
Abstract
We describe a founder mutation in the gene encoding ganglioside-induced differentiation associated-protein 1 (GDAP1), leading to amino acid change p.H123R, as a common cause of autosomal dominant axonal Charcot-Marie-Tooth (CMT2) neuropathy in Finland. The mutation explains up to 14 % of CMT2 in Finland, where most patients with axonal neuropathy have remained without molecular diagnosis. Only three families out of 28 were found to carry putative disease mutations in the MFN2 gene encoding mitofusin 2. In addition, the MFN2 variant p.V705I was commonly found in our patients, but we provide evidence that this previously described mutation is a common polymorphism and not pathogenic. GDAP1-associated polyneuropathy caused predominantly a mild and slowly progressive phenotype. Besides distal leg muscle weakness, most patients showed mild proximal weakness, often with asymmetry and pes cavus. Our findings broaden the understanding of GDAP1 mutations in CMT2 phenotypes and provide support for the use of whole-exome sequencing in CMT gene diagnostics.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Charcot-Marie-Tooth disease (CMT) is the most common inherited neurologic disease with prevalence of 1 in 2500 [1, 2]. The diagnosis is based on clinical and electrophysiological findings. In the demyelinating form (CMT1), nerve conduction velocity (NCV) is decreased, whereas in the axonal form (CMT2), NCV is normal or only slightly reduced. Some patients have intermediate CMT with features of both demyelination and axonal degeneration. Clinically, CMT often starts with distal muscle weakness predominantly in the lower limbs, which is commonly accompanied by sensory defects, loss of deep tendon reflexes, and skeletal abnormalities including pes cavus, hammertoes, and scoliosis.
The inheritance of CMT can be autosomal dominant (AD), recessive (AR), or X-linked. A precise molecular diagnosis is often pursued, since it can clarify the inheritance pattern and family planning decisions, in addition to establishing the natural history and prognosis of the disease. Furthermore, a molecular diagnosis can allow genotype-specific therapy in the future [1]. However, finding the causative mutation is not straight-forward because of the heterogeneous genetic background of the disease. CMT results from mutations in more than 40 different genes. Also, an estimated 30–50 CMT disease genes still remain to be identified [3].
For CMT2, nearly 100 probable disease-causing mutations have been identified in more than 10 genes according to the Inherited Peripheral Neuropathies Mutation Database. Of the known CMT2 disease genes, MFN2 encoding mitofusin 2 is the most common, being mutated in about 20 % of patients [4, 5]. Up to 50 distinct mutations have been found in the MFN2 gene, but no single mutation is considered highly prevalent. The large number of infrequent mutations in the different disease genes prevents simple subtyping of CMT2 patients based on genotype–phenotype correlations. As a further complication, significant clinical variability has been observed within CMT families [1, 2]. The phenotypic variability could in part be explained by modifying genetic factors; however, no such factors have been identified thus far.
Next-generation sequencing methods have proved efficient for CMT disease mutation identification [6, 7]. Whole-exome sequencing is already more cost-effective than direct sequencing of a large number of individual candidate disease genes. Therefore, we set out to test the efficiency of exome sequencing in elucidating the genetic causes of CMT in Finland. In the Finnish population, CMT1 patients are likely to receive a molecular diagnosis [8], whereas the genetic causes of CMT2 are largely unknown. In this study, we find that a heterozygous mutation in the gene encoding ganglioside-induced differentiation associated-protein 1 (GDAP1), causing an amino acid change p.H123R, is a common cause of autosomal dominant CMT2 in Finland, whereas MFN2 mutations occur less frequently in our patients than described in other European populations. We present the phenotypic variability of the GDAP1 founder mutation in patients from four families and compare it to the phenotypes of our MFN2 patients.
Materials and methods
Patients
Blood samples were collected from Finnish CMT2 patients diagnosed at the Helsinki University Central Hospital (HUCH) and their family members. Samples were taken in accordance with the Declaration of Helsinki, and all participants gave written informed consent. The study was approved by the HUCH ethics review board (dnro 399/E9/07). The diagnosis was based on clinical examination and electrophysiological studies. Selected patients underwent sural nerve biopsy, muscle biopsy, or MRI of muscle or brain. Members from 28 families participated in the study. The disease showed autosomal dominant inheritance in 19 families and 9 patients were sporadic with no known family history of CMT2.
DNA sequencing
DNA was extracted from peripheral blood using standard techniques. For whole-exome sequencing, target-enrichment was done using NimbleGen Sequence Capture 2.1 M Human Exome v2.0 array followed by sequencing with the Illumina Genome Analyzer-IIx platform with 2 × 82 bp paired end reads. The variant calling pipeline of the Finnish Institute of Molecular Medicine was used for the reference genome alignment and variant calling [9]. The PCR amplification and direct sequencing of the GDAP1 exon 3 with flanking intronic sequences were performed with primers 5′-TCTGGTGCATCAGGCCATTT-3′ and 5′- TCCGACTGGTTCATGGATCG-3′.
Haplotype analysis
We determined the disease-associated haplotypes of patients with the GDAP1 mutation by analyzing the sizes of four microsatellite markers surrounding the GDAP1 gene (D8S286, D8S551, D8S1144, D8S548). For PCR amplification, forward primers 5′-labeled with the fluorescent dye FAM were used. Fragments were size-separated on an ABI3730XL DNA analyzer, and genotyping results were analyzed with the GeneMapper 4.0 software (Applied Biosystems).
Results
Genetic findings
The participating patients and members of 28 families were examined clinically and electrophysiological testing was conducted. The patients were initially screened for mutations in MFN2 according to our genetic testing practice of patients with suspected CMT2. Putative MFN2 mutations were identified in three families (Fig. 1). One of the mutations c.280C > T (RefSeq NM_014874.3), leading to amino acid change p.R94W, has been previously reported [10]. The other two variants, c.1144 G > C [p.A382P] and c.1150C > T [p.R384W], were novel and not present in 104 Finnish control chromosomes, 1000Genomes database, or NHLBI GO Exome Sequencing Project database. It is not conclusive whether the latter variant is pathogenic since the amino acid residue R384 is not conserved (Fig. 3a), and only one affected family member was available for studying segregation (Family B, Fig. 1). In addition, a previously reported dominant mutation in MFN2 leading to amino acid change p.V705I [11, 12] has been frequently detected during MFN2 screening in Finland (unpublished observation). Among the 28 families studied here, the variant was present in 6 families (21 %). The p.V705I is, however, not likely to be a causative disease mutation because it is a common variant in the general Finnish population (allele frequencies 0.026 in 1000Genomes (n = 77) and 0.047 in our control material (n = 157)). Also, Val705 is not a highly conserved residue since for example in the mouse MFN2 protein, it is actually an isoleucine (Fig. 3a). Whether the MFN2 variant can modify the disease phenotype is however not known.

Reduced pedigrees of the families with MFN2 mutations. Individuals who were genetically tested are marked with the presence (positive sign) or absence (minus sign) of the respective MFN2 amino acid change. Only affected family members were heterozygous for the mutation
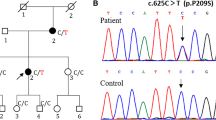
To further elucidate the genetic causes of CMT2 in Finland, we used whole-exome sequencing in members from four families. Single patients were selected for exome sequencing from family 3 (patient III-4) and family 7 (patient III-2) (Fig. 4). The exome data for these samples were filtered as follows using a candidate gene approach: (1) Common variants, i.e., variants found with more than 0.5 % frequency in the 1000Genomes project were excluded, (2) Variants located outside genes were excluded, (3) Heterozygous variants were selected, (4) Variants in previously known CMT2 genes were selected for closer examination, (5) Variants altering splice sites or amino acids were listed (Fig. 2). This analysis resulted in direct recognition of a heterozygous GDAP1 variant c.368A > G (RefSeq NM_018972.2) leading to p.H123R amino acid change in both families. This mutation has been reported only recently in a GDAP1 screen of a multi-ethnic cohort where it was actually found in a dominant family from Northern Finland and as a de novo mutation in a Tunisian patient [13]. The same study confirmed that 376 control subjects including 96 from Finland did not carry the mutation. We tested additional 370 Finnish control chromosomes and did not identify any carriers. The His123 of the GDAP1 protein is a highly conserved amino acid (Fig. 3b) supporting its pathogenicity. We then screened all our participating families for the mutation and identified two more dominant families with the same mutation (Fig. 4). Thus, 4 out of 28 CMT2 families (14 %) participating in our study carried the same GDAP1 mutation. The mutation segregated fully with the disease in all family members whose samples were available for the study. The exome analysis also identified that the patient III-2 from family 7 had the MFN p.V705I variant (Fig. 2), which is unlikely to be pathogenic as discussed above.

Exome data analysis focusing on known CMT2 genes. The number of variants for each filtering step are shown. The selected variants were the following: (1) rare variants with a frequency of 0.5 % or less in the 1000 Genomes project database; (2) variants that were inside genes; (3) variants that were heterozygous; (4) variants in known CMT2 genes; (5) variants that altered an amino acid or splice sites. As a result, GDAP1 p.H123R mutation was identified in both samples and the polymorphic MFN2 p.V705I change in the sample from family 7

Alignments of MFN2 (a) and GDAP1 (b) proteins from various species showing amino acid conservation. The amino acid position that was found to be changed in patients is indicated (arrow)

Reduced pedigrees of the families with GDAP1 mutation. For individuals who were genetically tested, the presence of GDAP1 p.H123R (H123R+) is indicated. In family 7, the presence or absence of MFN2 p.V705I (V705I+, and V705I−, respectively) is indicated in addition to the GDAP1 variant. The mutation segregated fully with the disease in the tested individuals. Individuals marked with a question mark had minor symptoms consistent with neuropathy but have not been clinically examined
The identification of a rare mutation in several families who originate from a genetic isolate suggests a founder effect. We therefore constructed disease haplotypes in the families and found that all families shared the same disease haplotype (Table 1). The patient IV-1 from family 20 shared only part of the common disease haplotype with her father, indicating that crossing over had occurred between the GDAP1 gene and D8S1144. Subsequent genealogical studies of the families traced their origin to Oulu region locating in Northern Finland.
For the single patients from the two other families (index patients from families 1 and 2) that participated in the exome-sequencing, the above-described primary filtering strategy that focused on known CMT2 genes did not result in any findings. We therefore performed exome-sequencing on samples of one additional affected family member per family. For family 1 index patient, the additional sample came from his father's cousin, and for family 2 index patient, the additional sample was from his cousin's son. The filtering was done using the following steps: (1) Variants outside genes were excluded, (2) Only heterozygous variants on autosomes were selected, (3) Common variants, i.e., variants found with more than 0.5 % frequency in the 1000Genomes project were excluded, (4) Nonsynonomous changes were selected, (5) The SIFT Tool was used to predict damaging effects of the variants, (6) Variants that were shared by the two affected family members were selected, (7) In-house database of exome data from Finnish individuals (n = 50) was used to exclude variants that were commonly found in the study population (Supplementary Figure 1). The use of the in-house database in filtering was justified because of the high incidence of population-specific variants in the genetic isolate, and it was used as a last step because of practical reasons. Our unbiased filtering approach thus focused on identifying very rare heterozygous variants that had a predicted damaging effect on protein function and were shared by the two affected family members. The identified candidate variants are listed in Supplementary Table 1. None of the candidates were evident CMT2-causing variants and thus no conclusion of the genetic cause of CMT2 in these two families could be drawn based on these findings, but further exome sequencing studies by us and others may confirm that one of the listed variants is in fact disease-causing. The reasons for why the actual disease mutations may not be included in the list are for example poor sequencing coverage or the mutation type being other than missense or because of inaccuracy in damage prediction. The remaining 22 families have not been studied by exome sequencing.
Clinical variability of the patients with GDAP1 p.H123R mutation
Clinical picture of nearly all the families with the GDAP1 mutation showed gradually progressing symptoms starting in lower limbs with onset between ages 10 and 57 (Table 2). Typical finding in the patients included distal muscle weakness and pes cavus, which was present in nearly all affected. Motor symptoms were most prominent findings, and overall somatosensory defects were less marked. Interestingly, at presentation, nearly all had evidence of proximal muscle weakness with some asymmetry. Plasma creatine kinase (P-CK) values were normal or mildly elevated. The muscle pattern involvement was further studied in family 5 with muscle MRI, and the most prominent changes were seen distally in posterior leg compartment with bilateral atrophy and fatty infiltration in gastrocnemius and soleus muscles. Proximally vastus lateralis at left side and semimembranosus muscles bilaterally showed similar but much milder changes.
Electroneuromyography (ENMG) results were available for ten patients carrying the GDAP1 mutation at the age range of 16 to 58 years. ENMG was done twice for one patient and three times for two patients. All studied patients had at least some signs of polyneuropathy, and the overall degree of polyneuropathy ranged from slight to severe. In all cases, the type of the polyneuropathy was classified as sensory-motor and axonal. First signs were seen during young adulthood as a reduction of sensory nerve action potential (SNAP) amplitudes in the leg sensory nerves. Some degree of atrophy in distal muscles had usually developed in this phase of disease. During the progression, reduction of motor amplitudes in the legs and SNAP amplitudes in the hands were seen. Motor amplitudes in the arms were reduced only in severe cases. Some asymmetry in the motor and SNAP amplitudes was usual. No remarkable decrease in the motor or sensory conduction velocities were seen in these patients except in one case with nerve entrapment (III-5 of family 3). Needle electromyography (EMG) findings varied from chronic neurogenic changes in motor unit potential morphology to active denervation defined by the occurrence of fibrillation potentials. In all cases, EMG findings began distally and spread proximally during the progression of polyneuropathy.
The clinical picture was somewhat different in family 7 where the affected members were carrying the MFN2 variant p.V705I in addition to the GDAP1 mutation. One affected (IV-3) developed muscle weakness already at childhood and only distal leg muscles were involved. He also had extensive myopia (−11.5/−11.75), but otherwise normal neuro-ophthalmological findings, Octopus fields and visual evoked potential (VEP). At the age of 16, his ENMG showed severe distal sensory-motor polyneuropathy with axonal predominance and slight demyelination. At the age of 17, his muscle MRI showed prominent bilateral muscle atrophy, and fatty infiltration in tibialis anterior muscles, and pathological intramuscular edema in right soleus muscle. P-CK was clearly elevated (over 1000 U/L). One of MFN2 variant carriers in the same family (IV-2) who was negative for the GDAP1 mutation had normal clinical findings in a careful neurological examination at the age of 20 and also completely normal ENMG and quantitative sensory test (QST) results.
In family 20, the currently 34-year-old proband (IV-1) was subjectively asymptomatic but showed mild proximal lower leg weakness, pes cavus, absent tendon reflexes and slight chronic distal sensory-motor axonal polyneuropathy findings in the ENMG study. Her muscle MRI was completely normal at the age of 34. She is currently successfully treated by Botox for spasmoid dysphonia and inability to speak with loud voice.
Clinical features of patients with MFN2 mutations
In general, the patients with MFN2 mutations had a more severe clinical outcome compared to the patients with the GDAP1 mutation, and they presented neuropathic symptoms already in childhood or early adulthood (Table 2). Compared with GDAP1 patients, the ENMG findings of the three patients with MFN2 mutations showed some tendency to a more severe neuropathy. The disease begun at a younger age and had more mixed-type characteristics: slight demyelination associated with axonal predominance. Involvement of the optic nerve was studied in the two families with novel MFN2 mutations. Mild bilateral optic nerve thinning was detected in brain MRI of the patient with the MFN2 p.A382P (II-2 of family A), but the patient with the MFN2 p.R384W (II-2 of family B) had normal brain MRI. VEP analysis was normal in both cases showing no major optic nerve involvement in association with these MFN2 mutations. Muscle biopsy of the patient with the MFN2 p.A382P showed signs of denervation, and in sural nerve biopsy, moderately strong axonal neuropathy was evident with only scant onion bulb formations as signs of demyelination, confirming axonal type of neuropathy.
Discussion
We report the identification of GDAP1 p.H123R as a common cause of autosomal dominant CMT2 (AD-CMT2) in Finland and describe the phenotypic characteristics that are associated with this founder mutation. We found the GDAP1 p.H123R in 14 % of families, which is a remarkably higher percentage than the previously reported frequency of about 1 % for all GDAP1 mutations in a large cohort of AD-CMT2 patients [13]. An Italian study has reported a 27 % GDAP1 mutation frequency in their dominant CMT2 families, with each family carrying a private mutation [14]. Our study is the first description of a highly prevalent single GDAP1 mutation causing AD-CMT2 in a particular ethnic group.
Recessive GDAP1 mutations were originally found in both demyelinating and axonal CMT [15, 16], but dominant mutations causing axonal neuropathy have also been reported [13, 14, 17–20]. More than 40 disease-causing GDAP1 mutations have been described to date, of which 10 are dominant. Recessive GDAP1 mutations often cause aggressive, early-onset disease, where vocal cord paralysis and diaphragmatic dysfunction are prevalent features [21]. In contrast, many of the patients carrying dominant GDAP1 mutations have had clearly milder symptoms [20], albeit with large variability [13, 20]. The previously reported Finnish family with GDAP1 (p.H123R) mutation consisted of mutation carriers in three generations of which those in the first generation were described as unaffected [13]. The three described patients had an age of onset between 3 and 32 years. Our patients presented with an even larger range for the age of onset (10 to 57), and all our mutation carriers were affected. In both studies, changes in proximal muscles were detected in addition to distal lower limb muscle weakness. In our patients, ENMG revealed chronic sensory-motor axonal polyneuropathy with distal predominance. In most of the cases, the degree of polyneuropathy was slight or moderate, it had some degree of asymmetry, and a tendency to progress slowly during the course of the adulthood. Interestingly, one patient showed vocal cord involvement although her other clinical manifestations were relatively mild.
The genetics of CMT in Finland has previously been addressed in one comprehensive study [8], which investigated a heterogeneous cohort of patients with CMT1 or 2, or with one of the related neuropathies Déjérine-Sottas syndrome (DSS) and hereditary neuropathy with liability to pressure palsies (HNPP). The study found GJB1 mutations in X-linked CMT, in addition to PMP22 or MPZ mutations in CMT1, but the CMT2 patients remained without a molecular diagnosis. We report here MFN2 mutations in 11 % of our dominant CMT2 families, which together with the identified GDAP1 mutation explain 25 % of CMT2 in Finland. The MFN2 variant p.V705I has been suspected as the major cause of dominant CMT2 in Finland as it is a common finding in our patients and has been reported as pathogenic in several studies [11, 12]. We, however, conclude that this MFN2 variant is highly unlikely to be a disease-causing mutation because it is a common polymorphism in the Finnish population and the amino acid Val705 is poorly conserved in the otherwise preserved MFN2 protein. Interestingly, however, one of our GDAP1 patients who also carried the MFN2 p.V705I variant had atypically severe polyneuropathy with early onset and with slight signs of demyelination compared to the other GDAP1 patients. This may suggest that the MFN2 variant can modify the phenotype caused by a GDAP1 mutation, which could be mediated by the function of both proteins in mitochondrial dynamics.
Anchored to the outer mitochondrial membrane, the GDAP1 protein may function in mitochondrial dynamics or otherwise regulate mitochondrial function. Mitochondrial dynamics, i.e., fusion and fission, allow the cell to reorganize the mitochondrial network according to its needs, a process that is essential for mitochondrial function and cell survival [22]. Neuronal cells appear to be particularly sensitive to defects in this process [23], perhaps owing to their need to reorganize the mitochondria in order to send them along the axon. Overexpression of GDAP1 has been shown to promote mitochondrial fission [24], but the mechanism of action is not known, partly due to a lack of an ortholog in yeast, a frequently used model system for mitochondrial dynamics. The protein has similarity to glutathione-S-transferase (GST) [25]. Several of the disease mutations have been tested for pathogenic effects on cultured cells [13, 19, 26]. Recessive mutations were shown to ablate the protein's ability to induce mitochondrial fragmentation [26]. The His123 residue is in the protein's α-loop domain located on the cytoplasmic side between GST-N and GST-C domains [25]. Four other dominant mutations have been found in or near the α-loop domain, three of which have been tested in cultured cells: p.R120W, p.T157P, and p.A156G exert dominant negative effects by blocking mitochondrial fusion in hybrid cells with differentially labeled mitochondria [13, 26]. Moreover, GDAP1 was found to bind tubulin, and this interaction was strongly increased in the α-loop mutants, meaning that they could interfere with mitochondria–cytoskeleton interactions [27]. His123 is a highly conserved residue. Based on the earlier studies, it is conceivable that a conversion of histidine to arginine at this position might cause problems with mitochondrial dynamics, deficient cell respiration, or an inappropriate binding of mitochondria to the microtubule cytoskeleton that inhibits mitochondrial mobility.
In conclusion, our study identified the first strongly enriched AD-CMT2 mutation in Finland. We recommend GDAP1 p.H123R testing at an early stage of evaluation of Finnish patients with hereditary axonal neuropathy. For CMT diagnostics in general, exome sequencing appeared useful for disease mutation identification in known disease genes, although a targeted resequencing approach may be sufficient and more cost-effective in clinical practice. The identification of novel dominant CMT2 disease genes using exome sequencing is complicated and requires combined efforts of sequencing many families and family members. Another challenge of CMT2 diagnostics remains to be the phenotypic variability also demonstrated in this study. Although the patients with MFN2 mutations had more severe polyneuropathy, which begun at a younger age and had more mixed type characteristics than the patients with the GDAP1 mutation, these genetic patient groups were not clearly identifiable by the ENMG study. The findings of this study broaden our understanding of the clinical outcome of GDAP1 mutations in AD-CMT2 and may provide a unique opportunity for treatment testing in patients from a homogeneous genetic population with identical disease mutation.
References
Patzko A, Shy ME (2011) Update on Charcot-Marie-Tooth disease. Curr Neurol Neurosci Rep 11(1):78–88
Szigeti K, Lupski JR (2009) Charcot-Marie-Tooth disease. Eur J Hum Genet 17(6):703–710
Braathen GJ, Sand JC, Lobato A, Hoyer H, Russell MB (2011) Genetic epidemiology of Charcot-Marie-Tooth in the general population. Eur J Neurol 18(1):39–48
Saporta AS, Sottile SL, Miller LJ, Feely SM, Siskind CE, Shy ME (2011) Charcot-Marie-Tooth disease subtypes and genetic testing strategies. Ann Neurol 69(1):22–33
Zuchner S, Mersiyanova IV, Muglia M et al (2004) Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet 36(5):449–451
Lupski JR, Reid JG, Gonzaga-Jauregui C et al (2010) Whole-genome sequencing in a patient with Charcot-Marie-Tooth neuropathy. N Engl J Med 362(13):1181–1191
Choi BO, Koo SK, Park MH, Rhee H, Yang SJ, Choi KG, Jung SC, Kim HS, Hyun YS, Nakhro K, Lee HJ, Woo HM, Chung KW (2012) Exome sequencing is an efficient tool for genetic screening of Charcot-Marie-Tooth Disease. Hum Mutat (in press)
Silander K, Meretoja P, Juvonen V, Ignatius J, Pihko H, Saarinen A, Wallden T, Herrgard E, Aula P, Savontaus ML (1998) Spectrum of mutations in Finnish patients with Charcot-Marie-Tooth disease and related neuropathies. Hum Mutat 12(1):59–68
Sulonen AM, Ellonen P, Almusa H, Lepisto M, Eldfors S, Hannula S, Miettinen T, Tyynismaa H, Salo P, Heckman C, Joensuu H, Raivio T, Suomalainen A, Saarela J (2011) Comparison of solution-based exome capture methods for next generation sequencing. Genome Biol 12(9):R94
Zuchner S, De Jonghe P, Jordanova A, Claeys KG, Guergueltcheva V, Cherninkova S, Hamilton SR, Van Stavern G, Krajewski KM, Stajich J, Tournev I, Verhoeven K, Langerhorst CT, de Visser M, Baas F, Bird T, Timmerman V, Shy M, Vance JM (2006) Axonal neuropathy with optic atrophy is caused by mutations in mitofusin 2. Ann Neurol 59(2):276–281
Braathen GJ, Sand JC, Lobato A, Hoyer H, Russell MB (2010) MFN2 point mutations occur in 3.4 % of Charcot-Marie-Tooth families. An investigation of 232 Norwegian CMT families. BMC Med Genet 11:48
Engelfried K, Vorgerd M, Hagedorn M, Haas G, Gilles J, Epplen JT, Meins M (2006) Charcot-Marie-Tooth neuropathy type 2A: novel mutations in the mitofusin 2 gene (MFN2). BMC Med Genet 7:53
Zimon M, Baets J, Fabrizi GM, Jaakkola E, Kabzinska D, Pilch J, Schindler AB, Cornblath DR, Fischbeck KH, Auer-Grumbach M, Guelly C, Huber N, De Vriendt E, Timmerman V, Suter U, Hausmanowa-Petrusewicz I, Niemann A, Kochanski A, De Jonghe P, Jordanova A (2011) Dominant GDAP1 mutations cause predominantly mild CMT phenotypes. Neurology 77(6):540–548
Crimella C, Tonelli A, Airoldi G, Baschirotto C, D’Angelo MG, Bonato S, Losito L, Trabacca A, Bresolin N, Bassi MT (2010) The GST domain of GDAP1 is a frequent target of mutations in the dominant form of axonal Charcot Marie Tooth type 2 K. J Med Genet 47(10):712–716
Baxter RV, Ben Othmane K, Rochelle JM, Stajich JE, Hulette C, Dew-Knight S, Hentati F, Ben Hamida M, Bel S, Stenger JE, Gilbert JR, Pericak-Vance MA, Vance JM (2002) Ganglioside-induced differentiation-associated protein-1 is mutant in Charcot-Marie-Tooth disease type 4A/8q21. Nat Genet 30(1):21–22
Cuesta A, Pedrola L, Sevilla T, Garcia-Planells J, Chumillas MJ, Mayordomo F, LeGuern E, Marin I, Vilchez JJ, Palau F (2002) The gene encoding ganglioside-induced differentiation-associated protein 1 is mutated in axonal Charcot-Marie-Tooth type 4A disease. Nat Genet 30(1):22–25
Claramunt R, Pedrola L, Sevilla T, Lopez de Munain A, Berciano J, Cuesta A, Sanchez-Navarro B, Millan JM, Saifi GM, Lupski JR, Vilchez JJ, Espinos C, Palau F (2005) Genetics of Charcot-Marie-Tooth disease type 4A: mutations, inheritance, phenotypic variability, and founder effect. J Med Genet 42(4):358–365
Chung KW, Kim SM, Sunwoo IN, Cho SY, Hwang SJ, Kim J, Kang SH, Park KD, Choi KG, Choi IS, Choi BO (2008) A novel GDAP1 Q218E mutation in autosomal dominant Charcot-Marie-Tooth disease. J Hum Genet 53(4):360–364
Cassereau J, Chevrollier A, Gueguen N, Malinge MC, Letournel F, Nicolas G, Richard L, Ferre M, Verny C, Dubas F, Procaccio V, Amati-Bonneau P, Bonneau D, Reynier P (2009) Mitochondrial complex I deficiency in GDAP1-related autosomal dominant Charcot-Marie-Tooth disease (CMT2K). Neurogenetics 10(2):145–150
Sivera R, Espinos C, Vilchez JJ, Mas F, Martinez-Rubio D, Chumillas MJ, Mayordomo F, Muelas N, Bataller L, Palau F, Sevilla T (2010) Phenotypical features of the p.R120W mutation in the GDAP1 gene causing autosomal dominant Charcot-Marie-Tooth disease. J Peripher Nerv Syst 15(4):334–344
Sevilla T, Jaijo T, Nauffal D, Collado D, Chumillas MJ, Vilchez JJ, Muelas N, Bataller L, Domenech R, Espinos C, Palau F (2008) Vocal cord paresis and diaphragmatic dysfunction are severe and frequent symptoms of GDAP1-associated neuropathy. Brain 131(Pt 11):3051–3061
Detmer SA, Chan DC (2007) Functions and dysfunctions of mitochondrial dynamics. Nat Rev Mol Cell Biol 8(11):870–879
Chen H, McCaffery JM, Chan DC (2007) Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell 130(3):548–562
Niemann A, Ruegg M, La Padula V, Schenone A, Suter U (2005) Ganglioside-induced differentiation associated protein 1 is a regulator of the mitochondrial network: new implications for Charcot-Marie-Tooth disease. J Cell Biol 170(7):1067–1078
Marco A, Cuesta A, Pedrola L, Palau F, Marin I (2004) Evolutionary and structural analyses of GDAP1, involved in Charcot-Marie-Tooth disease, characterize a novel class of glutathione transferase-related genes. Mol Biol Evol 21(1):176–187
Niemann A, Wagner KM, Ruegg M, Suter U (2009) GDAP1 mutations differ in their effects on mitochondrial dynamics and apoptosis depending on the mode of inheritance. Neurobiol Dis 36(3):509–520
Estela A, Pla-Martin D, Sanchez-Piris M, Sesaki H, Palau F (2011) Charcot-Marie-Tooth-related gene GDAP1 complements cell cycle delay at G2/M phase in Saccharomyces cerevisiae fis1 gene-defective cells. J Biol Chem 286(42):36777–36786
Acknowledgments
The authors would like to thank the patients and their families for participation in the study. Riitta Lehtinen and Annika Stormbom are thanked for technical help. We also acknowledge the exome capture, sequencing and variant calling pipeline analysis performed by the Institute for Molecular Medicine Finland FIMM. The authors wish to thank the following funding sources for support: Sigrid Jusélius Foundation, the Finnish Neuromuscular Disorders Association, and the Academy of Finland.
Web Resources
Inherited Peripheral Neuropathies Mutation Database (www.molgen.ua.ac.be/CMTmutations/Mutations/)
1000Genomes (www.1000Genomes.org)
NHLBI GO Exome Sequencing Project (evs.gs.washington.edu/EVS/)
SIFT Genome (sift.jcvi.org/)
Author information
Authors and Affiliations
Corresponding author
Additional information
Mari Auranen and Emil Ylikallio contributed equally.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
Filtering steps of the exome data using the unbiased approach. From each of the families 1 and 2, two related CMT2 patients were subjected to exome sequencing. The number of variants for each filtering step are shown. The selected variants were the following: (1) Intragenic, (2) Heterozygous on autosomes, (3) Rare in the 1000Genomes project (<0.5 % frequency), (4) Nonsynonomous, (5) Predicted damaging by the SIFT Tool, (6) Shared by the two affected family members, (7) Not present in our in-house database of exome data from Finnish individuals (n = 50). (JPEG 61 kb)
ESM 2
(PDF 6 kb)
Rights and permissions
About this article
Cite this article
Auranen, M., Ylikallio, E., Toppila, J. et al. Dominant GDAP1 founder mutation is a common cause of axonal Charcot-Marie-Tooth disease in Finland. Neurogenetics 14, 123–132 (2013). https://doi.org/10.1007/s10048-013-0358-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10048-013-0358-9