
In a recent study published in North China, two unrelated patients (patient 1 and patient 2) were found to carry the H256R variant in the GDAP1 gene in a compound heterozygous state with another mutant allele [15]. Patient 1 was found to carry a de novo H256R mutation while the parental origin of patient 2 was not available. The phenotypes of the H256R mutations were found to be similar sharing common characteristics such as an early age of onset, common initial symptoms, a moderate disease severity and belonging to the axonal subtype. This study also observed the H256R variant to be the most frequent mutation in their series suggesting that it might be a common mutation in GDAP1 associated Chinese CMT patients. Based on the combined observations from this series and our study, the frequent occurrence of this variant in the Chinese population has led us to speculate that this is a common mutation amongst the Chinese CMT cohorts associated with GDAP1. In addition, this mutation has also been reported in GDAP1 associated CMT disease in other Chinese CMT populations [16, 19].
To date, more than 80 GDAP1 variants have been implicated in the pathogenesis of CMT (http://www.hgmd.cf.ac.uk/ac/index.php), excluding those identified in this study. These include missense, non-sense, frame-shift, deletions, mutations generating truncated and non-functional proteins and those altering the splice sites in the GDAP1 transcripts [22, 23]. Overall, exon 6 is the most common exon to be affected followed by Exon 3. Mutations are mostly located in the GST domains of the protein indicating their important roles in the protein function. Most frequent protein consequences are missense followed by frame-shift, nonsense and splice site mutations.
GDAP1 variants were identified in 5 families out of 306 CMT cohorts in our study, corresponding to a frequency distribution of 1.63%. A recently published study in North China has reported a GDAP1 mutation frequency rate of 2.37% [15]. Our study, however, included a larger number of patients enrolled (306) as compared to this study (169). Combining the data on the prevalence rate as identified in previous studies in Japan [24], Taiwan [19], China [15] and present study, we find that the mean frequency distribution rate of GDAP1 variants in the Asian population is 1.3% (20/1541), a range of (1–2.8%). This figure is quite similar to the data from previous studies conducted in the US (1.1%) [25] and the UK (0.7%) [26], but very low as compared to the findings described in studies from European countries and the mediterranean basin [27,28,29,30,31,32].
Some specific pathogenic GDAP1 variants have frequently been observed and reported in certain geographic areas and ethnic backgrounds. These include the nonsense variant S194X reported in Spain, Morocco and Tunisia [7, 8, 10, 33]; Q163X reported mainly in Spain [8], missense variant R120W reported mainly in Belgium [34, 35] and L239F reported across Eastern and Central Europe [36,37,38,39]. This distribution has mainly been attributed to the founder effect and the high rate of consanguinity among population belonging to this region. We show the frequency distribution of GDAP1 variants as described in some previous studies in Table 2.
Affected individuals from the recessive families were found to have an axonal (AR-CMT2K) form of neuropathy. They shared common clinical characteristics with an early age of onset and walking difficulties as the most prominent initial clinical manifestation. These patients presented with a moderate disease severity and phenotype, contrary to previous findings [40, 41] but consistent with studies from Taiwan [19] and China [15]. None of the patients were wheelchair dependent. No evidence of vocal cord paresis or diaphragmatic weakness [40, 42] and hoarseness [43] was observed. In comparison to the previously reported patients with vocal cord paresis and diaphragmatic weakness, patients in our study presented with a similar clinical phenotype with an early age of disease onset, walking difficulties as the most common and initial clinical symptom, presence of foot deformities, variable degree of sensory deficits and reduced or absent deep tendon reflexes.
However, the disease severity and progression in the former was more intense and rapid. Most patients developed functional disability early in life and were wheelchair dependent by the second or third decade. Some patients presented with hypotonia, hoarseness of voice and dysphonia associated with significant weakness and wasting of proximal muscles of upper and lower limbs. The most striking difference was the absence of wheel chair dependence in the patients from our series.
Patients belonging to the AD family shared a common clinical presentation of mild weakness and atrophy of the distal lower limbs, foot deformities and gait instability, with a later age of onset and milder phenotype as compared to the recessive families [13, 14]. All affected members in the family were found to carry the V219G variant. The proband, however, had an earlier age of onset, severe phenotype and a rapid progression of symptoms compared to the rest of the affected individuals in the family, as opposed to previous reports [35, 41]. In addition to the dominant variant V219G, she also carried a novel variant Q38R, which was inherited from the father who was found to be normal and healthy by history and clinical examination. Due to his unwillingness to do the tests, data on EMG/NCV was not available. Nerve conduction study of the proband demonstrated an axonal (AD-CMT2K) form of neuropathy.
Given the mild symptoms and slow progression in other affected members, the presence of V219G alone does not explain the severe symptoms in the proband. To add to this, this variant has previously been reported in a Korean family along with the presence of another mutant allele, demonstrating a similar clinical picture in the proband and associated with a severe clinical phenotype and rapid disease progression [18], findings consistent with our study. Considering these observations, we strongly assume that the V219G variant when present with another mutant allele has a more pronounced and severe phenotypic effect and hence, the disease severity in the proband from our study is probably due to the co-existence of the novel variant Q38R, which may have caused an additional effect over the dominant variant V219G. In addition, evaluation of Charcot–Marie–Tooth examination score (CMTES) in the proband revealed a higher score (9) as compared to the other affected individuals (range 3–5) potentially reflecting this phenotypic variability.
Nonetheless, we cannot exclude the possibility of unidentified genetic and/or epigenetic factors and environmental entities that may have contributed to the phenotype. We thus observed a marked intra-familial variability within the family, which is consistent with previous findings [27, 44]. We have included two separate videos of the proband and her mother, who upon clinical examination was found to have a mild weakness of the distal lower limbs, a normal gait but with reduced pace and a slightly decreased knee reflex bilaterally. We hope that the videos will be very helpful and add to the paper. Please refer to the supplementary material for the videos. As this study utilized the application of a targeted multi-gene panel approach, the data obtained in this family could not be extensively analyzed for the investigation of larger deletions/insertions.
This study demonstrates the presence of an axonal form of CMT associated with GDAP1 variants. The predominance of this axonal subtype is synonymous to previous reports published in North China [15] and Japan [24]. A similar observation has been documented in literatures published in the past [24, 25, 29, 31]. Moreover, this finding can be supported by the observation of a summary of all GDAP1 variants and their phenotypes reported so far (see Supplementary material).
Conclusion
Our study broadens the genetic spectrum of CMT associated with GDAP1 mutations with the identification of novel GDAP1 variants. We detected the presence of an axonal (AD-CMT2K and AR-CMT2K) form of neuropathy in all affected individuals, indicating that axonal CMT is more common than demyelinating CMT in GDAP1-associated CMT disease. Recessive variants presented with an earlier age of onset and severe clinical manifestations as compared to the dominant ones, findings consistent with previous studies. Marked phenotypic variability was observed in the dominant family. In addition, we believe that the H256R mutation is a common mutation in GDAP1-associated CMT Chinese cohorts. A larger number of cases with CMT disease associated with GDAP1 mutations and a comprehensive genetic, clinical, electro-physiologic and pathological evaluation will be needed to further understand and elucidate the complex and heterogeneous correlation between the genotypes and the clinical phenotypes and determine a more approximate and accurate frequency of distribution.
Abbreviations
- GDAP1:
-
Ganglioside-induced differentiation-associated protein 1
- CMT:
-
Charcot–Marie–Tooth
- AR:
-
Autosomal Recessive
- AD:
-
Autosomal dominant
- NGS:
-
Next generation sequencing
- ACMG:
-
American College of Medical Genetics
- MNCV:
-
Motor nerve conduction velocity
- CMAP:
-
Compound muscle action potential
- CMTNS:
-
Charcot–Marie–Tooth neuropathy score
- MLPA:
-
Multiplex ligation probe amplification
- OMIM:
-
Online mendelian inheritance in man
- PROVEAN:
-
Protein variation effect analyzer
- VUS:
-
Variation of uncertain significance
- PCR:
-
Polymerase chain reaction
- HGVS:
-
Human Genome Variation Society
- CMTES:
-
Charcot–Marie–Tooth examination score
- EMG:
-
Electromyography
References
Harding AE, Thomas PK (1980) The clinical features of hereditary motor and sensory neuropathy types I and II. Brain 103(2):259–280
Skre H (1974) Genetic and clinical aspects of Charcot–Marie–Tooth’s disease. Clin Genet 6(2):98–118
Dyck PJ, Lambert EH (1968) Lower motor and primary sensory neuron diseases with peroneal muscular atrophy. I. Neurologic, genetic, and electrophysiologic findings in hereditary polyneuropathies. Arch Neurol 18(6):603–618
Davis CJ, Bradley WG, Madrid R (1978) The peroneal muscular atrophy syndrome: clinical, genetic, electrophysiological and nerve biopsy studies. I. Clinical, genetic and electrophysiological findings and classification. J Genet Hum 26(4):311–349
Pedrola L et al (2008) Cell expression of GDAP1 in the nervous system and pathogenesis of Charcot–Marie–Tooth type 4A disease. J Cell Mol Med 12(2):679–689
Niemann A et al (2005) Ganglioside-induced differentiation associated protein 1 is a regulator of the mitochondrial network: new implications for Charcot–Marie–Tooth disease. J Cell Biol 170(7):1067–1078
Baxter RV et al (2002) Ganglioside-induced differentiation-associated protein-1 is mutant in Charcot–Marie–Tooth disease type 4A/8q21. Nat Genet 30(1):21–22
Cuesta A et al (2002) The gene encoding ganglioside-induced differentiation-associated protein 1 is mutated in axonal Charcot–Marie–Tooth type 4A disease. Nat Genet 30(1):22–25
Sevilla T et al (2003) Clinical, electrophysiological and morphological findings of Charcot–Marie–Tooth neuropathy with vocal cord palsy and mutations in the GDAP1 gene. Brain 126(Pt 9):2023–2033
Azzedine H et al (2003) Variability of disease progression in a family with autosomal recessive CMT associated with a S194X and new R310Q mutation in the GDAP1 gene. Neuromuscul Disord 13(4):341–346
Boerkoel CF et al (2003) CMT4A: identification of a Hispanic GDAP1 founder mutation. Ann Neurol 53(3):400–405
Claramunt R et al (2005) Genetics of Charcot–Marie–Tooth disease type 4A: mutations, inheritance, phenotypic variability, and founder effect. J Med Genet 42(4):358–365
Chung KW et al (2008) A novel GDAP1 Q218E mutation in autosomal dominant Charcot–Marie–Tooth disease. J Hum Genet 53(4):360–364
Cassereau J et al (2009) Mitochondrial complex I deficiency in GDAP1-related autosomal dominant Charcot–Marie–Tooth disease (CMT2K). Neurogenetics 10(2):145–150
Fu J, Dai S, Lu Y, Wu R, Wang Z, Yuan Y, Lv H (2017) Similar clinical, pathological, and genetic features in Chinese patients with autosomal recessive and dominant Charcot-Marie-Tooth disease type 2K. Neuromuscul Disord 27(8):760–765
Zhang RX et al (2004) Mutation analysis of ganglioside-induced differentiation associated protein-1 gene in Chinese Charcot–Marie–Tooth disease. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 21(3):207–210
Abe A et al (2011) Molecular diagnosis and clinical onset of Charcot–Marie–Tooth disease in Japan. J Hum Genet 56(5):364–368
Chung KW et al (2011) Two recessive intermediate Charcot–Marie–Tooth patients with GDAP1 mutations. J Peripher Nerv Syst 16(2):143–146
Lin KP et al (2011) The mutational spectrum in a cohort of Charcot–Marie–Tooth disease type 2 among the Han Chinese in Taiwan. PLoS ONE 6(12):e29393
Pareyson D, Marchesi C (2009) Diagnosis, natural history, and management of Charcot–Marie–Tooth disease. Lancet Neurol 8(7):654–667
Murphy SM et al (2011) Reliability of the CMT neuropathy score (second version) in Charcot–Marie–Tooth disease. J Peripher Nerv Syst 16(3):191–198
Cassereau J et al (2011) A locus-specific database for mutations in GDAP1 allows analysis of genotype–phenotype correlations in Charcot–Marie–Tooth diseases type 4A and 2K. Orphanet J Rare Dis 6:87
Cassereau J et al (2011) Mitochondrial dysfunction and pathophysiology of Charcot–Marie–Tooth disease involving GDAP1 mutations. Exp Neurol 227(1):31–41
Yoshimura A, Yuan JH, Hashiguchi A, Hiramatsu Y, Ando M, Higuchi Y, Nakamura T, Okamoto Y, Matsumura K, Hamano T, Sawaura N, Shimatani Y, Kumada S, Okumura Y, Miyahara J, Yamaguchi Y, Kitamura S, Haginoya K, Mitsui J, Ishiura H, Tsuji S, Takashima H (2017) Clinical and mutational spectrum of Japanese patients with Charcot-Marie-Tooth disease caused by GDAP1 variants. Clin Genet 92(3):274–280
Saporta AS et al (2011) Charcot–Marie–Tooth disease subtypes and genetic testing strategies. Ann Neurol 69(1):22–33
Murphy SM et al (2012) Charcot–Marie–Tooth disease: frequency of genetic subtypes and guidelines for genetic testing. J Neurol Neurosurg Psychiatry 83(7):706–710
Crimella C et al (2010) The GST domain of GDAP1 is a frequent target of mutations in the dominant form of axonal Charcot Marie Tooth type 2K. J Med Genet 47(10):712–716
Auranen M et al (2013) Dominant GDAP1 founder mutation is a common cause of axonal Charcot–Marie–Tooth disease in Finland. Neurogenetics 14(2):123–132
Sivera R et al (2013) Charcot–Marie–Tooth disease: genetic and clinical spectrum in a Spanish clinical series. Neurology 81(18):1617–1625
Zimon M et al (2015) Unraveling the genetic landscape of autosomal recessive Charcot–Marie–Tooth neuropathies using a homozygosity mapping approach. Neurogenetics 16(1):33–42
Pezzini I et al (2016) GDAP1 mutations in Italian axonal Charcot–Marie–Tooth patients: phenotypic features and clinical course. Neuromuscul Disord 26(1):26–32
Manganelli F et al (2014) Charcot–Marie–Tooth disease: frequency of genetic subtypes in a Southern Italy population. J Peripher Nerv Syst 19(4):292–298
De Sandre-Giovannoli A et al (2003) Phenotypic and genetic exploration of severe demyelinating and secondary axonal neuropathies resulting from GDAP1 nonsense and splicing mutations. J Med Genet 40(7):e87
Ammar N et al (2003) Identification of novel GDAP1 mutations causing autosomal recessive Charcot–Marie–Tooth disease. Neuromuscul Disord 13(9):720–728
Zimon M et al (2011) Dominant GDAP1 mutations cause predominantly mild CMT phenotypes. Neurology 77(6):540–548
Rougeot C et al (2008) Clinical, electrophysiological and genetic studies of two families with mutations in the GDAP1 gene. Neuropediatrics 39(3):184–187
Kabzinska D et al (2006) Early onset Charcot–Marie–Tooth disease caused by a homozygous Leu239Phe mutation in the GDAP1 gene. Acta Myol 25(1):34–37
Kabzinska D et al (2010) L239F founder mutation in GDAP1 is associated with a mild Charcot–Marie–Tooth type 4C4 (CMT4C4) phenotype. Neurogenetics 11(3):357–366
Barankova L et al (2007) GDAP1 mutations in Czech families with early-onset CMT. Neuromuscul Disord 17(6):482–489
Sevilla T et al (2008) Vocal cord paresis and diaphragmatic dysfunction are severe and frequent symptoms of GDAP1-associated neuropathy. Brain 131(Pt 11):3051–3061
Kabzinska D et al (2014) A severe recessive and a mild dominant form of Charcot–Marie–Tooth disease associated with a newly identified Glu222Lys GDAP1 gene mutation. Acta Biochim Pol 61(4):739–744
Stojkovic T et al (2004) Vocal cord and diaphragm paralysis, as clinical features of a French family with autosomal recessive Charcot–Marie–Tooth disease, associated with a new mutation in the GDAP1 gene. Neuromuscul Disord 14(4):261–264
Sahin-Calapoglu N et al (2009) Novel GDAP1 mutation in a Turkish family with CMT2K (CMT2K with novel GDAP1 mutation). Neuromolecular Med 11(2):106–113
Manganelli F et al (2012) A novel autosomal dominant GDAP1 mutation in an Italian CMT2 family. J Peripher Nerv Syst 17(3):351–355
Schabhüttl M, Wieland T, Senderek J, Baets J, Timmerman V, De Jonghe P, Reilly MM, Stieglbauer K, Laich E, Windhager R, Erwa W, Trajanoski S, Strom TM, Auer-Grumbach M (2014) Whole-exome sequencing in patients with inherited neuropathies: outcome and challenges. J Neurol 261(5):970–982
DiVincenzo C, Elzinga CD, Medeiros AC, Karbassi I, Jones JR, Evans MC, Braastad CD, Bishop CM, Jaremko M, Wang Z, Liaquat K, Hoffman CA, York MD, Batish SD, Lupski JR, Higgins JJ (2014) The allelic spectrum of Charcot-Marie-Tooth disease in over 17,000 individuals with neuropathy. Mol Genet Genomic Med 2(6):522–529
Acknowledgements
We strongly acknowledge and appreciate the participation and cooperation of all the patients and their families. This study was supported by The National Natural Science Foundation of China (81771366) The Science Foundation of Health and Family Planning Commission of Hunan Province (A2017001) and The Natural Science Foundation of Hunan Province (2017JJ2365). In addition, further support was received from The National Key Plan for Scientific Research and Development of China (no. 2016YFC1306000).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that they have no conflict of interest.
Ethical approval
This study was approved by the ethics committee of the The Third Xiangya Hospital and Xiangya Hospital and was performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments.
Informed consent
Written informed consent was obtained from all participants and/or their parents prior to their inclusion in this study.
Additional information
Papers of particular interests (* denoting special interest) and (** denoting outstanding interest) have been highlighted.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Pakhrin, P.S., Xie, Y., Hu, Z. et al. Genotype–phenotype correlation and frequency of distribution in a cohort of Chinese Charcot–Marie–Tooth patients associated with GDAP1 mutations. J Neurol 265, 637–646 (2018). https://doi.org/10.1007/s00415-018-8743-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-018-8743-9