
Abstract
At present, epigenetic markers have been extensively studied in various fields and have a high value in forensic medicine due to their unique mode of inheritance, which does not involve DNA sequence alterations. As an epigenetic phenomenon that plays an important role in gene expression, non-coding RNAs (ncRNAs) act as key factors mediating gene silencing, participating in cell division, and regulating immune response and other important biological processes. With the development of molecular biology, genetics, bioinformatics, and next-generation sequencing (NGS) technology, ncRNAs such as microRNA (miRNA), circular RNA (circRNA), long non-coding RNA (lncRNA), and P-element induced wimpy testis (PIWI)-interacting RNA (piRNA) are increasingly been shown to have potential in the practice of forensic medicine. NcRNAs, mainly miRNA, may provide new strategies and methods for the identification of tissues and body fluids, cause-of-death analysis, time-related estimation, age estimation, and the identification of monozygotic twins. In this review, we describe the research progress and application status of ncRNAs, mainly miRNA, and other ncRNAs such as circRNA, lncRNA, and piRNA, in forensic practice, including the identification of tissues and body fluids, cause-of-death analysis, time-related estimation, age estimation, and the identification of monozygotic twins. The close links between ncRNAs and forensic medicine are presented, and their research values and application prospects in forensic medicine are also discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Epigenetics is the study of changes in gene expression that do not involve alterations in the DNA sequence [1]. The study of epigenetic changes is of interest in forensic medicine due to their unique mode of inheritance. At present, epigenetics has been widely studied, and epigenetic phenomena mainly include DNA methylation [2], non-coding RNA regulation [3], histone modification [4], X chromosome inactivation [5], chromatin remodeling [6], and genomic imprinting [7]. In these processes, non-coding RNAs (ncRNAs) act as key factors mediating gene silencing, participating in cell division, and regulating immune response and other important biological processes. As a research hotspot, ncRNAs are currently being extensively studied in various fields and have become an essential part of cancer research [8]. In addition, novel and extensive research has been conducted on ncRNAs in the fields of inflammation [9], immunity [10], cardiovascular diseases [11], metabolic diseases [12], psychiatric disorders [13], as well as reproduction and development of plants and animals [14, 15].
Forensic medicine is an applied discipline that uses medical and other related knowledge to provide scientific basis and evidence for criminal investigations, civil disputes, medical disputes, and other related fields involving law, to achieve justice and maintain social harmony and stability. It mainly includes forensic pathology, forensic clinical science, forensic genetics, forensic toxicology, forensic psychiatry, and other sub-disciplines. Although DNA genetic markers have led to great success in forensic research and applications, RNA molecules have not received much attention in forensic medicine due to their instability and susceptibility to degradation [16]. Until 2011, the European DNA Profiling Group (EDNAP) collaborated with many forensic laboratories to explore and demonstrate the extraordinary application potential of messenger RNA (mRNA) in the identification of body fluid stains [17,18,19,20,21,22,23]. Since then, RNA molecules have been widely studied in the field of forensic medicine. With the development of molecular biology, genetics, and detection technology, ncRNAs such as microRNA (miRNA), circular RNA (circRNA), long non-coding RNA (lncRNA), and P-element induced wimpy testis (PIWI)-interacting RNA (piRNA) are gradually being shown to have great potentials in relation to the practice of forensic medicine. MiRNAs provide new strategies and methods for the identification of tissues and body fluids, cause-of-death analysis, time-related estimation, age estimation, and the identification of monozygotic twins. Since the discovery of miRNAs in 1993, their role in physiological and pathological processes has been continuously revealed, and the potential of miRNA analysis in forensic medicine has been explored in the last decade. In 2010, Courts et al. [24] conducted a review on the biological functions and tissue and cell specificity of miRNA and the role of miRNA analysis in forensic medicine, demonstrating its potential for application in forensic medicine. In 2020, Rocchi et al. [25] reviewed the application of miRNA in the identification of body fluids, wound vitality, drowning, monozygotic twins, time of death determination, anti-doping, and sepsis. In our review, we will provide more recent advances on the application of miRNA in the practice of forensic medicine, especially cause-of-death analysis, estimation of time since deposition, and age estimation, which have not been previously reviewed.
To demonstrate the close link between non-coding RNAs and forensic medicine, the research progress and application status of ncRNAs, mainly miRNA, and other ncRNAs such as circRNA, lncRNA, and piRNA, in the practice of forensic medicine have been reviewed in this article. Moreover, the research value and application prospects of ncRNAs in the identification of tissues and body fluids, cause-of-death analysis, time-related estimation, age estimation, and identification of monozygotic twins have been discussed. The selected articles included in this review are original research articles or reviews in English, are available in PubMed (https://pubmed.ncbi.nlm.nih.gov/), and that discuss ncRNA as a biomarker in forensic practice.
Classification and detection methods of non-coding RNAs
Classification of non-coding RNAs
Unlike coding RNAs, ncRNAs usually do not have the function of encoding proteins, that is, such RNAs are not translated into proteins after transcription. Initially, the biological significance of ncRNAs was not well understood [26], unlike mRNAs that have been the focus of research [27,28,29]. It is only after the Human Genome Project and the Encyclopedia of DNA Elements (ENCODE) project were conducted that ncRNAs started gaining interest. Studies have shown that at least 80% of the human genome is transcribed into ncRNAs, and as research evolves, their important role in biological processes is gradually being revealed [30, 31]. In addition, the ENCODE project has made remarkable contributions to the discovery of ncRNAs, making it attract increasing attention [32, 33].
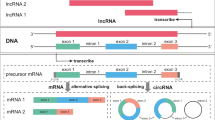
The different types of ncRNAs are continuously being discovered due to advances in molecular biology, molecular genetics, bioinformatics, and detection methods. Generally, according to the biological functions of ncRNAs, they can be classified into housekeeping ncRNAs, which are essential for maintaining basic cellular functions, and regulatory ncRNAs, which play regulatory roles in cells. The former mainly includes ribosomal RNA (rRNA), which binds to proteins to generate ribosomes; transfer RNA (tRNA), which carries and transports amino acids; small nuclear RNA (snRNA), which constitutes spliceosomes; small nucleolar RNA (snoRNA), which guides RNA chemical modification; and telomerase RNA (TR), which participates in the synthesis of chromosome ends. The latter mainly includes lncRNA, which is involved in various cellular regulatory processes; miRNA, which regulates gene expression; piRNA, which forms complexes with members of the PIWI protein family to regulate gene silencing; circRNA, which is rich in miRNA binding sites and acts as miRNA sponges; small interfering RNA (siRNA), which induces efficient and specific degradation of RNA; and enhancer RNA (eRNA), which is associated with gene expression near enhancers. In contrast, Dahariya et al. [34] divided ncRNAs into structural ncRNAs and regulatory RNAs. In addition, according to the length of ncRNAs, they can be divided into small ncRNAs with less than 200 nucleotides, also known as short ncRNAs, and long ncRNAs with more than 200 nucleotides. Meanwhile, other scholars have also classified ncRNAs differently based on length [34,35,36]. In this review, ncRNAs are classified according to length into short ncRNAs (< 50 nucleotides) such as miRNA, siRNA, and piRNA; medium ncRNAs (50–200 nucleotides) such as tRNA, snRNA, and snoRNA; and long ncRNAs (> 200 nucleotides) such as lncRNA. The classification of ncRNAs is shown in Fig. 1.

The main classification methods of non-coding RNAs mentioned above based on biological function and length
In addition, recent studies have shown that some circRNAs may be translated. Sun et al. [37] developed the CircCode tool to explore the translation potential of circRNA. Miao et al. [38] summarized relevant studies to demonstrate the ability of circRNA in coding proteins. The study provided evidence on the direct translation of endogenous circRNA. A few circRNAs contain internal ribosome entry sites, which give them translation ability and have been shown to serve as protein templates. Focusing on the expression and function of circRNA, Misir et al. [39] provided an overview of the progress of research on the biogenesis, function, and molecular mechanism of circRNA. Thus, it remains unclear whether circRNA should still be classified as ncRNA. In this review, circRNA was classified into ncRNA, which is the generally accepted view.
Common detection methods of non-coding RNAs
The development of detection methods has promoted the study of the unique molecular and biological characteristics and the expression features of ncRNAs, providing a theoretical basis for their application in forensic practice. The detection methods of ncRNAs can be classified according to whether they involve qualitative or quantitative determination, the throughput size, the dimension of the interaction between ncRNAs and DNA, RNA, or protein, and other aspects. This review summarizes the common detection methods of ncRNAs in forensic medicine, and Table 1 shows the common detection methods of ncRNAs in forensic medicine and their advantages and disadvantages.
Quantitative real-time polymerase chain reaction
Quantitative real-time polymerase chain reaction (qPCR) can perform the quantitative analysis of the starting template, and monitor the PCR process in real-time through changes in fluorescent signals after the addition of fluorescent substances to the PCR reaction system. Moreover, it can sensitively quantify low abundance ncRNA molecules expressed and is currently the most used technique for detecting RNA expression. The fluorescent substances used in qPCR are mainly divided into fluorescent probes, which are more specific, and fluorescent dyes, which are simpler and easier to use. The main types of qPCR include stem-loop qPCR [40], polyadenylation-based qPCR [41], and primer extension methods [42].
RNA sequencing
RNA sequencing (RNA-seq) is an important tool for transcriptomics research, which can detect the overall transcriptional activity at the single nucleotide level and provide comprehensive information on the transcriptome by analyzing the structure and expression levels of existing transcripts and discovering unknown transcripts. Based on the principle of sequencing by synthesis or semiconductor sequencing, RNA-seq is utilized to investigate transcriptional activity or to detect the expression and abundance of certain RNA molecules by massively parallel sequencing (MPS) techniques. The current MPS technology platform mainly includes Illumina’s Solexa sequencing technology [43] and Thermo Scientific’s ion semiconductor sequencing [44].
Microarray
Microarray is a hybridization-based method for high-throughput detection of ncRNA expression, which can obtain the expression of known loci at the transcriptome level in a short time [45]. The microarray can immobilize thousands of nucleic acid probes, such as miRNA, circRNA, and piRNA, on tiny solid-phase vectors and analyze them by scanning the hybridization signal intensity for detection [46]. Microarray is currently a powerful means to detect expression abundance within the genome, but it depends on known genomic information and can be accompanied by false positive results; thus, it is generally used for preliminary screening [47, 48].
Non-coding RNAs in the identification of tissues and body fluids
Accurately identifying of source of DNA samples from tissues or body fluids is crucial in the practice of forensic medicine, as it can provide clues and evidence for the investigation of criminal cases and also reduces the difficulty of the inspection work. The identification of tissues and body fluids can provide evidence to solve the problem of species and the source of the materials, which is conducive for determining the investigation direction and reconstructing the crime scene. The traditional methods of identifying tissues and body fluids mostly rely on chemical or physical methods, which are sometimes subject to cross-reactivity and false positive results and have limited specificity. Blood, saliva, semen, and vaginal fluids commonly found at crime scenes should be screened and identified using non-destructive and precise methods to obtain critical information for the reconstruction of crime scenes. In recent years, there has been an intensified focus on exploring the molecular characteristics and functions of ncRNAs, and the application of ncRNAs in the identification of tissues and body fluids has become a research hotspot in the field of forensic medicine.
Compared with mRNA, miRNAs are not easily degraded due to their short length, and have tissue specificity, which makes them extremely valuable in the identification of degraded materials or complex mixtures. In addition, research on the application of ncRNAs such as circRNA and piRNA in the identification of tissues and body fluids is also being actively conducted, favorably promoting the application of ncRNAs as epigenetic markers in the practice of forensic medicine. The ncRNA markers related to the identification of tissues and body fluids mentioned in this review are summarized in Table 2.
MicroRNA in the identification of tissues and body fluids
Due to its small molecular weight and rich tissue specificity, miRNA has become a promising biomarker in the identification of tissues and body fluids. Recently, Glynn et al. [49] assessed the relevant literature to investigate the future potential of miRNAs in the identification of tissue and body fluids. Unlike their review, we focus on the discovery of effective markers, the stability of markers, and the construction of inferential models for miRNA in the identification of tissues and body fluids, presenting new research findings from the last 3 years.
In 2009, Hanson et al. [50] examined 452 miRNA markers in peripheral blood, saliva, semen, vaginal secretions, and menstrual blood by reverse transcription-qPCR (RT-qPCR), and finally screened out differentially expressed markers that require as low as 50 pg of total RNA for identification in body fluids. Among them, miR-451 and miR-16 were mostly differentially expressed in peripheral blood, miR-135b and miR-10b in semen, miR-658 and miR-205 in saliva, miR-124a and miR-372 in vaginal secretions, and miR-412 and miR-451 in menstrual blood. This study is the first to explore the value of miRNA in forensic body fluids identification, indicating that miRNA is promising for the identification of tissues and body fluids in forensic practice. Then, Hanson et al. [51] provided detailed procedures for improving the success of analyzing degraded samples using the previously identified nine miRNA markers. Zubakov et al. [52] used the microarray platform to analyze 718 human miRNAs derived from saliva, semen, vaginal secretion, venous blood, and menstrual blood. The result revealed that miR-20a, miR-106a, miR-185, and miR-144 were useful for venous blood distinction, and miR-135a, miR-10a, miR-507, miR-943, and miR-891a for semen identification. Park et al. [53] examined more than 1700 miRNAs by microarray and screened for specific markers using Shannon Entropy and Q-statistics, and identified eight miRNAs for the identification of blood (miR-484 and miR-182), semen (miR-2392 and miR-3197), saliva (miR-223 and miR-145), and vaginal secretions (miR-1260b and miR-654–5p).
Thereafter, Wang et al. [54] developed an accurate model for the analysis of miRNA expression data to reduce the impact of technology platforms and statistical methods on the accuracy of miRNA markers for the identification of tissues and body fluids. The team analyzed the expression abundance of three miRNAs in different body fluids by RT-qPCR and the efficiency-calibrated model of relative expression ratios, demonstrating that miR-16 is specific in venous blood and detectable at 50 pg of total RNA. This study suggested that to identify miRNAs in body fluids, they should be highly expressed and should have constant expression levels in individuals. Future studies should focus on finding the best miRNA markers for body fluid identification and analyzing the effects of environmental factors such as temperature, humidity, contamination, and ultraviolet radiation on the stability of miRNAs in vitro. The team then used MPS to screen for miRNA biomarkers at the genome-wide level and identified 6 and 19 potential specific miRNA biomarkers in blood and saliva, respectively [55]. The study demonstrated that miRNA expression patterns in different body fluids can be measured at the genome-wide level for application in forensic practice. Seashols-Williams et al. [56] examined the relative levels of miRNA in blood, semen, vaginal fluids, menstrual blood, saliva, urine, feces, and sweat using NGS technology. The study found that miR-200b, miR-1246, miR-320c, miR-10b-5p, miR-26b, and miR-891a had body fluid specificity, and also identified potentially normalized markers including let-7 g and let-7i. Sauer et al. [57] conducted a comprehensive study on body fluids to recognize a single source sample. This study showed that hsa-miR891a-5p could be a potential marker for semen identification, and that hsa-miR-144-3p could differentiate between blood and non-blood samples. In addition, the combination of hsa-miR-144-3p and hsa-miR-203a-3p could be used to distinguish between venous and menstrual blood samples, and hsa-miR-203a-3p and hsa-miR-124-3p could be used to distinguish between saliva and vaginal secretion samples. There is still a challenge in distinguishing between venous blood and menstrual blood in mixtures, as well as saliva and vaginal secretion. Sirker et al. [58] used receiver operating characteristics to analyze 19 target microRNAs in blood, saliva, semen, vaginal secretions, menstrual blood, and skin. The most stably expressed genes in samples were miR-26b, miR-92, miR-484, and miR-144, which are regarded as preselected endogenous controls. In their study, they finally confirmed the use of miR-10b, miR-203, miR-374, miR-451, and miR-943 to identify five forensically relevant body fluids and skin. Fujimoto et al. [59] examined the relative expressions of miR-144-3p, miR-451a-5p, miR-888-5p, miR-891a-5p, miR-203a-3p, miR-223-3p, and miR-1260b by RT-qPCR and constructed a partial least squares discriminant analysis (PLS-DA) model. The results showed that the model could be used to identify venous blood, saliva, semen, and vaginal secretions. Dorum et al. [60] detected miRNA expression in blood, semen, saliva, vaginal secretions, menstrual blood, and skin by the miRNome MPS method, and built an optimally performing model consisting of nine miRNA markers based on partial least squares (PLS) and linear discriminant analysis. Mayes et al. [61] used RT-qPCR to examine mRNA and miRNA in blood and semen under different conditions such as heat, humidity, and sunlight, showing that miRNA markers are more stable and can be used for the identification of body fluids that prove to be challenging. Li et al. [62] investigated the stability of mRNA and miRNA in blood samples by RT-qPCR and multiplex PCR system to detect their expression under different environments such as light and dark, humidity, high temperature, and ribonuclease A (RNase A). Their study showed that humidity and RNase A have a greater effect on stability, and that miRNA is more stable than mRNA, which could be used for the identification of body fluids in aged and environmentally contaminated samples. In addition, Li et al. [63] examined the expression of 12 miRNAs in peripheral blood, menstrual blood, semen, saliva, and vaginal secretions by qPCR. The team constructed inferential models using two probabilistic methods, Naive Bayes and PLS-DA, showing that probabilistic methods have great potential for miRNA-based identification of body fluids, and that the selection of reference genes is important. Iroanya et al. [64] studied the stability of miR-451a, miR-10b, and miR-205 in blood, semen, and saliva samples under indoor, outdoor, 4 °C storage, and − 80 °C freezing conditions. The study showed that the stability of each miRNA marker is highest in the freezing group, and miR-451a, miR-10b, and miR-205 were reliable biomarkers for the identification of blood, semen, and saliva, respectively. In the future, more miRNA markers can be studied in different populations, diverse environments, broader age ranges, and multiple body fluids.
Sauer et al. [65] evaluated the differential expressions of 15 preselected miRNAs in the skin, skeletal muscle, heart muscle, kidney, lung, liver, and brain tissues by a methodologically validated qPCR procedure. It was shown that miR-206, miR-208b-3p, miR-205-5p, miR-122-5p, and miR-219a-5p could identify organ tissues such as skeletal muscle, heart, skin, liver, and brain, respectively. This study successfully proposed the first method based on miRNA biomarkers for organ tissue identification, which is compatible and complementary with forensic DNA analysis and can be useful in the identification of forensic field samples such as mixtures, aged and degraded samples, or trace samples.
As advancements in miRNA marker research continue, developing precise and consistent identification models for tissues and bodily fluids will greatly enhance the applicability of miRNAs in forensic practices. He et al. [66] established a stepwise strategy to discriminate peripheral blood from menstrual blood based on Fisher’s discriminant function, which first discriminates between blood and non-blood samples using miR-451, and then discriminates between peripheral blood and menstrual blood using miR-203, miR-205, and miR-214. Subsequently, He et al. [67] constructed a mathematical model using the same strategy, which consisted of miR-451a, miR-205-5p, miR-203-3p, miR-214-3p, miR-1445p, miR-654-5p, miR-888-5p, miR-891a-5p, and miR-124a-3p. This nine-miRNA-marker discriminant analysis model can be used for identifying vaginal secretions, saliva, semen, menstrual blood, and peripheral blood. The results showed that there is no significant difference in the expression of these markers in different amounts of total miRNA, which is beneficial for the identification of a small number of samples. However, the model is only suitable for the identification of samples from a single component of body fluids and not for mixed samples. Liu et al. [68] constructed an identification model by combining miRNA markers and kernel density estimation (KDE), whose kernel function was a radial basis function. By comparing multiple classification algorithms and combinations of 10 miRNAs, miR-451a, miR-891a-5p, miR-144-5p, and miR-203a-3-3p combined with KDE were found to be the most accurate and stable models for identifying vaginal secretions, saliva, semen, menstrual blood, and peripheral blood. This study evaluated the performance of different machine learning methods in constructing body fluid identification models for the first time and fully demonstrated that identification models constructed by suitable classification algorithms can significantly improve accuracy. Wang et al. [69] constructed a model to differentiate peripheral blood from menstrual blood samples based on the copy number ratio of miR-451a/miR-21-5p, which showed high sensitivity and specificity. Bamberg et al. [70] constructed a simultaneous mRNA/miRNA detection method to reduce the effects of degradation and tissue specificity, and achieved the combined application of multiple detections with the two markers. Rhodes et al. [71] constructed a ten-fold cross-validated quadratic discriminant analysis model based on let-7 g, let-7i, miR-200b, miR-320c, miR-10b, and miR-891a to identify vaginal secretions, semen, saliva, urine, feces, menstrual secretions, and blood, among which blood and feces were identified with 100% accuracy. The study provided a new method for model construction of tissues and body fluids identification, but the accuracy of identification in mixed samples still needed to be improved. Wei et al. [72] examined and validated miRNA expression levels in peripheral blood, menstrual blood, saliva, semen, and vaginal secretions by high-throughput sequencing and RT-qPCR. This study screened the most stable endogenous reference gene miR-320a-3p by geNorm, NormFinder, BestKeeper, and ΔCq algorithms, providing a method for screening endogenous reference genes.
CircRNA in the identification of tissues and body fluids
Sequencing studies have shown that circRNA can be abundantly, stably, and conservatively expressed throughout the life cycle and are highly tissue-specific and developmental stage-specific, as well as resistant to degradation by ribonuclease R (RNase R)[73, 74]. The closed-loop structure of circRNA renders them more stable and less susceptible to degradation, making them highly advantageous in the identification of aged or degraded samples. Song et al. [75] first explored the circRNA expression profiles of common samples such as venous blood, menstrual blood, saliva, semen, and vaginal secretions by Arraystar Human circRNA Array. The study showed that circRNA expression profiles were significantly different in venous blood, semen, and saliva, while the expression patterns were similar in menstrual blood and vaginal secretions. Thus, circRNA is expected to be a biological marker for body fluids identification. Zhang et al. [76] improved the sensitivity and accuracy of the assay by combining the linear and circular transcripts of the peripheral blood-specific marker ALAS2 and the menstrual blood-specific marker MMP7. In this study, circRNA was successfully included in mRNA profiling for body fluids identification, providing a new idea of circRNA application for this forensic practice.
Liu et al. [77] characterized the circRNA expression levels of 45 genes specifically expressed in 5 tissues and body fluids and further identified 38 circRNA markers from 14 genes. These biomarkers with circular transcripts have been validated in six body fluids, including HBA and ALAS2 in blood, MMP7 and MMP10 in menstrual blood, HTN3 in saliva, SPINK5, SERPINB3, ESR1, and CYP2B7P1 in vaginal secretions, TGM4, KLK3, and PRM2 in sperm, and SLC22A6 and MIOX in urine, all with expression specificity. It was shown that the inclusion of circRNA in the mRNA profile facilitates the construction of a multiplex analysis system for the identification of tissues and body fluids. Subsequently, Liu et al. [78] combined 14 tissue-specific mRNA markers with circRNA expression, 2 high expression abundance mRNA markers, and 2 housekeeping genes to construct a multiplex analysis system (F18plex system). The system was applied to the identification of urine, semen, vaginal secretions, saliva, menstrual blood, and peripheral blood, and had good sensitivity and specificity in the identification of mixture, degraded, and aged samples. The team also combined the one-step multiplex reverse transcription PCR (RT-PCR) method with the F18plex system to complete the identification of 0.1 ng total RNA in peripheral blood and semen, and 1 ng total RNA in menstrual blood, vaginal secretions, saliva, and urine [79]. This provided a reliable and cost-effective method for body fluids identification and could be used as tissue-specific biomarkers to simplify and perform simultaneous analysis.
PIWI-interacting RNA in the identification of tissues and body fluids
piRNA is a class of ncRNAs expressed in a wide range of cells, which generally functions by forming specific RNA–protein complexes and plays roles in many important biological processes, including cell proliferation, differentiation, and survival [80]. In addition, piRNA is closely associated with transposon silencing and epigenetics. It is not only expressed in a wide range of cells but also has the characteristics of short length and tissue-specific expression [81, 82]. Studies have shown that its 2'-O-methylated 3'-end can stabilize RNA molecules, making piRNA a promising epigenetic marker [83]. Wang et al. [84] identified the expression levels of four piRNAs in vaginal secretions, menstrual blood, semen, saliva, and venous blood by RT-qPCR and determined that piR-55521 is specifically expressed in semen. In addition, it was shown that piR-61648, piR-43994, and piR-33151 are differentially expressed in at least two body fluids, suggesting that piRNA has the potential for tissue and body fluids identification and is expected to become a new class of epigenetic markers.
Non-coding RNAs in cause-of-death analysis
The cause of death is the core and first issue to be solved in traditional forensic medicine, which is of great significance. The analysis of the cause of death is the hot spot and difficult point of forensic medicine practice, and accurate judgment of the cause of death is crucial for conviction, sentencing, and justice, as well as for the fairness of judicial practice. At present, the routine methods of the cause-of-death analysis are autopsy and pathological examination. Meanwhile, with the continuous development of science and technology, molecular techniques are gradually being applied to cause-of-death analysis. In the process of human death, tissue metabolism varies depending on the cause of death, among which the types and expression levels of ncRNAs will change in the relevant tissues. Based on these differences, we can provide valuable information on the cause of death. Currently, ncRNAs are widely studied in cause-of-death analysis, including mechanical asphyxia, sudden cardiac death (SCD), and suicide, and are expected to be new epigenetic markers that can be applied in forensic practice. Table 3 summarizes the research and application of ncRNAs in the analysis of these causes of death.
MicroRNA in the mechanical asphyxia
Under hypoxia, cell biological processes such as cell cycle and repair of DNA damage are markedly altered, thus affecting the expression of miRNAs involved in the metabolic regulation of hypoxia, with the brain and heart being the most sensitive to hypoxia. Studies have shown that hypoxia, as an important regulator of miRNA biosynthesis and function, can play a role in important biological processes such as transcription initiation, post-transcriptional processing, and post-transcriptional modification, thereby affecting the expression levels of miRNAs [85]. Schober et al. [86] analyzed the differences in miRNA expression between frontal cortex injury and natural death samples by genome-wide analysis and RT-qPCR. Two combinations of miR-138/miR744 and miR-195/miR-324-5p could completely distinguish between the two groups of samples and were not affected by biological age and postmortem time. It was shown that miRNA can be used as a marker of cerebellar hypoxia and a forensic marker for determining traumatic brain injury. Zeng et al. [87] examined the expression levels of miRNAs in brain and heart samples from mechanical asphyxia and compared them with samples from craniocerebral injury and hemorrhagic shock to identify differentially expressed miRNAs. Studies have shown that miR-122 is significantly downregulated in brain and heart samples from mechanical asphyxia and can be used as a biomarker of mechanical asphyxia.
Han et al. [88] showed that miR-3185 expression is significantly increased in cardiac tissues of deceased persons with mechanical asphyxia compared to those with craniocerebral injury, hemorrhagic shock, SCD, and poisoning. MiR-3185 and its target gene CYP4A11 could be used as markers of mechanical asphyxia in cardiac tissues. Subsequently, this team screened out markers associated with mechanical asphyxia, including DUSP, KCNJ2, miR-122, and miR-3185, and constructed a molecular prediction model to predict mechanical asphyxia [89]. Their study showed that the markers are not affected by factors such as age, temperature, and time of death, and the prediction model consisting of multiple markers could help to accurately infer whether mechanical asphyxia is the cause of death.
In addition, circRNAs are more stable and conserved than linear RNAs, and there is growing evidence that circRNAs play important roles in a variety of physiological processes [90]. Recently, Huang et al. [91] demonstrated new evidence of the interaction between circRNA and hypoxia, suggesting that hypoxia-induced circRNA is closely linked to cancer, angiogenesis, and energy metabolism, which provides a theoretical basis for screening suitable specific markers that can be used in mechanical asphyxia. However, there remains a gap in the study of circRNA as a forensic marker for mechanical asphyxia, and more experimental validations are needed.
Non-coding RNAs in SCD
MicroRNA in SCD
Cardiovascular disease is the leading cause of death worldwide, and SCD is one of the most common types. Due to the sudden and latent characteristics of SCD, it is very difficult for forensic pathologists to analyze and infer the cause of death. Studies have shown that miRNAs play an important role in cardiogenesis, cardiac function, and pathology, and are stably present in the progression of cardiovascular disease [92]. Thus, they have great potential as biomarkers of SCD.
Kakimoto et al. [93] performed deep sequencing of miRNA and RT-qPCR on autopsy samples of cardiac tissue from SCD with cardiac hypertrophy (SCH), compensated cardiac hypertrophy, and normal controls to determine the differences in miRNA expression. It was shown that miR-221 was significantly increased in SCD with SCH; thus, miR-221 could be a potentially useful marker for predicting and diagnosing SCH. Pinchi et al. [94] examined the expression levels of miR-1, miR-133, miR-208, and miR-499 in autopsy samples of SCD and acute myocardial infarction (AMI), and used samples from deceased patients without pathological cardiac involvement as controls. It was shown that miR-1 and miR-499 could accurately identify SCD and AMI, and different cardiac diseases could be distinguished by miRNA expression after autopsy.
Yan et al. [95] examined the expression levels of specific miRNAs including miR-133a-3p, miR-499a-5p, miR-223-3p, and miR-3113-5p in cardiac tissue from positive and negative autopsy samples of SCD, and used carbon monoxide poisoning and lethal injury samples as controls. It was shown that miR-133a-3p, miR-499a-5p, miR-223-3p, and miR-3113-5p are significantly increased in SCD samples and could be used to distinguish SCD from non-cardiac death. In addition, the combination of miR-3113-5p and miR-223-3p had the highest sensitivity and specificity in identifying positive and negative autopsy samples of SCD, and each pairwise combination of these miRNAs could effectively identify the specific cause of SCD.
Li et al. [96] divided autopsy samples of coronary artery disease-induced SCD (CAD-SCD) into two groups with or without obvious pathological features and used trauma victims as the control group to explore the value of specific miRNAs in the determination of the cause of death due to CAD-SCD. The results showed that miR-126-5p and miR-499a-5p are significantly downregulated in cardiac tissues of CAD-SCD and could be used as sensitive postmortem biomarkers for the diagnosis of CAD-SCD. In addition, the combined application of miR-126-5p and miR-499a-5p has high sensitivity and specificity in identifying whether a deceased person had experienced silent myocardial ischemia and is a valid marker for inferring the CAD condition.
CircRNA in SCD
CircRNA is widespread and abundant in the cardiovascular system, playing important roles in a variety of developmental and physiological or pathological processes, and its expression levels are tissue-specific and time-specific. Studies have shown that circRNA is expressed at higher levels and is significantly less sensitive to exonuclease activity than linear RNA. More importantly, it can be secreted and identified in cardiac diseases, which makes it a highly promising biomarker [97]. Tian et al. [98] explored the role of circNFIX and circSLC8A1 in the inference of the cause of death in SCD due to acute ischemic heart disease (IHD). The team found elevated levels of circSlc8a1 in IHD rat models and ischemia-hypoxia H9c2 cell models, while circNfix levels were elevated early in ischemia and then downregulated. The results suggested a time-dependent nature of both markers. The detection results in autopsy samples showed that circSLC8A1 has high sensitivity and specificity for myocardial infarction. In addition, decreased circNFIX levels could indicate ischemic myocardial injury, and it was negatively correlated with the degree of coronary artery stenosis. These studies suggested that circSLC8A1 and circNFIX can be used as new molecular markers for diagnosing SCD due to acute IHD.
Non-coding RNAs in suicide
Although the influencing factors of suicide are very complex, it has become a common cause of death in modern society. NcRNAs are abundantly expressed in the brain and are brain region-specific and cell type-specific. The expression level of ncRNA in the brain influences the development of psychiatric disorders, and its role in suicidal behavior is gradually being revealed [99, 100], demonstrating the potential of ncRNA as a biomarker for the analysis of the cause of death by suicide.
Long non-coding RNA in suicide
LncRNA is closely related to brain development and participates in the proliferation and differentiation of pluripotent stem cells. It also has a tissue-specific expression pattern that can change with age. At present, the role of lncRNA in psychiatric disorders is a research hot topic, and it also plays an important role in suicidal behavior [101]. Punzi et al. [102] showed that the expression level of LINC01268 is significantly increased in violent suicides compared to non-violent suicides and non-suicide deaths. Subsequently, the team used RNA sequencing data to demonstrate that LINC01268 expression is significantly increased in the dorsolateral prefrontal cortex in violent suicide [103]. The Brown-Goodwin questionnaire results indicated that the high expression of specific long intergenic non-coding RNA (lincRNA) in violent suicide is associated with aggressive behavior, and weighted gene co-expression network analysis revealed a strong correlation between LINC01268 and the immune-related gene P2RY13. This study suggested that LINC01268 affects emotion regulation, aggressive behavior, and violent suicide, and is a potential marker of violent suicides.
Cui et al. [99] performed microarray analysis and RT-qPCR on peripheral blood mononuclear cells (PBMC) from patients with major depression and normal controls. A total of six differentially expressed lncRNAs including NONHSAT142707, NONHSAT034045, ENST00000517573, NONHSAG045500, ENST00000566208, and TCONS_00019174 were identified as downregulated in patients with major depressive disorder (MDD). The study showed that these six downregulated lncRNA expression levels are negatively correlated with suicide risk in MDD patients and could be used as biomarkers of suicidal ideation in MDD patients. By genome-wide multiplex testing correction, Zhou et al. [104] screened differentially expressed lncRNAs by RNA sequencing in the rostral anterior cingulate cortex of 26 depressed suicides and 24 healthy controls and identified a total of 23 statistically significant differentially expressed lncRNAs, with 15 being upregulated and 8 being downregulated. Six of these lncRNAs (RP11-326I11.3, RP11-273G15.2, CTD-2647L4.4, CTC-487M23.5, RP1-269M15.3, and RP11-96D1.10) were each significantly associated with the expression of an antisense or overlapping protein-coding gene, which could serve as a cis target of the lncRNA. This study showed that lncRNAs are differentially expressed in the brains of depressed patients who died by suicide and provided new clues for the analysis of the cause of death by suicide.
MicroRNA in suicide
Among ncRNAs, miRNA functionality is strongly linked to the relevance of psychiatric disorders, and its role in suicidal behavior has been extensively studied. Studies have shown that overall miRNA expression is downregulated in suicide patients with MDD [105]. The team then screened two upregulated miRNAs including miR-376a and miR-625 and six downregulated miRNAs including miR-152, miR-34a, miR-330-3p, miR-181a, and miR-133b, which were differentially expressed in suicide and non-suicide subjects. Among these, miR-152 was found to be closely related to suicide according to the significance values [106]. Afterward, the group focused on how pro-inflammatory cytokine genes are regulated in suicidal behavior [100] and found increased expression of miR-19a-3p in MDD patients who died by suicide and in PBMCs of MDD patients with suicidal ideation, suggesting that miR-19a-3p may contribute to cytokine dysregulation in suicidal individuals.
Lopez et al. [107] analyzed miRNA expression in the prefrontal cortex of individuals who committed suicide and controls by RT-qPCR and found miR-34c-5p, miR-320c, miR-139-5p, and miR-195 to be significantly upregulated in suicide victims. This study suggested that there is a significant correlation between these miRNAs and the expression levels of the polyamine genes SAT1 and SMOX, and that the dysregulation of these genes in the brains of suicidal individuals may be influenced by post-transcriptional regulation of miRNAs to cause suicidal behavior. In another study, Lopez et al. [108] detected significant differences in the expression of miR-425-3p, miR-24-3p, miR-146a-5p, and miR-146b-5p by RT-qPCR in the ventrolateral prefrontal cortex of suicidal individuals with MDD and healthy controls, and all these miRNAs were upregulated in the brain tissue of suicidal individuals. In addition, through bioinformatics and functional in vitro experiments, they found that all four miRNAs are also associated with the MAPK/Wnt signaling pathway that is closely related to suicidal behavior.
MicroRNA in other causes of death
In addition to the above-mentioned causes of death, miRNAs have been explored in the analysis of sudden infant death syndrome (SIDS), drowning, and acute spinal cord injury, but the relevant studies are few or in an initial stage, and more studies are still needed to further validate their roles in the analysis of these causes of death.
Courts et al. [109] examined the differential miRNA expression in heart and brain tissues from SIDS decedents and controls by real-time fluorescence qPCR and successfully found significant upregulation of miR-1 in heart tissues and let-7b in brain tissues of SIDS decedents. This study suggests that organ-specific miRNA dysregulation may be related to the pathogenesis of SIDS, and that miR-1 and let-7b may be used as biomarkers of SIDS to provide a reference for the cause-of-death analysis.
Yu et al. [110] conducted experiments in freshwater and seawater drowning models of mice and analyzed the expression of miRNAs in the brain using bioinformatics screening. In their study, eight specific miRNAs (miR-6394, miR-706, miR-30c-1-3p, miR-6238, miR-494-3p, miR-669 h-3p, miR-135a-1-3p, and miR-5109) were examined, and they all showed increased expressions in the freshwater drowning model and decreased expressions in the seawater drowning model. The expression of miR-706 was higher and statistically different in the freshwater group than in the seawater and control groups. Studies have shown that miR-706 can be used as a potential biomarker of drowning to provide clues for crime scene investigation.
Recently miRNAs have attracted considerable interest in the field of forensic pathology due to their tissue and body fluid specificity, disease specificity, and because miRNA research is less costly. These features make miRNAs ideal candidate markers for forensic practice. Pinchi et al. [111] described specific miRNAs that can provide indications for understanding crime scene investigation and postmortem pathology processes in acute spinal cord injury. The study suggested that the specificity and sensitivity of miRNAs can be utilized during autopsy to help determine the cause of death; thus, miRNAs may become a reliable forensic biomarker for the diagnosis of acute spinal cord injury in the future.
Non-coding RNAs in time-related estimation
Time-related estimations such as estimation of time since death (TSD), wound age estimation, and estimation of time since deposition are of great significance in forensic practice. It plays an important role in determining the time of the crime, judging the sequence of injury, delineating the scope of the investigation, analyzing the nature of cases, and verifying the alibi, which can provide a scientific basis for forensic identification and ensure the justice and fairness of judicial practice. Traditional time-related estimation mostly relies on physical, chemical, morphological, histological, immunohistochemical, and imaging methods, but these methods are susceptible to environmental and individual differences and are prone to false positive results. In addition, the lack of standardized protocols and the dependence on personnel reduce the reliability of identification results. Recently, the role of ncRNA in a variety of physiological and pathological processes has been extensively investigated, and its potential for application in time-related estimation has also attracted the attention of forensic scientists. The study of ncRNA in time-related estimation, especially in wound age estimation and estimation of time since deposition, is a relatively new field. Although almost all related studies are at an early stage, the combination of multiple methods can also provide more reliable results and has great potential for application.
Non-coding RNAs in the estimation of TSD
The inference of the TSD or postmortem interval (PMI) has been a major challenge in forensic medicine since ancient times, which can be generally classified into early PMI estimation, late PMI estimation, and PMI estimation of skeletonized body. Traditional methods are mostly based on physicochemical methods such as body temperature, postmortem phenomena, and the condition of stomach contents, and forensic anthropology and forensic entomology also provide multiple methods. However, these methods are influenced by a variety of internal and external factors such as environmental temperature, humidity, and individual differences. Due to the widespread existence of ribonucleases, it is generally believed that RNA is more easily degraded postmortem or in vitro than DNA. However, studies have shown that some ncRNAs such as miRNA and circRNA have high stability. Therefore, research on sensitive and specific ncRNA markers that can be used for the estimation of TSD has attracted extensive attention.
MiRNAs have tissue and body fluid specificity and higher resistance to degradation compared to mRNAs, which have promising applications in the estimation of TSD/ PMI. Wang et al. [112] investigated the degradation patterns of miRNAs as a way of exploring the potential value of miRNA applications for PMI estimation. The team explored the changes of miR-122, miR-133, miR-150, miR-195, and miR-206 in the brain, liver, heart muscle, and skeletal muscle of mice within 48 h postmortem. The study showed that miR-133 and miR-206 start to degrade significantly 24 h after death, and miRNAs remain relatively stable in liver tissues within 24 h postmortem. Lv et al. [113] examined the gene transcript abundances in rat spleen tissue by RT-qPCR and studied the feasibility of using miRNAs, U6 snRNA, 18S rRNA, and mRNAs to determine PMI. The study showed by the geNorm algorithm that miRNAs are less affected by PMI and temperature and are suitable endogenous control markers. GAPDH and ACTB are more suitable for early PMI estimation due to their rapid degradation after death, while 18S rRNA has a unique degradation pattern and is suitable for estimating PMI over a longer period. Thereafter, the team constructed mathematical models for PMI determination using the transcript levels of lung and muscle tissue samples from rats and humans at three different temperatures from 0 to 144 h postmortem [114]. In their study, RPS29, U6 snRNA, 5S rRNA, miR200c, and miR-195 were selected as reference markers for lung tissue, and RPS29, 5S rRNA, miR-206, and miR-1 were selected as reference markers for muscle tissue. In addition, ACTB and GAPDH were significantly correlated with PMI. The mathematical model had multi-temperature and multi-indicator characteristics and was validated in human samples, which greatly improved the accuracy and reliability of the model and provided a practical tool for forensic practice. Lv et al. [115] screened PMI markers in samples collected from the heart, liver, and brain of human cadavers as well as in mouse heart and liver samples. 5S rRNA, miR-1, and miR-133a were selected as the best reference markers because they showed high stability over 5 days or more. In contrast, miR-122, which is a liver-specific marker, started to degrade at higher temperatures; thus, only 5S rRNA was selected as an endogenous control marker for the liver. They concluded that ncRNA can be used as an epigenetic marker to accurately determine PMI in human samples, but for very short PMI more sensitive markers are needed for estimation. In addition, Pasaribu et al. [116] provided a strategy to screen the literature for miRNA related to PMI research and demonstrated the potential value of miRNA in PMI.
The high stability, abundance, and tissue specificity of circRNA makes it potentially valuable in late PMI estimation. The short hairpin structure of snRNA and its lack of nuclease degradation make snRNA highly correlated with late PMI. In the quantitative detection of ncRNAs, reference genes facilitate the standardization and normalization of data, and genes with stable expression levels are often the better choice. Tu et al. [117] performed a stability assessment of tissue-specific reference genes by geNorm and NormFinder algorithms. Reference genes suitable for the estimation of PMI were screened out from 11 candidate genes, including miR-133a, circ-AFF1, and LC-LRP6 in skeletal muscle tissues, miR-122, circ-AFF1, and LC-Ogdh in liver tissues, and 18S rRNA, miR-122, and miR-133a in heart tissues. The study demonstrated that miRNA and circRNA are more stable than other types of RNAs as reference genes for the estimation of PMI. Subsequently, Tu et al. [118] constructed mathematical models applicable to the three tissues by studying PMI estimation markers in postmortem skeletal muscle, liver, and heart tissues of mice. Among them, miR-133a and circ-AFF1 were used as reference markers for skeletal muscle tissues, miR-122, circ-AFF1, and LC-Ogdh for liver tissues, and 18S rRNA, miR-122, and miR-133a for heart tissues. It was shown that both U6 snRNA and Rps18 are suitable biomarkers for heart and liver tissues, while both U6 snRNA and ACTB are suitable biomarkers for skeletal muscle tissues.
It is challenging to apply appropriate ncRNA markers for PMI estimation when decomposing or destructive cadavers and skeletonized bodies are involved. Na et al. [119] detected miRNA levels in patella samples collected from cadavers, and the expressions of let-7e and miR-16 decreased with the increase in PMI. Studies have shown that let-7e and miR-16 could be used as specific markers for PMI estimation, and their expression levels could be used to estimate PMI over several months. In addition, the differences in ncRNA marker expression levels in postmortem samples may be related to sampling sites. Kim et al. [120] examined the effect of sampling sites on miRNA levels by measuring miRNA expression in blood from different collection points of cardiac and non-cardiac death samples, such as the external iliac vein, inferior vena cava, and coronary sinus. It was shown that the cardiac-specific miR-208b and miR-1 markers in postmortem blood differ depending on the sampling sites and are not related to PMI. Therefore, it is important to consider the tissue specificity of miRNAs in forensic applications and to pay attentions to the differences in sampling sites.
Currently, ncRNAs show potential for application in the estimation of PMI, but there are still many challenges to overcome before they can be effectively applied in forensic practice. Table 4 summarizes the available research on ncRNAs for TSD estimation. It is important to identify the ideal endogenous reference gene, which should have a stable expression level and not be affected by the cause of death, health status, PMI, and other individual parameters. However, the expression level and specificity of ncRNAs are different between animal models and human models, and many death factors are more difficult to replicate in the models. Therefore, the lack of human data is one of the biggest limitations for the application of ncRNAs in PMI estimation. With the discovery of more effective epigenetic markers and the comprehensive consideration of various factors, the combination of various tissues and multiple ncRNA markers will provide a powerful forensic tool for PMI estimation.
MicroRNA in wound age estimation
The time of injury occurrence is an important issue in forensic medicine, which can provide a scientific basis for the practice of forensic medicine, such as the time of the crime, the sequence of injury, the analysis of the nature of the case, and the reconstruction of the crime scene. Traditional methods of wound age estimation such as morphology, histology, and immunohistochemistry have limited the accuracy of wound age estimation due to the lack of standardization and dependence on personnel. Therefore, it is still a challenge to make accurate wound age estimation. Generally, wound age estimation can be divided into wound age estimation of cadavers and wound age estimation of living bodies, and current research on wound age estimation involves organs and tissues such as the skin, brain, muscle, and bone.
Since miRNAs are involved in the physiological and pathological processes from injury to wound healing, wound age estimation has become an innovative application area of miRNAs [121, 122]. Neri et al. [123] investigated the expression of miRNA in skin samples from the site of hanging and revealed the difference in miRNA expression in wounds before and after death. Compared with the control group, miR-214a-3p, miR-103a-3p, and miR-92a-3p were significantly upregulated in samples of subjects who died by hanging. In addition, studies have shown that miRNAs are differentially expressed over time in wounds, showing potential for application in wound age estimation [124]. Bertero et al. [125] showed that miR-483-3p starts to increase at 3 days after wound formation and peaks at 6 to 7 days. Wang et al. [126] detected miRNA expression profiles in granulation tissue on the 7th day of wound healing by microarray analysis and RT-qPCR. It was found that the expressions of miR-203, miR-21, and miR-31 are upregulated by 2.5, 3.1, and 17.2 times, respectively, while the expression of miR-249 is downregulated by 2 times. Etich et al. [127] revealed changes in expression levels of miRNA during skin wound healing by RT-qPCR and bioinformatics tools. Among them, the expression levels of miR-204 and miR-205 were downregulated from day 5 to day 10 and day 1 to day 7, respectively, while the expression level of miR-31 was significantly upregulated after day 5. Chang et al. [128] investigated the role of miR-126 in human skin wound healing and detected the expression of miR-126 in skin wounds by RT-qPCR. The results showed that miR-126 expression significantly increases with time on day 1 after injury (inflammatory phase) and day 7 after injury (proliferative phase), respectively.
In a study on wound age estimation of burns, Lyu et al. [129] analyzed 24 differentially expressed miRNAs in a mouse model of antemortem burn skin through microarray analysis, among which 19 miRNAs were significantly upregulated and 5 miRNAs were significantly downregulated. Zhang et al. [130] investigated miR-711 and miR-183-3p expression levels in mouse and postmortem human burned skin by RT-qPCR. The study showed that miR-711 and miR-183-3p levels in antemortem burn areas were elevated until 120 h after death, and that postmortem burns do not induce changes in miR-711 and miR-183-3p expression levels in mouse skin, suggesting that these two miRNAs are potential biomarkers for distinguishing antemortem from postmortem burns. In contrast, the levels of these two miRNAs were also elevated in human burn skin at 48 h of autopsy and correlated with the severity of burns. In addition, since miRNAs play an important role in the process of fracture healing, they may provide a new approach for determining the wound age of fracture injury [131, 132].
Current miRNA-related studies have opened up new directions and promising prospects for their use as wound age estimation markers [133]. Table 5 summarizes the research on miRNA in wound age estimation. However, few studies have been conducted, and almost all of them are in the early stages. The challenge lies in how to select standardized samples to obtain reliable experimental data, so as to improve the feasibility, effectiveness, and accuracy of miRNA as a tool for wound age estimation. In addition, one method alone is unlikely to provide the credibility required for forensic cases, and combining histological, immunohistochemical, and genetic analysis may become a new tool for precise diagnosis.
Non-coding RNAs in the estimation of time since deposition
Estimating the time since deposition (TsD) of a crime scene and determining the biological traces formed before and after the crime event is one of the challenges faced by forensic investigators. A more accurate estimate of the time of the crime can provide valuable information for verifying witness statements, identifying the number of suspects, and assessing the suspects’ alibis. Estimating the TsD is helpful in determining the time of the crime and has great value in the practice of forensic medicine. At present, there are several methods to determine the deposition time of body fluids, including physical, chemical, and biological methods, among which research on the deposition time of bloodstains is relatively abundant. NcRNA has the characteristics of body fluid specificity and relative stability; thus, it has a good application prospect in the estimation of time since deposition. At present, there are relatively few studies on the application of ncRNA markers to estimate the TsD, and more relevant studies are still needed.
In 2005, Anderson et al. [134] used RT-qPCR to examine the expression of ACTB and 18 s rRNA in eight human dried blood samples and constructed linear functions that could be used to infer the time since deposition of blood traces over 150 days. Lech et al. [135] evaluated whether miRNA-142-5p and miRNA-541, two significant diurnal expression patterns in the vitreous humor, could be used to estimate the deposition time of bloodstains. Although the results showed that these markers cannot be applied to the estimation of bloodstain deposition time, they provide insights regarding the methods and experience for such studies. Alshehhi et al.[136] examined the relative expression ratio of saliva-specific markers and semen-specific markers by RT-qPCR and developed a method to estimate the deposition time of body fluids, in which miRNA and U6 snRNA could be used as reference genes. Wei et al. [137] detected the relative expression levels of bloodstains in indoor and outdoor environments by RT-qPCR and constructed a mathematical model based on circ-0001445, ALAS2, and HBB markers. Their study showed that the relative expression levels of circ-0001445 varied over time and that circRNA could be used as a potential marker for TsD estimation. In addition, different environments significantly affect the relative expression levels of some blood markers, but sex differences do not affect the estimation of bloodstain deposition time. This study was the first to use circRNA for TsD estimation and initially explored the practical value of circRNA in estimating the deposition time of bloodstains.
Non-coding RNAs in age estimation
Estimation of age is widely used in forensic practice and plays an important role in the identification of corpse sources, conviction and sentencing, and the determination of criminal capacity. Age estimation initially relied on bones and teeth, then based on biomolecular alterations of DNA or proteins such as Signal joint T-cells receptor excision DNA circles, telomere shortening, mitochondrial DNA deletion, aspartic acid racemization, and advanced glycation end-products, which developed the molecular framework of forensic estimation of age [138]. With the development of epigenetics, DNA methylation also provides a powerful marker for forensic applications in age estimation [139]. Currently, forensic anthropology methods are the main methods used for age estimation, but they are often affected by individual nutritional differences, environment, disability, and other factors. More importantly, they are often confounded by trace detection. Currently, age-associated ncRNAs as an epigenetic marker for age estimation may provide a new direction and idea to meet the practice of forensic medicine.
MicroRNA in age estimation
As early as 2010, Noren Hooten et al. [140] evaluated more than 800 miRNAs by RT-qPCR, and the expression abundance of most miRNAs decreased with age. The investigators screened miR-103, miR-107, miR-128, miR-130a, miR-155, miR-24, miR-221, miR-496, and miR-1538, a total of nine miRNAs that are significantly reduced in older adults, and showed that the reduction of miRNAs is independent of race and sex. Their study revealed that the expression of miRNAs is closely related to human age, and miRNAs are expected to be epigenetic markers for age estimation. Rubie et al. [141] revealed that miR-486 and mechanistic target of rapamycin (mTOR) protein levels are negatively correlated in PBMCs, and miR-486 levels are higher in older individuals than in younger individuals. It was shown that miR-496 is involved in aging regulation through the insulin/mTOR pathway and that the expression levels of miRNAs are correlated with increasing age. Huan et al.[142] identified 127 miRNAs that are differentially expressed with age in 5221 adult samples and constructed a linear age prediction model based on the expression levels of 80 miRNAs. The team used the differences between the predicted and actual age of miRNAs as indicators of biological aging, which predict all-cause mortality and correlate with coronary heart disease and hypertension. The study showed that cell type has little effect on the correlation between miRNA expression and age, and that age prediction models could be constructed based on a large sample size and a wide age range of whole-blood miRNA expression profiles.
CircRNA in age estimation
Wang et al. [143] screened age-associated circRNAs by false discovery rate, lasso regression, and support vector machine analysis, and their analysis revealed that circRNA levels were upregulated with age. Twenty-eight circRNAs were validated by RT-qPCR, and five circRNAs were finally selected to construct age prediction models. By comparing with five modeling methods, the regression tree model and random forest regression model had the best mean average error, reaching 8.767 and 9.126 years, respectively. In addition, it was shown that the mean average error of the age prediction model is significantly smaller in male age prediction than that in females, showing the influence of sex factors on circRNA markers. Their study is the first to construct age prediction models using circRNAs as biological markers, showing that circRNA as an epigenetic marker with potential application in age estimation has promising research prospects.
Recently, the role of ncRNAs in the aging process has been revealed and is continuously being studied. Moreover, ncRNAs are involved in various key cellular processes and are closely associated with age [144, 145]. At present, the expression levels of ncRNAs show age specificity and have great potential for application in the practice of forensic medicine. However, there are few relevant studies, and more questions need to be addressed. The variation of ncRNAs in the life cycle of individuals, the influence of gender factors on markers, the number and age range of study samples, and the methods of constructing prediction models are the key elements that need to be further explored.
MicroRNA in the identification of monozygotic twins
Currently, forensic genetics mainly applies short tandem repeat, single nucleotide polymorphism, and insertion/deletion genetic markers for identification, such as genetic relationship and personal identification. However, monozygotic (MZ) twins are two embryos derived from the same zygote through cleavage, having the same DNA sequence. Thus, conventional methods cannot distinguish MZ twins. Ensuring the identification of potential offenders and obtaining accurate identification results in criminal cases or parentage testing is an urgent problem for forensic scientists. As a type of ncRNA widely existing in eukaryotic cells, miRNA participates in a variety of biological processes and has the characteristics of low molecular weight, relative stability, abundant quantity, and tissue specificity. In recent years, many studies have shown that disease-related miRNAs are differentially expressed in MZ twins, which provides a theoretical basis for the application of miRNAs as an epigenetic marker in identifying MZ twins [146,147,148].
Fang et al. [149] performed a genome-wide analysis of miRNAs in the blood of four pairs of MZ twins by MPS, but only 14% of the examined miRNAs were differentially expressed. Moreover, RT-qPCR was used to verify the six most abundant differentially expressed miRNAs, and only miR-451a was differentially expressed among all MZ twins. Xiao et al. [150] analyzed the miRNA expression profiles in the blood of seven pairs of MZ twins by microarray. A total of 545 miRNAs were differentially expressed, and only miR-142-3p and miR-3653-3p were differentially expressed in six pairs of MZ twins. The authors further validated 10 differentially expressed miRNAs using RT-qPCR in 18 pairs of MZ twins, demonstrating their application value in the identification of MZ twins.
At present, research on miRNA applications in forensic practice is insufficient, and most studies have relatively small sample sizes. Moreover, the tissue specificity of miRNAs makes it necessary for researchers to explore the related markers for different kinds of materials, and it is not yet possible to provide miRNA markers for the identification of MZ twins. Therefore, the application potential of miRNAs in the identification of MZ twins remains to be explored.
Conclusions
As an epigenetic phenomenon that plays an important role in gene expression, the function and mechanism of ncRNAs in the physiological and pathological processes of different systems, organs, tissues, and cells are constantly being revealed. Moreover, with the development of molecular biology, molecular genetics, and bioinformatics, the advantages of low molecular weight, relative stability, abundant quantity, and tissue specificity possessed by ncRNAs are gradually revealed. This provides a richer theoretical basis and more promising research direction for the application of ncRNAs in the practice of forensic medicine, among which ncRNAs show great potential and a good prospect in the identification of tissues and body fluids, cause-of-death analysis, time-related estimation, estimation of age, and identification of monozygotic twins. Figure 2 illustrates the research on ncRNAs and their application in forensic practices.

Various research and potential applications of microRNA (miRNA), circRNA, long non-coding RNA (lncRNA), and PIWI-interacting RNA in the practice of forensic medicine
In addition, the development of detection technologies such as NGS also provides a powerful tool for the study and exploration of ncRNAs. Notably, single-cell RNA sequencing (scRNA-seq) has been widely used to detect the expression patterns of ncRNAs in cells with different tissue types, disease states, and samples from different periods. Recent studies comparing the differences between scRNA-seq and single-nucleus RNA sequencing (snRNA-seq) for various tissues and disease types have shown that snRNA-seq is more advantageous in frozen and difficult-to-dissociate samples [151,152,153]. Moreover, snRNA-seq can reduce the effects of enzymatic digestion and mechanical stress. Thus, we can obviously obtain more molecular numbers through scRNA-seq. Due to their advantages in the study of cell heterogeneity, they may provide new tools and methods for the application of ncRNAs in forensic practice, especially in the identification of monozygotic twins, the identification of tissues and body fluids, and age estimation.
Currently, miRNA has achieved significant results in related research, but some emerging ncRNAs such as circRNA, piRNA, and lncRNA are still in the exploratory stage for forensic applications. The ideal ncRNA markers should be specific, sensitive, stable, non-destructive, universal, and efficient. However, changes in biological processes, age, sex differences, population differences, health status, and other factors will affect the expression levels of ncRNAs. To exploit the application potential of ncRNAs in forensic practice, it is crucial to establish stable identification models. Moreover, the development of methods with short identification time and strong identification ability is still a highly valuable research direction for application.
There are still many studies on non-coding RNAs relying mainly on animal models, and studies with large samples are limited by ethical and moral factors. However, there are differences in the expression level and specificity of ncRNAs between animal models and humans. Thus, the lack of human data is one of the biggest limitations for the application of ncRNAs in the practice of forensic medicine. The development of ncRNA databases such as miRNA and circRNA databases may provide a basis for the discovery of specific markers and the exploration of modeling methods. In addition, with the continuous development of biological technology, the functions of ncRNA in different organs, tissues, and cells will be further revealed, which will provide a richer theoretical basis and a more promising research direction for their application in the practice of forensic medicine.
At present, ncRNAs are closely connected with forensic medicine, which provides powerful tools and methods for the practice of forensic medicine. It has unique advantages and extremely high application value in solving problems such as the identification of tissues and body fluids, cause-of-death analysis, time-related estimation, age estimation, and the identification of monozygotic twins. It is believed that the application potential of ncRNAs in forensic medicine will be explored more in-depth, providing more novel and effective means for the practice of forensic medicine.
Data availability
Not applicable.
References
Gayon J (2016) From Mendel to epigenetics: history of genetics. C R Biol 339:225–230. https://doi.org/10.1016/j.crvi.2016.05.009
de Mendoza A, Nguyen TV, Ford E et al (2022) Large-scale manipulation of promoter DNA methylation reveals context-specific transcriptional responses and stability. Genome Biol 23:163. https://doi.org/10.1186/s13059-022-02728-5
Li J, Xue Y, Amin MT et al (2020) ncRNA-eQTL: a database to systematically evaluate the effects of SNPs on non-coding RNA expression across cancer types. Nucleic Acids Res 48:D956–D963. https://doi.org/10.1093/nar/gkz711
Zhu Z, Han Z, Halabelian L et al (2021) Identification of lysine isobutyrylation as a new histone modification mark. Nucleic Acids Res 49:177–189. https://doi.org/10.1093/nar/gkaa1176
Werner JM, Ballouz S, Hover J, Gillis J (2022) Variability of cross-tissue X-chromosome inactivation characterizes timing of human embryonic lineage specification events. Dev Cell 57:1995-2008.e5. https://doi.org/10.1016/j.devcel.2022.07.007
Belk JA, Yao W, Ly N et al (2022) Genome-wide CRISPR screens of T cell exhaustion identify chromatin remodeling factors that limit T cell persistence. Cancer Cell 40:768–86.e7. https://doi.org/10.1016/j.ccell.2022.06.001
Weinberg-Shukron A, Ben-Yair R, Takahashi N et al (2022) Balanced gene dosage control rather than parental origin underpins genomic imprinting. Nat Commun 13:4391. https://doi.org/10.1038/s41467-022-32144-z
Imani S, Zhang X, Fu S et al (2018) Non-coding RNAs in Cancer. In: Fu J, Imani S (ed) Epigenetics in Cancer, 1st edn. Science Press, Beijing, pp 104–184
Ashrafizadeh M, Zarrabi A, Mostafavi E et al (2022) Non-coding RNA-based regulation of inflammation. Semin Immunol 101606. https://doi.org/10.1016/j.smim.2022.101606
Liu X, Li Y, Jiang X et al (2022) Long non-coding RNA: multiple effects on the differentiation, maturity and cell function of dendritic cells. Clin Immunol 245:109167. https://doi.org/10.1016/j.clim.2022.109167
Shah AM, Giacca M (2022) Small non-coding RNA therapeutics for cardiovascular disease. Eur Heart J 43:4548–4561. https://doi.org/10.1093/eurheartj/ehac463
Beucher A, Miguel-Escalada I, Balboa D et al (2022) The HASTER lncRNA promoter is a cis-acting transcriptional stabilizer of HNF1A. Nat Cell Biol 24:1528–1540. https://doi.org/10.1038/s41556-022-00996-8
Fagan SG, Helm M, Prehn JHM (2021) tRNA-derived fragments: a new class of non-coding RNA with key roles in nervous system function and dysfunction. Prog Neurobiol 205:102118. https://doi.org/10.1016/j.pneurobio.2021.102118
Chen X, Rechavi O (2022) Plant and animal small RNA communications between cells and organisms. Nat Rev Mol Cell Biol 23:185–203. https://doi.org/10.1038/s41580-021-00425-y
Liu N, Xu Y, Li Q et al (2022) A lncRNA fine-tunes salicylic acid biosynthesis to balance plant immunity and growth. Cell Host Microbe 30:1124–38.e8. https://doi.org/10.1016/j.chom.2022.07.001
Bauer M (2007) RNA in forensic science. Forensic Sci Int Genet 1:69–74. https://doi.org/10.1016/j.fsigen.2006.11.002
Haas C, Hanson E, Bar W et al (2011) mRNA profiling for the identification of blood–results of a collaborative EDNAP exercise. Forensic Sci Int Genet 5:21–26. https://doi.org/10.1016/j.fsigen.2010.01.003
Haas C, Hanson E, Anjos MJ et al (2012) RNA/DNA co-analysis from blood stains–results of a second collaborative EDNAP exercise. Forensic Sci Int Genet 6:70–80. https://doi.org/10.1016/j.fsigen.2011.02.004
Haas C, Hanson E, Anjos MJ et al (2013) RNA/DNA co-analysis from human saliva and semen stains–results of a third collaborative EDNAP exercise. Forensic Sci Int Genet 7:230–239. https://doi.org/10.1016/j.fsigen.2012.10.011
Haas C, Hanson E, Anjos MJ et al (2014) RNA/DNA co-analysis from human menstrual blood and vaginal secretion stains: results of a fourth and fifth collaborative EDNAP exercise. Forensic Sci Int Genet 8:203–212. https://doi.org/10.1016/j.fsigen.2013.09.009
Haas C, Hanson E, Banemann R et al (2015) RNA/DNA co-analysis from human skin and contact traces–results of a sixth collaborative EDNAP exercise. Forensic Sci Int Genet 16:139–147. https://doi.org/10.1016/j.fsigen.2015.01.002
Ingold S, Dorum G, Hanson E et al (2018) Body fluid identification using a targeted mRNA massively parallel sequencing approach - results of a EUROFORGEN/EDNAP collaborative exercise. Forensic Sci Int Genet 34:105–115. https://doi.org/10.1016/j.fsigen.2018.01.002
Ingold S, Dorum G, Hanson E et al (2020) Body fluid identification and assignment to donors using a targeted mRNA massively parallel sequencing approach - results of a second EUROFORGEN / EDNAP collaborative exercise. Forensic Sci Int Genet 45:102208. https://doi.org/10.1016/j.fsigen.2019.102208
Courts C, Madea B (2010) Micro-RNA - a potential for forensic science? Forensic Sci Int 203:106–111. https://doi.org/10.1016/j.forsciint.2010.07.002
Rocchi A, Chiti E, Maiese A, Turillazzi E, Spinetti I (2020) MicroRNAs: An Update of Applications in Forensic Science. Diagnostics (Basel) 11. https://doi.org/10.3390/diagnostics11010032
Huttenhofer A, Schattner P, Polacek N (2005) Non-coding RNAs: hope or hype? Trends Genet 21:289–297. https://doi.org/10.1016/j.tig.2005.03.007
Wilusz CJ, Wilusz J (2004) Bringing the role of mRNA decay in the control of gene expression into focus. Trends Genet 20:491–497. https://doi.org/10.1016/j.tig.2004.07.011
Faure G, Ogurtsov AY, Shabalina SA, Koonin EV (2016) Role of mRNA structure in the control of protein folding. Nucleic Acids Res 44:10898–10911. https://doi.org/10.1093/nar/gkw671
Cerezo M, Robert C, Liu L, Shen S (2021) The role of mRNA translational control in tumor immune escape and immunotherapy resistance. Cancer Res 81:5596–5604. https://doi.org/10.1158/0008-5472.CAN-21-1466
Djebali S, Davis CA, Merkel A et al (2012) Landscape of transcription in human cells. Nature 489:101–108. https://doi.org/10.1038/nature11233
Consortium EP (2012) An integrated encyclopedia of DNA elements in the human genome. Nature 489:57–74. https://doi.org/10.1038/nature11247
Harrow J, Frankish A, Gonzalez JM et al (2012) GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res 22:1760–1774. https://doi.org/10.1101/gr.135350.111
Derrien T, Johnson R, Bussotti G et al (2012) The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res 22:1775–1789. https://doi.org/10.1101/gr.132159.111
Dahariya S, Paddibhatla I, Kumar S, Raghuwanshi S, Pallepati A, Gutti RK (2019) Long non-coding RNA: Classification, biogenesis and functions in blood cells. Mol Immunol 112:82–92. https://doi.org/10.1016/j.molimm.2019.04.011
Esteller M (2011) Non-coding RNAs in human disease. Nat Rev Genet 12:861–874. https://doi.org/10.1038/nrg3074
Asim MN, Ibrahim MA, Imran Malik M, Dengel A, Ahmed S (2021) Advances in computational methodologies for classification and sub-cellular locality prediction of non-coding RNAs. Int J Mol Sci 22. https://doi.org/10.3390/ijms22168719
Sun P, Li G (2019) CircCode: a powerful tool for identifying circRNA coding ability. Front Genet 10:981. https://doi.org/10.3389/fgene.2019.00981
Miao Q, Ni B, Tang J (2021) Coding potential of circRNAs: new discoveries and challenges. PeerJ 9:e10718. https://doi.org/10.7717/peerj.10718
Misir S, Wu N, Yang BB (2022) Specific expression and functions of circular RNAs. Cell Death Differ 29:481–491. https://doi.org/10.1038/s41418-022-00948-7
Chen C, Ridzon DA, Broomer AJ et al (2005) Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res 33:e179. https://doi.org/10.1093/nar/gni178
Shi R, Chiang VL (2005) Facile means for quantifying microRNA expression by real-time PCR. Biotechniques 39:519–525. https://doi.org/10.2144/000112010
Raymond CK, Roberts BS, Garrett-Engele P, Lim LP, Johnson JM (2005) Simple, quantitative primer-extension PCR assay for direct monitoring of microRNAs and short-interfering RNAs. RNA 11:1737–1744. https://doi.org/10.1261/rna.2148705
Bentley DR, Balasubramanian S, Swerdlow HP et al (2008) Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456:53–59. https://doi.org/10.1038/nature07517
Rothberg JM, Hinz W, Rearick TM et al (2011) An integrated semiconductor device enabling non-optical genome sequencing. Nature 475:348–352. https://doi.org/10.1038/nature10242
Plomin R, Schalkwyk LC (2007) Microarrays. Dev Sci 10:19–23. https://doi.org/10.1111/j.1467-7687.2007.00558.x
Li W, Ruan K (2009) MicroRNA detection by microarray. Anal Bioanal Chem 394:1117–1124. https://doi.org/10.1007/s00216-008-2570-2
Mathew R, Mattei V, Al Hashmi M, Tomei S (2020) Updates on the Current Technologies for microRNA Profiling. Microrna 9:17–24. https://doi.org/10.2174/2211536608666190628112722
Li S, Teng S, Xu J et al (2019) Microarray is an efficient tool for circRNA profiling. Brief Bioinform 20:1420–1433. https://doi.org/10.1093/bib/bby006
Glynn CL (2020) Potential applications of microRNA profiling to forensic investigations. RNA 26:1–9. https://doi.org/10.1261/rna.072173.119
Hanson EK, Lubenow H, Ballantyne J (2009) Identification of forensically relevant body fluids using a panel of differentially expressed microRNAs. Anal Biochem 387:303–314. https://doi.org/10.1016/j.ab.2009.01.037
Hanson EK, Ballantyne J (2013) Circulating microRNA for the identification of forensically relevant body fluids. Methods Mol Biol 1024:221–234. https://doi.org/10.1007/978-1-62703-453-1_18
Zubakov D, Boersma AW, Choi Y, van Kuijk PF, Wiemer EA, Kayser M (2010) MicroRNA markers for forensic body fluid identification obtained from microarray screening and quantitative RT-PCR confirmation. Int J Legal Med 124:217–226. https://doi.org/10.1007/s00414-009-0402-3
Park JL, Park SM, Kwon OH et al (2014) Microarray screening and qRT-PCR evaluation of microRNA markers for forensic body fluid identification. Electrophoresis 35:3062–3068. https://doi.org/10.1002/elps.201400075
Wang Z, Luo H, Pan X, Liao M, Hou Y (2012) A model for data analysis of microRNA expression in forensic body fluid identification. Forensic Sci Int Genet 6:419–423. https://doi.org/10.1016/j.fsigen.2011.08.008
Wang Z, Zhou D, Cao Y et al (2016) Characterization of microRNA expression profiles in blood and saliva using the Ion Personal Genome Machine((R)) System (Ion PGM System). Forensic Sci Int Genet 20:140–146. https://doi.org/10.1016/j.fsigen.2015.10.008
Seashols-Williams S, Lewis C, Calloway C et al (2016) High-throughput miRNA sequencing and identification of biomarkers for forensically relevant biological fluids. Electrophoresis 37:2780–2788. https://doi.org/10.1002/elps.201600258
Sauer E, Reinke AK, Courts C (2016) Differentiation of five body fluids from forensic samples by expression analysis of four microRNAs using quantitative PCR. Forensic Sci Int Genet 22:89–99. https://doi.org/10.1016/j.fsigen.2016.01.018
Sirker M, Fimmers R, Schneider PM, Gomes I (2017) Evaluating the forensic application of 19 target microRNAs as biomarkers in body fluid and tissue identification. Forensic Sci Int Genet 27:41–49. https://doi.org/10.1016/j.fsigen.2016.11.012
Fujimoto S, Manabe S, Morimoto C et al (2019) Distinct spectrum of microRNA expression in forensically relevant body fluids and probabilistic discriminant approach. Sci Rep 9:14332. https://doi.org/10.1038/s41598-019-50796-8
Dorum G, Ingold S, Hanson E et al (2019) Predicting the origin of stains from whole miRNome massively parallel sequencing data. Forensic Sci Int Genet 40:131–139. https://doi.org/10.1016/j.fsigen.2019.02.015
Mayes C, Houston R, Seashols-Williams S, LaRue B, Hughes-Stamm S (2019) The stability and persistence of blood and semen mRNA and miRNA targets for body fluid identification in environmentally challenged and laundered samples. Leg Med (Tokyo) 38:45–50. https://doi.org/10.1016/j.legalmed.2019.03.007
Li Z, Chen D, Wang Q et al (2021) mRNA and microRNA stability validation of blood samples under different environmental conditions. Forensic Sci Int Genet 55:102567. https://doi.org/10.1016/j.fsigen.2021.102567
Li Z, Lv M, Peng D et al (2021) Feasibility of using probabilistic methods to analyse microRNA quantitative data in forensically relevant body fluids: a proof-of-principle study. Int J Legal Med 135:2247–2261. https://doi.org/10.1007/s00414-021-02678-w
Iroanya OO, Olutunde OT, Egwuatu TF, Igbokwe C (2022) Stability of selected microRNAs in human blood, semen and saliva samples exposed to different environmental conditions. Forensic Sci Int 336:111338. https://doi.org/10.1016/j.forsciint.2022.111338
Sauer E, Extra A, Cachee P, Courts C (2017) Identification of organ tissue types and skin from forensic samples by microRNA expression analysis. Forensic Sci Int Genet 28:99–110. https://doi.org/10.1016/j.fsigen.2017.02.002
He H, Ji A, Zhao Y et al (2020) A stepwise strategy to distinguish menstrual blood from peripheral blood by Fisher’s discriminant function. Int J Legal Med 134:845–851. https://doi.org/10.1007/s00414-019-02196-w
He H, Han N, Ji C et al (2020) Identification of five types of forensic body fluids based on stepwise discriminant analysis. Forensic Sci Int Genet 48:102337. https://doi.org/10.1016/j.fsigen.2020.102337
Liu Y, He H, Xiao ZX et al (2021) A systematic analysis of miRNA markers and classification algorithms for forensic body fluid identification. Brief Bioinform 22. https://doi.org/10.1093/bib/bbaa324
Wang G, Wang Z, Wei S et al (2022) A new strategy for distinguishing menstrual blood from peripheral blood by the miR-451a/miR-21-5p ratio. Forensic Sci Int Genet 57:102654. https://doi.org/10.1016/j.fsigen.2021.102654
Bamberg M, Bruder M, Dierig L, Kunz SN, Schwender M, Wiegand P (2022) Best of both: a simultaneous analysis of mRNA and miRNA markers for body fluid identification. Forensic Sci Int Genet 59:102707. https://doi.org/10.1016/j.fsigen.2022.102707
Rhodes C, Lewis C, Szekely J et al (2022) Developmental validation of a microRNA panel using quadratic discriminant analysis for the classification of seven forensically relevant body fluids. Forensic Sci Int Genet 59:102692. https://doi.org/10.1016/j.fsigen.2022.102692
Wei S, Hu S, Han N et al (2023) Screening and evaluation of endogenous reference genes for miRNA expression analysis in forensic body fluid samples. Forensic Sci Int Genet 63:102827. https://doi.org/10.1016/j.fsigen.2023.102827
Memczak S, Jens M, Elefsinioti A et al (2013) Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 495:333–338. https://doi.org/10.1038/nature11928
Jeck WR, Sorrentino JA, Wang K et al (2013) Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 19:141–157. https://doi.org/10.1261/rna.035667.112
Song F, Luo H, Xie M, Zhu H, Hou Y (2017) Microarray expression profile of circular RNAs in human body fluids. Forensic Sci Int: Genet Suppl Ser 6:e55–e56. https://doi.org/10.1016/j.fsigss.2017.09.005
Zhang Y, Liu B, Shao C et al (2018) Evaluation of the inclusion of circular RNAs in mRNA profiling in forensic body fluid identification. Int J Legal Med 132:43–52. https://doi.org/10.1007/s00414-017-1690-7
Liu B, Song F, Yang Q et al (2019) Characterization of tissue-specific biomarkers with the expression of circRNAs in forensically relevant body fluids. Int J Legal Med 133:1321–1331. https://doi.org/10.1007/s00414-019-02027-y
Liu B, Yang Q, Meng H et al (2020) Development of a multiplex system for the identification of forensically relevant body fluids. Forensic Sci Int Genet 47:102312. https://doi.org/10.1016/j.fsigen.2020.102312
Yang Q, Liu B, Zhou Y et al (2021) Evaluation of one-step RT-PCR multiplex assay for body fluid identification. Int J Legal Med 135:1727–1735. https://doi.org/10.1007/s00414-021-02535-w
Ponnusamy M, Yan KW, Liu CY, Li PF, Wang K (2017) PIWI family emerging as a decisive factor of cell fate: an overview. Eur J Cell Biol 96:746–757. https://doi.org/10.1016/j.ejcb.2017.09.004
Ross RJ, Weiner MM, Lin H (2014) PIWI proteins and PIWI-interacting RNAs in the soma. Nature 505:353–359. https://doi.org/10.1038/nature12987
Fu A, Jacobs DI, Zhu Y (2014) Epigenome-wide analysis of piRNAs in gene-specific DNA methylation. RNA Biol 11:1301–1312. https://doi.org/10.1080/15476286.2014.996091
Simon B, Kirkpatrick JP, Eckhardt S et al (2011) Recognition of 2’-O-methylated 3’-end of piRNA by the PAZ domain of a Piwi protein. Structure 19:172–180. https://doi.org/10.1016/j.str.2010.11.015
Wang S, Wang Z, Tao R et al (2019) The potential use of Piwi-interacting RNA biomarkers in forensic body fluid identification: a proof-of-principle study. Forensic Sci Int Genet 39:129–135. https://doi.org/10.1016/j.fsigen.2019.01.002
Nallamshetty S, Chan SY, Loscalzo J (2013) Hypoxia: a master regulator of microRNA biogenesis and activity. Free Radic Biol Med 64:20–30. https://doi.org/10.1016/j.freeradbiomed.2013.05.022
Schober K, Ondruschka B, Dressler J, Abend M (2015) Detection of hypoxia markers in the cerebellum after a traumatic frontal cortex injury: a human postmortem gene expression analysis. Int J Legal Med 129:701–707. https://doi.org/10.1007/s00414-014-1129-3
Zeng Y, Lv Y, Tao L et al (2016) G6PC3, ALDOA and CS induction accompanies mir-122 down-regulation in the mechanical asphyxia and can serve as hypoxia biomarkers. Oncotarget 7:74526–36. https://doi.org/10.18632/oncotarget.12931
Han L, Zhang H, Zeng Y et al (2020) Identification of the miRNA-3185/CYP4A11 axis in cardiac tissue as a biomarker for mechanical asphyxia. Forensic Sci Int 311:110293. https://doi.org/10.1016/j.forsciint.2020.110293
Han L, Li W, Hu Y et al (2021) Model for the prediction of mechanical asphyxia as the cause of death based on four biological indexes in human cardiac tissue. Sci Justice 61:221–226. https://doi.org/10.1016/j.scijus.2021.02.003
Liu CX, Chen LL (2022) Circular RNAs: Characterization, cellular roles, and applications. Cell 185:2016–2034. https://doi.org/10.1016/j.cell.2022.04.021
Huang Q, Yang J, Goh RMW, You M, Wang L, Ma Z (2022) Hypoxia-induced circRNAs in human diseases: from mechanisms to potential applications. Cells 11. https://doi.org/10.3390/cells11091381
Barwari T, Joshi A, Mayr M (2016) MicroRNAs in cardiovascular disease. J Am Coll Cardiol 68:2577–2584. https://doi.org/10.1016/j.jacc.2016.09.945
Kakimoto Y, Tanaka M, Hayashi H, Yokoyama K, Osawa M (2018) Overexpression of miR-221 in sudden death with cardiac hypertrophy patients. Heliyon 4:e00639. https://doi.org/10.1016/j.heliyon.2018.e00639
Pinchi E, Frati P, Aromatario M et al (2019) miR-1, miR-499 and miR-208 are sensitive markers to diagnose sudden death due to early acute myocardial infarction. J Cell Mol Med 23:6005–6016. https://doi.org/10.1111/jcmm.14463
Yan F, Chen Y, Ye X et al (2021) miR-3113-5p, miR-223-3p, miR-133a-3p, and miR-499a-5p are sensitive biomarkers to diagnose sudden cardiac death. Diagn Pathol 16:67. https://doi.org/10.1186/s13000-021-01127-x
Li L, He X, Liu M, Yun L, Cong B (2022) Diagnostic value of cardiac miR-126-5p, miR-134-5p, and miR-499a-5p in coronary artery disease-induced sudden cardiac death. Front Cardiovasc Med 9:944317. https://doi.org/10.3389/fcvm.2022.944317
Wang W, Wang Y, Piao H et al (2019) Circular RNAs as potential biomarkers and therapeutics for cardiovascular disease. PeerJ 7:e6831. https://doi.org/10.7717/peerj.6831
Tian M, Xue J, Dai C, Jiang E, Zhu B, Pang H (2021) CircSLC8A1 and circNFIX can be used as auxiliary diagnostic markers for sudden cardiac death caused by acute ischemic heart disease. Sci Rep 11:4695. https://doi.org/10.1038/s41598-021-84056-5
Cui X, Niu W, Kong L et al (2017) Long noncoding RNA expression in peripheral blood mononuclear cells and suicide risk in Chinese patients with major depressive disorder. Brain Behav 7:e00711. https://doi.org/10.1002/brb3.711
Wang Q, Roy B, Turecki G, Shelton RC, Dwivedi Y (2018) Role of Complex epigenetic switching in tumor necrosis factor-alpha upregulation in the prefrontal cortex of suicide subjects. Am J Psychiatry 175:262–274. https://doi.org/10.1176/appi.ajp.2017.16070759
Yoshino Y, Dwivedi Y (2020) Non-coding RNAs in psychiatric disorders and suicidal behavior. Front Psychiatry 11:543893. https://doi.org/10.3389/fpsyt.2020.543893
Punzi G, Ursini G, Shin JH, Kleinman JE, Hyde TM, Weinberger DR (2014) Increased expression of MARCKS in post-mortem brain of violent suicide completers is related to transcription of a long, noncoding, antisense RNA. Mol Psychiatry 19:1057–1059. https://doi.org/10.1038/mp.2014.41
Punzi G, Ursini G, Viscanti G et al (2019) Association of a noncoding RNA postmortem with suicide by violent means and in vivo with aggressive phenotypes. Biol Psychiatry 85:417–424. https://doi.org/10.1016/j.biopsych.2018.11.002
Zhou Y, Lutz PE, Wang YC, Ragoussis J, Turecki G (2018) Global long non-coding RNA expression in the rostral anterior cingulate cortex of depressed suicides. Transl Psychiatry 8:224. https://doi.org/10.1038/s41398-018-0267-7
Smalheiser NR, Lugli G, Rizavi HS, Torvik VI, Turecki G, Dwivedi Y (2012) MicroRNA expression is down-regulated and reorganized in prefrontal cortex of depressed suicide subjects. PLoS One 7:e33201. https://doi.org/10.1371/journal.pone.0033201
Smalheiser NR, Lugli G, Zhang H, Rizavi H, Cook EH, Dwivedi Y (2014) Expression of microRNAs and other small RNAs in prefrontal cortex in schizophrenia, bipolar disorder and depressed subjects. PLoS One 9:e86469. https://doi.org/10.1371/journal.pone.0086469
Lopez JP, Fiori LM, Gross JA et al (2014) Regulatory role of miRNAs in polyamine gene expression in the prefrontal cortex of depressed suicide completers. Int J Neuropsychopharmacol 17:23–32. https://doi.org/10.1017/S1461145713000941
Lopez JP, Fiori LM, Cruceanu C et al (2017) MicroRNAs 146a/b-5 and 425–3p and 24–3p are markers of antidepressant response and regulate MAPK/Wnt-system genes. Nat Commun 8:15497. https://doi.org/10.1038/ncomms15497
Courts C, Grabmuller M, Madea B (2013) Dysregulation of heart and brain specific micro-RNA in sudden infant death syndrome. Forensic Sci Int 228:70–74. https://doi.org/10.1016/j.forsciint.2013.02.032
Yu S, Na JY, Lee YJ, Kim KT, Park JT, Kim HS (2015) Forensic application of microRNA-706 as a biomarker for drowning pattern identification. Forensic Sci Int 255:96–101. https://doi.org/10.1016/j.forsciint.2015.06.011
Pinchi E, Frati A, Cantatore S et al (2019) Acute spinal cord injury: a systematic review investigating miRNA families involved. Int J Mol Sci 20. https://doi.org/10.3390/ijms20081841
Wang H, Mao J, Li Y et al (2013) 5 miRNA expression analyze in post-mortem interval (PMI) within 48h. Forensic Sci Int: Genet Suppl Ser 4:e190–e191. https://doi.org/10.1016/j.fsigss.2013.10.098
Lv YH, Ma KJ, Zhang H et al (2014) A time course study demonstrating mRNA, microRNA, 18S rRNA, and U6 snRNA changes to estimate PMI in deceased rat’s spleen. J Forensic Sci 59:1286–1294. https://doi.org/10.1111/1556-4029.12447
Lv YH, Ma JL, Pan H et al (2016) RNA degradation as described by a mathematical model for postmortem interval determination. J Forensic Leg Med 44:43–52. https://doi.org/10.1016/j.jflm.2016.08.015
Lv YH, Ma JL, Pan H et al (2017) Estimation of the human postmortem interval using an established rat mathematical model and multi-RNA markers. Forensic Sci Med Pathol 13:20–27. https://doi.org/10.1007/s12024-016-9827-4
Pasaribu RS, Auerkari EI, Suhartono AW, Auerkari P (2023) A small RNA, microRNA as a potential biomolecular marker to estimate post mortem interval in forensic science: a systematic review. Int J Legal Med. https://doi.org/10.1007/s00414-023-03015-z
Tu C, Du T, Shao C, Liu Z, Li L, Shen Y (2018) Evaluating the potential of housekeeping genes, rRNAs, snRNAs, microRNAs and circRNAs as reference genes for the estimation of PMI. Forensic Sci Med Pathol 14:194–201. https://doi.org/10.1007/s12024-018-9973-y
Tu C, Du T, Ye X, Shao C, Xie J, Shen Y (2019) Using miRNAs and circRNAs to estimate PMI in advanced stage. Leg Med (Tokyo) 38:51–57. https://doi.org/10.1016/j.legalmed.2019.04.002
Na JY (2020) Estimation of the post-mortem interval using microRNA in the bones. J Forensic Leg Med 75:102049. https://doi.org/10.1016/j.jflm.2020.102049
Kim SY, Jang SJ, Jung YH, Na JY (2021) Difference in microRNA levels in the post-mortem blood from different sampling sites: a proof of concept. J Forensic Leg Med 78:102124. https://doi.org/10.1016/j.jflm.2021.102124
Lang H, Zhao F, Zhang T et al (2017) MicroRNA-149 contributes to scarless wound healing by attenuating inflammatory response. Mol Med Rep 16:2156–2162. https://doi.org/10.3892/mmr.2017.6796
De Simone S, Giacani E, Bosco MA et al (2021) The role of miRNAs as new molecular biomarkers for dating the age of wound production: a systematic review. Front Med (Lausanne) 8:803067. https://doi.org/10.3389/fmed.2021.803067
Neri M, Fabbri M, D’Errico S et al (2019) Regulation of miRNAs as new tool for cutaneous vitality lesions demonstration in ligature marks in deaths by hanging. Sci Rep 9:20011. https://doi.org/10.1038/s41598-019-56682-7
Maiese A, Manetti AC, Iacoponi N et al (2022) State-of-the-art on wound vitality evaluation: a systematic review. Int J Mol Sci 23. https://doi.org/10.3390/ijms23136881
Bertero T, Gastaldi C, Bourget-Ponzio I et al (2011) miR-483-3p controls proliferation in wounded epithelial cells. FASEB J 25:3092–3105. https://doi.org/10.1096/fj.10-168401
Wang T, Feng Y, Sun H et al (2012) miR-21 regulates skin wound healing by targeting multiple aspects of the healing process. Am J Pathol 181:1911–1920. https://doi.org/10.1016/j.ajpath.2012.08.022
Etich J, Bergmeier V, Pitzler L, Brachvogel B (2017) Identification of a reference gene for the quantification of mRNA and miRNA expression during skin wound healing. Connect Tissue Res 58:196–207. https://doi.org/10.1080/03008207.2016.1210606
Chang L, Liang J, Xia X, Chen X (2019) miRNA-126 enhances viability, colony formation, and migration of keratinocytes HaCaT cells by regulating PI3 K/AKT signaling pathway. Cell Biol Int 43:182–191. https://doi.org/10.1002/cbin.11088
Lyu HP, Cheng M, Liu JC et al (2018) Differentially expressed microRNAs as potential markers for vital reaction of burned skin. J Forensic Sci Med 4:15
Zhang K, Cheng M, Xu J et al (2022) MiR-711 and miR-183-3p as potential markers for vital reaction of burned skin. Forensic Sci Res 7:503–509. https://doi.org/10.1080/20961790.2020.1719454
Liu W, Li L, Rong Y et al (2020) Hypoxic mesenchymal stem cell-derived exosomes promote bone fracture healing by the transfer of miR-126. Acta Biomater 103:196–212. https://doi.org/10.1016/j.actbio.2019.12.020
Li X, Zhong Z, Ma E, Wu X (2021) Identification of miRNA regulatory networks and candidate markers for fracture healing in mice. Comput Math Methods Med 2021:2866475. https://doi.org/10.1155/2021/2866475
Manetti AC, Maiese A, Baronti A et al (2021) MiRNAs as new tools in lesion vitality evaluation: a systematic review and their forensic applications. Biomedicines 9. https://doi.org/10.3390/biomedicines9111731
Anderson S, Howard B, Hobbs GR, Bishop CP (2005) A method for determining the age of a bloodstain. Forensic Sci Int 148:37–45. https://doi.org/10.1016/j.forsciint.2004.04.071
Lech K, Ackermann K, Wollstein A, Revell VL, Skene DJ, Kayser M (2014) Assessing the suitability of miRNA-142-5p and miRNA-541 for bloodstain deposition timing. Forensic Sci Int Genet 12:181–184. https://doi.org/10.1016/j.fsigen.2014.06.008
Alshehhi S, Haddrill PR (2019) Estimating time since deposition using quantification of RNA degradation in body fluid-specific markers. Forensic Sci Int 298:58–63. https://doi.org/10.1016/j.forsciint.2019.02.046
Wei Y, Wang J, Wang Q, Cong B, Li S (2022) The estimation of bloodstain age utilizing circRNAs and mRNAs biomarkers. Forensic Sci Int 338:111408. https://doi.org/10.1016/j.forsciint.2022.111408
Freire-Aradas A, Phillips C, Lareu MV (2017) Forensic individual age estimation with DNA: from initial approaches to methylation tests. Forensic Sci Rev 29:121–144
Vidaki A, Kayser M (2018) Recent progress, methods and perspectives in forensic epigenetics. Forensic Sci Int Genet 37:180–195. https://doi.org/10.1016/j.fsigen.2018.08.008
Noren Hooten N, Abdelmohsen K, Gorospe M, Ejiogu N, Zonderman AB, Evans MK (2010) microRNA expression patterns reveal differential expression of target genes with age. PLoS One 5:e10724. https://doi.org/10.1371/journal.pone.0010724
Rubie C, Kolsch K, Halajda B et al (2016) microRNA-496 - a new, potentially aging-relevant regulator of mTOR. Cell Cycle 15:1108–1116. https://doi.org/10.1080/15384101.2016.1158360
Huan T, Chen G, Liu C et al (2018) Age-associated microRNA expression in human peripheral blood is associated with all-cause mortality and age-related traits. Aging Cell 17. https://doi.org/10.1111/acel.12687
Wang J, Wang C, Wei Y et al (2022) Circular RNA as a potential biomarker for forensic age prediction. Front Genet 13:825443. https://doi.org/10.3389/fgene.2022.825443
Nikolajevic J, Ariaee N, Liew A, Abbasnia S, Fazeli B, Sabovic M (2022) The role of microRNAs in endothelial cell senescence. Cells 11. https://doi.org/10.3390/cells11071185
Lettieri-Barbato D, Aquilano K, Punziano C, Minopoli G, Faraonio R (2022) MicroRNAs, long non-coding RNAs, and circular RNAs in the redox control of cell senescence. Antioxidants (Basel) 11. https://doi.org/10.3390/antiox11030480
Abu-Halima M, Weidinger J, Poryo M et al (2019) Micro-RNA signatures in monozygotic twins discordant for congenital heart defects. PLoS One 14:e0226164. https://doi.org/10.1371/journal.pone.0226164
Tuncer SB, Erdogan OS, Erciyas SK et al (2020) miRNA expression profile changes in the peripheral blood of monozygotic discordant twins for epithelial ovarian carcinoma: potential new biomarkers for early diagnosis and prognosis of ovarian carcinoma. J Ovarian Res 13:99. https://doi.org/10.1186/s13048-020-00706-8
Bresciani E, Squillace N, Orsini V et al (2022) miRNA expression profiling in subcutaneous adipose tissue of monozygotic twins discordant for HIV Infection: validation of differentially expressed miRNA and bioinformatic analysis. Int J Mol Sci 23. https://doi.org/10.3390/ijms23073486
Fang C, Zhao J, Liu X et al (2019) MicroRNA profile analysis for discrimination of monozygotic twins using massively parallel sequencing and real-time PCR. Forensic Sci Int Genet 38:23–31. https://doi.org/10.1016/j.fsigen.2018.09.011
Xiao C, Pan C, Liu E et al (2019) Differences of microRNA expression profiles between monozygotic twins’ blood samples. Forensic Sci Int Genet 41:152–158. https://doi.org/10.1016/j.fsigen.2019.05.003
Wu H, Kirita Y, Donnelly EL, Humphreys BD (2019) Advantages of single-nucleus over single-cell RNA sequencing of adult kidney: rare cell types and novel cell states revealed in fibrosis. J Am Soc Nephrol 30:23–32. https://doi.org/10.1681/ASN.2018090912
Slyper M, Porter CBM, Ashenberg O et al (2020) A single-cell and single-nucleus RNA-Seq toolbox for fresh and frozen human tumors. Nat Med 26:792–802. https://doi.org/10.1038/s41591-020-0844-1
Ding J, Adiconis X, Simmons SK et al (2020) Systematic comparison of single-cell and single-nucleus RNA-sequencing methods. Nat Biotechnol 38:737–746. https://doi.org/10.1038/s41587-020-0465-8
Acknowledgements
The authors thank people in the Research Center for Preclinical Medicine, Southwest Medical University.
Funding
This work was funded by the Technology Project Foundation of Luzhou City (No. 2021-SYF-37), the Teaching Reform Project of Postgraduate Education in Southwest Medical University (No. YJG202222), and partially funded by the National Natural Science Foundation of China (Nos. 81672887).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Research involving human participants and/or animals
Not applicable.
Informed consent
Not applicable.
Ethical approval
Not applicable.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Song, B., Qian, J. & Fu, J. Research progress and potential application of microRNA and other non-coding RNAs in forensic medicine. Int J Legal Med 138, 329–350 (2024). https://doi.org/10.1007/s00414-023-03091-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00414-023-03091-1