
Abstract
Glioblastoma is the most common primary brain tumor and has a dismal prognosis. The development of central necrosis represents a tipping point in the evolution of these tumors that foreshadows aggressive expansion, swiftly leading to mortality. The onset of necrosis, severe hypoxia and associated radial glioma expansion correlates with dramatic tumor microenvironment (TME) alterations that accelerate tumor growth. In the past, most have concluded that hypoxia and necrosis must arise due to “cancer outgrowing its blood supply” when rapid tumor growth outpaces metabolic supply, leading to diffusion-limited hypoxia. However, growing evidence suggests that microscopic intravascular thrombosis driven by the neoplastic overexpression of pro-coagulants attenuates glioma blood supply (perfusion-limited hypoxia), leading to TME restructuring that includes breakdown of the blood–brain barrier, immunosuppressive immune cell accumulation, microvascular hyperproliferation, glioma stem cell enrichment and tumor cell migration outward. Cumulatively, these adaptations result in rapid tumor expansion, resistance to therapeutic interventions and clinical progression. To inform future translational investigations, the complex interplay among environmental cues and myriad cell types that contribute to this aggressive phenotype requires better understanding. This review focuses on contributions from intratumoral thrombosis, the effects of hypoxia and necrosis, the adaptive and innate immune responses, and the current state of targeted therapeutic interventions.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glioblastoma (IDH-wild type, WHO grade 4) is the most frequent malignant brain tumor and has a dismal prognosis. The 5-year survival rate is only 5.6% and the median survival interval is 15 months from initial diagnosis [173]. By definition, glioblastoma is a high grade, infiltrating astrocytic glioma with one or more of the following features: (1) necrosis, (2) microvascular proliferation, or (3) the presence of specific genetic alterations (EGFR amplification, TERT promoter mutation, or the +7/−10 cytogenetic signature) [230]. Historically, the histologic presence of necrosis was the first recognized feature linked to poor prognosis among diffuse gliomas and it remained the sole criterion for establishing the diagnosis of glioblastoma as grade 4 for decades. Even today, it is recognized that nearly all patients with glioblastoma die after a brief period of accelerated tumor expansion following the onset of necrosis.
In fact, necrosis is a criterion of malignancy in many tumor types, highlighting its fundamental association with rapid growth and poor patient prognosis [28, 197]. The prevailing dogma passed along to explain the relationship of malignancy and necrosis has been that “cancer outgrows its blood supply,” as metabolic demands exceed supply during the rapid and uncontrolled cell division and tumor expansion (diffusion-limited hypoxia). While this explanation has been superficially satisfying, it has never been supported by evidence, it is counterintuitive on deeper inspection, and its perpetuation has precluded serious investigations into more plausible mechanisms that link malignant behavior to necrosis in a manner that might shed light towards potential therapies.
Several studies seem to contradict the malignant necrosis dogma and suggest that cancers grow in a manner that actively compromises their blood supply, leading to necrosis and augmented growth due to hypoxia- and tumor microenvironment (TME)-mediated mechanisms. This theory holds that tumors grow in a manner that attenuates local blood flow, leading to perfusion-limited hypoxia and necrosis. There is strong evidence to suggest that microscopic intravascular thrombosis within a tumor, most likely driven by the neoplastic overexpression of pro-coagulants, initiates or propagates hypoxia and necrosis that in turn causes TME restructuring in a manner that favors accelerated growth [71, 150, 199, 234, 241]. The spatial distribution of thrombosis in and around foci of necrosis is highly suggestive of an intimate relationship between the two, with thrombosis potentially causing necrotic development. Microscopic thrombosis can be identified in nearly all glioblastomas but is rarely found in lower grade gliomas without necrosis, which are characterized by sheet-like diffuse infiltration and grow more slowly (Fig. 1). The small number of diffusely infiltrative astrocytic gliomas that have thrombosis, but not necrosis, are also associated with poor prognosis, suggesting that it is a precursor to the development of necrosis and higher grade behavior [241].

Histopathology of glioma progression (H&E staining). Diffusely infiltrating astrocytic tumor without necrosis (histologic grade 3) shows a pattern of sheet-like infiltration by individual tumor cells within the brain parenchyma (a). The presence of intravascular thrombosis (arrow) within a diffuse glioma signals transition to the development of hypoxia, necrosis, and rapid progression. Note the perivascular clearing of neuropil, representing the initial stages of BBB breakdown, activation of perivascular TAMs, parenchymal disruption, and outward migration of glioma cells (b; see also Fig. 3 right panel). At later stages, the presence of intravascular thrombosis (arrow) is spatially associated with adjacent tumor necrosis (c; asterisk). As the necrotic focus enlarges (asterisk), it becomes surrounded by palisading cells that migrate radially outward in three dimensions (d; arrow indicates direction of movement; see also Fig. 2d, boxed region for corresponding MRI region, and Fig. 5)
Molecular genetic alterations driving progression among the diffuse gliomas are well characterized and have elucidated several molecular subtypes based on genomic alterations, epigenetic signatures or transcriptional class [1, 25, 27, 32, 153, 172, 181, 254]. Transcriptional classification has identified three robust subtypes among the IDH-wild-type GBMs (proneural, classical and mesenchymal) that appear to have distinct TME properties. Proneural (PN) tumors are enriched for PDGFRA, CDK4 and SOX2 amplification and display increased PI3K/AKT signaling [22, 181]. Despite PDGF signaling correlating with immune modulation in other solid tumors, glioblastoma displays a strictly proliferative association with PDGF expression [9]. Mesenchymal (MES) tumors contain inactivating mutations in NF1, increased MAPK signaling, and are enriched for endothelial markers and inflammatory infiltrates, especially the macrophage component [22, 170, 259]. Regarding pro-angiogenic signals, MES upregulate ADAM9 that enhances chemotactic factor shedding from tumor cells, cleaves the extracellular matrix (ECM) promoting invasion, and releases angiogenic factors from endothelial cells promoting microvascular hyperproliferation [178]. MES tumor cells also display increased MMP14 membrane localization that promotes ECM cleavage, endothelial and tumor cell invasion and contributes to the vascular abnormalities commonly seen in solid tumors [51]. Classical (CL) tumors are characterized by EGFR mutation and amplification, NOTCH pathway activation, and downregulation of both apoptotic and MAPK signaling pathways [22]. EGFR activation in combination with PTEN loss enhances VEGF expression to support angiogenesis [186], as well as CCL2 secretion that enriches TAM infiltration [4].
Despite transcriptional class differences, all glioblastoma subsets display accelerated progression following the onset of necrosis, indicating that it may be a shared final common pathway that represents an abrupt turning point towards rapid expansion [93, 202]. Of interest, cellular proliferation rates are not a prognostic factor once necrosis develops, indicating that other factors influence survival to a greater extent [19]. Most likely, accelerated growth of glioblastoma is due at least to some extent to hypoxia-induced expansion encouraged through TME dynamics [79]. There is no doubt that glioblastomas are highly heterogeneous, as recognized by the now outdated term “multiforme”. In addition to glioma cells of variable morphologies, differentiation states and stem-like features, glioblastomas also contain tumor-associated macrophages (TAMs), a variety of other immune cells, florid angiogenesis, entrapped native neural elements and reactive glia [15, 54, 107, 153, 191, 245]. TAMs consist of activated resident brain microglia and bone marrow derived monocytes (BMDMs), which differentiate into macrophages upon extravasation into the brain parenchyma. While TME restructuring following necrosis in glioblastoma appears to be an initiator of rapid tumor growth, appropriate animal models to establish the causal relationship between necrosis, TME alterations, and radial expansion are lacking. Indeed, many orthotopic patient-derived xenograft (PDX) mouse models do not develop necrosis [103, 132]. A recent study postulates that this arises in part from defective cross species chemokine signaling [40]. This review assesses the stages of TME-related changes that occur during disease progression in glioblastoma, highlighting the role of hypoxia and necrosis in modulating the immune response.
Thrombosis
The blood–brain barrier (BBB), comprised of brain microvascular endothelial cells, astrocytes, pericytes, oligodendrocytes and unique basement membrane, represents one of the most controlled vascular networks of any organ and its deterioration marks a dramatic change in disease progression among patients with diffuse gliomas. The BBB is largely intact in non-necrotic, lower grade diffuse gliomas and corresponds to the absence of contrast enhancement on MR imaging [255]. The enhancement pattern that becomes apparent in high-grade gliomas represents contrast agent seeping through the BBB and being retained in the brain tumor parenchyma (Fig. 2) [96]. Initial stages of contrast enhancement are often subtle and patchy and can be noted before the onset of necrosis. This likely represents the first stages of vascular pathology and barrier compromise (corresponding with endothelial hypertrophy) yet precedes the onset of severe hypoxia and necrosis that is associated with more extensive vascular proliferation (Fig. 3). Prior work has suggested that microscopic intravascular thrombosis arises at this early stage of glioblastoma progression and is responsible for initiating or propagating hypoxia. The classic MRI features of glioblastoma, with central necrosis surrounded by a rim of intense contrast enhancement and enveloped by T2 signal intensity, are noted later, once there is substantial loss of BBB integrity and extensive microvascular proliferation in and around the contrast-enhancing component (Fig. 2).

MRI images typical of glioma progression. Fluid-attenuated inversion recovery (FLAIR) (a, c) and T1 post-contrast (b, d) MRI. The outline of this histologic grade 3 diffusely infiltrative glioma is noted on FLAIR images (a, arrow). The tumor does not demonstrate central necrosis or contrast enhancement on post-contrast images (b, arrow, corresponding to histology in Fig. 1a). After the onset of hypoxia and necrosis, MRI demonstrates the tumor outline on FLAIR images (c, arrow), while T1 post-contrast images show the classic features glioblastoma, with a prominent region of central necrosis surrounded by a rim of intense contrast enhancement (d, arrow). The region of the box (d) corresponds to the histology in Fig. 1d, with the border region moving radially outward away from the tumor center

Vascular pathology disrupts the blood–brain barrier, limits perfusion, and reshapes the tumor microenvironment. Astrocyte end feet, pericytes, and endothelial cells in pre-thrombotic vessels form an intact blood–brain barrier (left) and provide a relatively immune privileged environment within the central nervous system. When intact, this barrier largely excludes circulating immune cells from the early tumor microenvironment (TME). Following thrombosis (right), astrocytes and pericytes detach from vascular endothelial cells, which also develop leaky junctions, permitting circulating BMDM to traverse the vascular wall and enter the TME. Vascular disruption reduces perfusion and leads to focal hypoxia, driving cells radially out from the area of nutrient deprivation
The link between cancer and thrombotic events is not new. Trousseau recognized well over a century ago that cancer patients exhibit significant systemic dysregulation of coagulation, resulting in frequent peripheral deep venous thromboses and embolic events [58, 150, 246]. This same tendency towards thrombosis is present within the neoplasm, where the causative pro-coagulants are highly expressed [210, 239]. Many investigations have focused on tissue factor (TF), the body’s most potent pro-coagulant, as the primary mediator of systemic coagulopathy [208, 209]. Notably, TF is significantly upregulated in gliomas and its levels correlate with tumor grade [203, 204]. Factors associated with malignant behavior in gliomas, such as EGFR overexpression, PTEN loss and hypoxia-induced early growth response gene (Egr)-1, have all been shown to upregulate TF expression by gliomas [203, 204, 241, 248]. The vascular leakiness that is noted by neuroimaging at early stages of malignant progression would allow circulating coagulation factors, including TF’s primary downstream effector, factor VIIa, to encounter TF. Interestingly, IDH mutant gliomas show significantly reduced TF levels as compared to IDH-wild-type gliomas, potentially related to their slower rate of malignant progression [249, 250]. Conversely, higher grade gliomas display dysfunctional coagulation/fibrinolysis regulatory pathways supporting local coagulation events within the tumor [266]. Increased levels of coagulation are also likely due to thrombin-protease-activated receptor 1 (PAR1) signaling, which is similarly upregulated in glioblastoma [60, 71, 126]. PAR1 localizes to astrocyte end feet where its binding to thrombin leads to a wide variety of downstream effects, including neuroinflammation and vascular pathology [71]. Thrombin-mediated PAR1 cleavage actives the G-protein-coupled receptor leading to Rho and phospholipase C activation and adenylyl cyclase inactivation [20, 30] and promotes VEGF secretion [99] while eliciting an immunosuppressive response [215]. Local VEGF accumulation around the BBB induces pericyte detachment, basement membrane degradation, vessel enlargement and leakiness, perpetuating the cycle of vascular pathology [237, 238]. During this process, glioblastomas also upregulate podoplanin, which enhances local platelet aggregation and has been implicated in systemic thrombosis through its cell surface expression by circulating glioblastoma cells [180, 199, 234]. Podoplanin binds C-type lectin-like receptor (CLEC)-2 on circulating platelets and induces clotting [180]. Concurrently, the emerging hypoxia upregulates plasminogen activator inhibitor 1 (PAI-1) and fibronectin in the perinecrotic niche and surrounding occluded vasculature, generating local pro-coagulant environments [207].
Upregulation of coagulation factors influences the TME in manners that may be unexpected, distinct from their role in thrombosis. For example, TF, factor VIIa (FVIIa), and factor XIIa (FXIIa), are capable of recruiting TAMs to the TME and inducing an immunosuppressive phenotype [69, 149]. The TF-FVIIa complex can trigger mitogenic, angiogenic, and cell survival signaling, as well as enhance a glioma stem cell (GSC) phenotype in certain contexts [248]. Podoplanin may have effects on dendritic cell mediated immunosuppression by binding to and activating CLEC-2, leading to platelet aggregation, enhancing local and distal thrombotic events and monocyte/macrophage recruitment to the area of vascular pathology, reflecting another mechanistic link between coagulation and tumor progression [146, 195, 199]. The prevention or suppression of early thrombotic events in glial neoplasms represents a strategy to slow down disease progression that results from hypoxia- and necrosis-driven TME changes.
Hypoxia
Hypoxia, a state of low oxygen availability, is a critical mediator of pathologic events, yet remains challenging to model and study within physiologically relevant experimental systems. Establishing hypoxic conditions, maintaining physiological gradients and monitoring of oxygen levels in vivo remain daunting prospects, yet recent advances in positron emission tomography (PET) and two-photon phosphorescence microscopy are encouraging [119, 228, 274]. Nevertheless, fluctuations in oxygen availability have profound effects on homeostasis, as well as disease onset and progression, and therefore must be a central consideration of any serious scientific pursuit of mechanisms relevant to glioma progression [224, 242]. We have suggested that vaso-occlusive mechanisms initiate and propagate the severe hypoxia that is present in nearly all high-grade diffuse gliomas and trigger the adaptive responses that lead to TME restructuring and tumor expansion. Hypoxia-inducible factors (HIFs) 1 and 2 are the predominant cellular oxygen sensors, and are upregulated under hypoxic conditions to activate a transcriptional program conducive to an adaptive response that allows cell survival under these conditions [164]. In the case of malignant gliomas, the response to hypoxia also results in events that favor disease progression (Fig. 4). Nuclear HIFs enhance glycolytic metabolism, cellular migration through a urokinase (uPA)-uPA receptor (uPAR) autocrine loop, and invasion through matrix metalloproteinases (MMPs)-2 and -9 secretion [18, 33, 72, 115, 127, 152, 160, 162, 164, 217, 283]. Enhanced glioma cell HIF expression leads to a distinct survival advantage in hypoxic and necrotic conditions [137, 152, 179, 257, 284] including therapeutic resistance through GSC enrichment [134, 251, 258, 276]. Furthermore, intratumoral HIF and other hypoxia-induced genes correlate with a more aggressive, pro-invasive and highly angiogenic phenotype across many solid tumors including glioblastoma [49, 64, 90, 115, 157, 189, 236, 253, 260, 271].

The hypoxic tumor microenvironment is highly coordinated and dynamic. Glioblastoma cells upregulate HIFs and subsequently stimulate MMPs to remodel the ECM, clotting factors to induce focus thrombosis, angiogenic factors that drive microvascular hyperproliferation, and immunomodulatory factors including damage-associated molecular patterns that form an immunosuppressive environment
The adaptive response to hypoxia also influences inflammatory and immune responses following the onset of necrosis. For example, HIF-1α modulates hypoxic T cell metabolism, facilitating Treg recruitment and an immunosuppressive phenotype [158]. Hypoxia increases TF secretion exacerbating focal vascular pathology, and Ras and PI3K-Akt signaling further enhance migration away from the hypoxic region [2, 204]. One study described p21 activated kinase (PAK) 1-dependent autophagy, linking hypoxia to glioblastoma tumorigenesis and radial expansion [62]. Hypoxic glioblastoma cells display not only altered DNA repair machinery, but are increasingly resistant to chemo- and radiation therapies [35, 45, 167]. Others have shown that hypoxia-induced epigenetic changes in histone deacetylase (HDAC) 3 activity and downstream transcription factors CCAAT enhancer binding protein beta (CEBPB) and JUN contribute to temozolomide (TMZ) resistance [66]. Intratumoral hypoxia also induces an inflammatory GSC phenotype that facilitates glioblastoma radial expansion [235]. Thus, acute and sustained hypoxia arising from vaso-occlusion and associated with necrosis has profound effects on disease progression and therapeutic resistance.
Necrosis
While the metabolic stress related to hypoxia contributes to a wide range of adaptive responses, as noted above, the associated development of necrosis also plays a pivotal role in reshaping the local brain tumor microenvironment. Although necrosis has historically been considered an unprogrammed, passive cell death response, work over the last 2 decades has uncovered specific signaling networks that regulate its development [281]. Separating the effects of necrosis and hypoxia may be difficult or impossible, and the classic histopathologic features of glioblastoma, including intratumoral thrombosis, microvascular proliferation, and neoplastic palisade formation around necrosis are intimately related to both (Fig. 5) [18, 191, 265]. Our prior work suggests that intravascular thrombosis causes vaso-occlusion, leading to nutrient deprivation and sustained hypoxia/anoxia that triggers cellular necrosis [202, 204, 241]. How these early hypoxic/necrotic events coordinate TME, reshaping is an area of active research. Necrotic cells are now known to release endogenous damage-associated molecular patterns (DAMPs), capable of recruiting TAMs or damage-associated microglia (DAM) to the TME [21, 50, 87], facilitating disease progression [120, 201] (Fig. 5). DAMP release normally initiates sterile inflammation to drive tissue repair yet when left unchecked can facilitate a chronic inflammatory state resulting in unwanted tissue damage, particularly in ischemia-related injuries [46, 110, 198]. Necrosis-associated DAMPs include adenosine/adenosine triphosphate (ATP) [17, 105, 128], biglycan [6, 200, 213], heparan sulfate [113, 272], heat shock proteins (HSPs) [11, 12, 109, 193, 252], high-mobility group box 1 (HMGB1) [94, 212, 222, 244, 282], hyaluronan (HA) [112, 214, 240], interleukin (IL)-1α [36, 59, 73, 125], IL-33 [31, 163, 216, 220], S100 proteins [42, 91, 92, 135], and versican [95, 121, 264]. Of interest, ATP, HA, HMGB1, IL-1α and S100 proteins are potent DAMPs that are enriched in brain and glioma tissues [10, 21, 73, 92, 94, 128, 182, 192, 221, 229, 247]. Extracellular adenosine binds to adenosine receptors on many immune cells including macrophages, driving initial inflammation, then inducing an M2-like immunosuppressive phenotype [128] and enhancing glioblastoma invasion [182]. HA cleavage from the ECM into small molecular weight fragments engage not only it’s canonical receptor, CD44, but also several Toll-like receptors (TLR2 and 4) known to mediate inflammatory responses [240] while simultaneously enhancing glioblastoma invasive capacity [37]. HMGB1 acts through both TLR4 and the receptor for advanced glycation end products (RAGE) to initiate pro-inflammatory cytokine release, recruiting bone marrow-derived monocytes (BMDMs) to sites of injury and contributing to the immunosuppressive TME [94, 101, 232]. Initial IL-1α release from necrotic cells draws neutrophils and BMDM in, followed by a second wave of IL-1α secretion from subsequently activated macrophages, further enhancing the pro-inflammatory microenvironment [59, 125]. S100 proteins also bind RAGE and attract BMDM to the TME, contributing to immune cell reprogramming and at the same time promoting tumor cell proliferation [87, 92, 135]. In addition to the generation of sterile inflammation, necrotic cellular pathology upregulates cell survival pathways to compensate for an increasingly inhospitable environment. Dramatic microenvironmental restructuring following necrosis enriches for distinct cellular subpopulations that thrive under these selective pressures.

Necrosis initiates a sterile inflammatory response that promotes glioma progression. Intratumoral thrombosis within the hypoxic/necrotic core forces glioblastoma cells to migrate towards a more hospitable locale. While most surviving perinecrotic glioblastoma cells migrate away from the necrotic core, an enriched stem-like phenotype is found in the highly hypoxic perinecrotic (palisading) niche. Meanwhile, blood–brain barrier disruption allows immune access to the tumor, monocyte influx and macrophage differentiation as those cells migrate towards the necrotic core. Simultaneously this emerging perivascular niche becomes enriched in stem-like cells, budding vessels, and myeloid-derived suppressor cells
Immune microenvironment
Microglia represent the largest phagocytic cell population in the brain under normal homeostatic conditions. They are unique to the brain and arise from immature yolk sac (Runx1+) progenitor cells between embryonic days 8.5 and 9.5 [68, 70, 83]. They are also among the most long-lived brain-resident cells, rivaling post-mitotic neuron life spans [280]. As mentioned, DAMs respond to DAMPs during brain injury [14, 50], representing an early and rapid innate immune response. In some disease states, DAMs play a neuroprotective role and hinder disease progression [120, 154]. However, sustained neuroinflammation and DAM reprogramming can result in neurotoxic events mediated not only by DAMs but also through modulating reactive astrocytes [13, 138]. Upon brain injury, stroke or tumorigenesis, the BBB becomes compromised leading to significant influx of circulating BMDM, as well as microglial activation [41, 168, 183, 262], and distinguishing these cell types and various activation states requires detailed analysis [77, 129]. In addition, TAM derived IL-1b exacerbates BBB defects, enhancing vascular edema and BBB leakiness [88]. These cell lineage determinations become crucial when determining how to counteract disease processes as BMDMs and microglia play differing roles in brain inflammatory responses [29, 55, 277]. A recent study utilizing a mouse model of pediatric high-grade glioma demonstrated that BMDMs, but not microglia are responsible for mediating the intratumoral immune response [206]. In addition, a single-cell RNA sequencing study revealed spatial and functional diversity among infiltrating microglia and BMDMs [130, 169]. These distinct subpopulations require informed consideration when designing therapeutic interventions to effectively target the malignant immune behaviors while preserving neuroprotective responses.
Despite advanced understanding of inflammation following traumatic brain injury and ischemia [46, 110, 262], mechanisms and therapeutic vulnerabilities of the sterile inflammatory response have not been well established in the glioblastoma TME. Following necrosis, TAMs represent the most abundant non-neoplastic cells within glioblastoma, accounting for 30–50% of all cells within the tumor mass (Fig. 6) [39, 79, 80]. TAMs are not passive bystanders, but rather actively promote tumor progression and modulate treatment responses [61, 98, 187, 278]. By the time a malignant brain tumor has developed severe hypoxia and central necrosis, the vast majority (> 80%) of TAMs derive from BMDMs, while the remainder are comprised of microglia [38, 80]. However, not all microglia respond to chemotactic/activating factors leaving residual undifferentiated tumor-associated microglia that appear as web-like immune surveillance cells enriched around the disease periphery [41, 227]. TAM density increases five- to tenfold following necrosis, mainly in hypoxic, perinecrotic zones [48, 156, 259]. Hypoxia induces TAM influx, activation then conversion from an anti-tumor (M1-like) to an immunosuppressive (M2-like) phenotype, promoting tumor progression [38, 39, 86, 169]. A recent TCGA pan-cancer study indicated that glioblastoma has a prominent TAM signature, with a highly immunosuppressive phenotype and suppressed Th1 lymphocytes [243]. Immune response genes are enriched in mesenchymal glioblastomas, indicating genomic background and transcriptional activities influence the TME [54, 111, 254, 285]. One study found increased immune cell infiltration, including TAMs and lymphocytes, in human mesenchymal glioblastomas compared to proneural and classic subtypes [118], while another found that classical glioblastomas display greater CD4+ and CD8+ T cell infiltration [40]. Analysis of TCGA glioblastoma data showed that allograft inflammatory factor 1 (AIF1), the gene encoding ionized calcium binding adaptor molecule 1 (IBA1), was significantly upregulated in mesenchymal glioblastomas compared to others [118]. Distribution within the TME—potentially related to the hypoxia gradient—also alters TAM behavior, as peripheral TAMs display pro-inflammatory signaling, homing circulating BMDMs to the TME [26, 130]. Myeloid-derived suppressor cells (MDSCs) are functionally similar to immunosuppressive TAMs but express specific cell surface markers such as CD33, CD14 and CD15 in humans or CD11b and protein gamma response 1 (Gr1) in mouse models [34, 175]. TAM-secreted CCL2 recruits MDSCs from circulation while GSC-secreted macrophage migration inhibitory factor (MIF) enhances their immunosuppressive activity [3, 34, 175]. The protective role has largely been attributed to enhanced MDSC programmed death-ligand 1 (PD-L1) expression that mitigates CD4+ T cell activity in and around the glioblastoma TME [56]. This active recruitment and reprogramming among immune subpopulations in and around the tumor create an increasingly complex, heterogeneous milieu that we are just beginning to recognize. Future investigations into the temporal and spatial dynamics will enable systematic interventions to reverse the immune privileged tumor state.

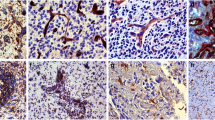
Enhanced tumor-associated macrophages (TAMs) in glioma progression (CD163 immunohistochemistry). In diffusely infiltrating gliomas that are low grade (histologic grade 2) and have an intact blood–brain barrier (BBB), there is a small population of inactive, flattened, perivascular CD163-positive TAMs (arrows) and only rare CD163-positive cells within the CNS parenchyma (a). With glioma progression and the development of hypoxia and BBB breakdown, there is activation and enlargement of the perivascular CD163-positive TAM population (arrow) and a large influx of CD163-positive TAMs from derived from circulating BMDM that traverse the BBB and infiltrate into the brain (b, corresponding to histology in Fig. 1b). With the development of necrosis, large numbers of CD163-positive TAMs are noted around necrosis (asterisk) and within the infiltrating component of the glioblastoma (c, corresponding to histology in Fig. 1c)
Glioblastoma exhibits far fewer infiltrating lymphocytes than other solid tumors, consisting largely of Tregs followed by CD3+ T helper cells, other CD4+ T cells and few CD8+ T cells [85, 268]. Importantly, glioblastoma T cell infiltration co-localizes with areas displaying vascular pathology, suggesting that thrombosis, vascular leakiness or angiogenesis may mediate T cell access to the CNS [47, 145]. Tregs respond to glioblastoma secreted CCL2 as well as GSC and dendritic cell (DC) produced indoleamine 2,3-dioxygenase (IDO), and their accumulation inversely correlates with survival [44, 114, 159, 176, 256]. In addition, TAMs upregulate T cell immunoglobulin- and mucin domain-containing molecule (TIM) 3 and TIM4 expression on infiltrating T cells, inducing Treg programming while simultaneously eliminating hypoxia-induced phosphatidylserine (PS) expressing CD8+ T cells in the glioblastoma TME [268, 275]. However, in neurodegeneration and traumatic brain injury, Tregs appear to enhance re-myelination and OPC proliferation while suppressing DAM and CD8+ T cell activity, providing a neuroprotective effect combating disease progression [124, 139, 270]. Given the differing roles that various immune subpopulations play in neurologic disorders, it is essential to properly identify and target those specific immune cells to harness the innate and adaptive immune system to counteract disease progression.
Glioma stem cells
Many recent reviews provide a comprehensive understanding of the history and significance of GSCs and their markers [131, 273]. Single cell RNAseq analysis and lineage tracing experiments reveal substantial inter- and intratumoral heterogeneity and inherent plasticity among GSC subpopulations [15, 53, 76, 107, 165, 245]. While terms and concepts related to GSCs vary considerably in the literature, most studies converge on the conclusion that GSC enrichment correlates with tumor grade, therapeutic resistance and recurrence [63, 131]. Most studies have also indicated that GSCs are enriched in specific biological niches, particularly in the hypoxic palisading cells around necrosis and within the immediate perivascular region [24, 79, 81, 116, 155]. Thus, establishing mechanistic links between TME enrichment of GSCs is highly relevant to the human disease and may have therapeutic implications. In particular, the perinecrotic niche contains a high density of neoplastic cells that show dramatic upregulation of hypoxia-inducible transcription factors and downstream targets, with a gradually diminishing hypoxic gradient extending beyond this zone [18, 20, 23, 164, 202, 265]. GSCs are enriched within this niche through a combination of hypoxic- and necrotic-driven chemotaxis and GSC phenotype enrichment [7, 18, 84, 102, 117, 134, 217, 226]. In turn, the GSC subpopulation facilitates TAM recruitment and subsequent immunosuppressive conversion along with microvascular hyperplasia surrounding the necrotic zones [63, 225, 253]. Within the perivascular niche, GSCs secrete chemotactic factors, such as VEGF, FGF, and PDGF, that disrupt the BBB and local vasculature; colony-stimulating factor (CSF) 1, periostin and stromal cell-derived factor (SDF) 1a that facilitate BMDM influx into the TME; and IL1 and IL6 that reprogram macrophages into an immunosuppressive phenotype [16, 61, 79, 89, 269, 279, 287]. A recent study found GSC-secreted extracellular vesicles reprogram local endothelial cells and identified potential pro-angiogenic miRNAs [147]. Others have suggested that bone marrow-derived mesenchymal stem cells recruited to the TME directly fuse with perivascular GSCs to drive neoangiogenesis in the expanding glioblastoma [231]. Endothelial cells secrete IL-8, which enhances the GSC phenotype and promotes glioblastoma expansion [155, 219] while also generating a positive feedback loop in which TAMs respond by producing tumor necrosis factor alpha (TNFα) that supports endothelial cell activation and microvascular proliferation [261]. In addition, these tumor-associated endothelial cells protect glioblastomas from radiation therapy [67, 78], chemotherapy [100], and angiogenic blockade [142]. GSCs accumulate within these tumor niches using them as safe havens and represent a critical subpopulation to address when developing future clinical approaches.
GSCs in both the perivascular and perinecrotic niche play a coordinated role in attracting and redirecting circulating monocytes towards the central necrotic region. The BBB disruption that occurs together with vascular pathology not only generates an ideal environment for one subset of GSCs; it also establishes an entry point for recruiting BMDMs into the tumor microenvironment [24, 177, 286]. While some BMDMs will remain in this niche, others proceed through the parenchymal space along the hypoxic gradient into the necrotic core. Upon arrival, tumor infiltrating TAMs again find themselves surrounded by GSCs in the perinecrotic niche, where there is a mutually beneficial relationship in an otherwise inhospitable environment [117, 217, 251]. The specific signaling interplay that enables this directed TAM relocation largely remains a mystery, in part due to difficulty in modeling these unique microenvironmental niches separated by a hypoxic gradient. Understanding this relationship could reveal divergent roles for these GSC subpopulations and enable differential immunotherapeutic based interventions aimed at disrupting complementary homing signals.
Therapeutic interventions
Therapeutic interventions for modulating macrophage activity across cancer types have been the subject of much investigation and review [5, 104, 108, 151, 185]. Here, we highlight recent advances in microenvironmental manipulation within the context of brain disease. Vascular pathology, GSC enrichment and immunosuppressive infiltrating immune cells all contribute to enhanced therapeutic resistance in glioblastoma and serve as rational broad targets for therapy [74, 106, 161, 218, 223, 251].
Glucocorticoids are time-tested immunomodulatory agents that are commonly employed at initial clinical presentation, perioperatively and during radiotherapy for patients with gliomas to diminish reactive edema and improve patient quality of life [52]. However, steroid-related side effects and toxicities necessitate short-term usage and dose de-escalation regimens. Both preclinical and retrospective clinical studies have suggested that corticosteroids may compromise immunotherapeutic efficacies and clinical outcomes [174, 184].
T cell-targeted immunotherapy has become the gold-standard approach to generating anti-tumor immunity in solid tumors. However, the early phase 3 immunotherapy trial targeting programmed cell death protein 1 (PD-1) in glioblastoma failed to improve overall patient survival (NCT02017717), which has been attributed to limited immune access to the TME [196].
Novel preclinical work shows that nanoscale immunoconjugates successfully penetrate the BBB to enhance T cell-targeted immunotherapy and overcome Treg-mediated immunosuppression [65]. Astonishingly, one study even found that anti-PD-1 therapy activated an anti-tumor immune response despite lacking conventional CD8 cytotoxic T cells in the TME [194]. These therapeutic adaptations emphasize the necessity for understanding TME interactions to inform effective clinical interventions.
A recently established macrophage-related gene signature [233], containing both macrophage and glioblastoma expressed genes, predicted therapeutic sensitivity more accurately than the previously published immune response signature [43] or the classical (EGFR amplified) signature. Other investigations demonstrated that CD74+ TAMs and MDSCs reduce clinical therapeutic efficacy [123, 267]. Given the unique immunology within glioblastomas, many interventions have been developed to inhibit TAM influx and/or conversion to an M2-like phenotype. These approaches upregulate IL-12 signaling, disrupt mammalian target of rapamycin (mTOR), colony-stimulating factor 1 receptor (CSF-1R), cyclin-dependent kinase (CDK), or phosphoinositide-3-kinase (PI3K) signaling, yielding mixed results with the most promising demonstrating resensitization to standard of care therapies and increased survival in animal glioblastoma models [8, 97, 133, 136, 140, 141, 144, 188, 190, 191, 263]. Still other studies show promising potential for exploiting the robust immune presence within glioblastoma. For instance, inhibiting proprotein convertases not only reduces immunosuppressive TAM polarization, but re-engages anti-tumoral activity to blunt glioblastoma expansion [205].
Due to treatment resistance inherent in GSC subpopulations, forced differentiation or directly targeting GSC phenotype promoting pathways have a substantial capacity to resensitize glioblastomas to conventional therapeutic approaches and extend time to recurrence. The perinecrotic niche protects GSCs through necrotic-driven DAMP signaling, which when obstructed eliminates these safe havens. This has been supported by the finding that disruption of adenosine signaling was capable of blunting GSC-driven migration and invasion, and that HMGB1 blockade was capable of reducing vascular permeability, neuroinflammation and edema [94, 166, 232]. Another approach seeks to diminish the GSC phenotype, targeting key transcriptional programs along the ERK1/2-SRY-box transcription factor 9 (SOX9), casein kinase (CK)2-signal transducer and activator of transcription (STAT)3, or SOX2-miR-126-3p axes resulting in cellular differentiation, decreased proliferation and invasion, increased apoptosis as well as enhanced susceptibility to radiation and TMZ therapies [75, 82, 143, 148, 211]. In addition, GSCs can give rise to drug refractory recurrent disease necessitating novel second-line therapies. One such study found CDK inhibitor-resistant glioblastomas are sensitive to c-MET/Trk dual inhibition, demonstrating effective sequential intervention modalities [171]. Other approaches exploit GSC-specific metabolism identifying a glycogen synthase kinase (GSK) 3β inhibitor, kenpaullone, and a pyrimidine synthesis inhibitor, 10580, which resensitize tumors to standard of care therapy [57, 122]. Future endeavors will continue to capitalize on these unique disease-related aspects to precisely target neurological and neuroinflammatory malregulation, further emphasizing the importance of understanding these microenvironmental pathways.
Conclusion
The brain TME contains a diversity of cell types, a complex vascular barrier, and unconventional stroma. Combined, these features, along with the access barriers imposed by the skull, make understanding dynamic microenvironmental changes of glioblastoma a challenging process, differing from neoplastic processes in other organs. The state of our current understanding suggests that the TME of diffuse gliomas is dramatically altered with the development of microscopic intravascular thrombosis at an early stage that is responsible for initiating or propagating a cascade that results in rapid disease progression. Glioblastomas display enhanced pro-coagulant activity, stemming from intrinsic genomic drivers (EGFR overexpression, PTEN loss) as well as hypoxia-induced signaling (Egf-1). These coagulant factors (TF, FVIIa, FXIIa) generate focal intravascular coagulation within the TME contributing to central necrosis, BBB disruption, radial progression, immune influx and modulation, which all combine to the advancement of disease. The resultant hypoxic gradient also enhances GSC survival mechanisms while reducing therapeutic efficacy, providing a challenging scenario for clinical intervention.
Prolonged and severe hypoxia cues the onset of necrosis that releases a variety of DAMPs (adenosine, HA, HMGB1, IL-1α, S100 proteins) that initiate sterile inflammation. Perhaps the most substantial TME feature that distinguishes glioblastoma from many other solid tumors and CNS diseases is the massive influx and reprogramming of the innate immune system. While in the past, some have suggested that glioblastoma is an immunologically “cold” tumor, more recent immunohistochemical, flow cytometric and transcriptional analyses have shown that the glioblastoma TME displays an abundance of infiltrating immune cells. Furthermore, hypoxia-induced signaling supports conversion of immune cells from an inflammatory to an immunosuppressive phenotype within the TME, including Treg recruitment, TAM immunomodulation, and MDSC localization to the perivascular niche. At the tumor periphery, MDSCs and DAMs play critical roles in excluding adaptive immune cells from the bulk tumor and represent potential barriers to current T cell focused immunotherapy that are becoming commonplace in other solid tumors.
Current efforts continue to explore spatial, temporal and cell-of-origin related contributions to immunomodulation among microglial and BMDM subpopulations of TAMs. The close spatial and temporal association between TAMs and GSCs in perivascular and perinecrotic niches is worthy of further study for their cooperation in the development of therapeutic resistance, disease progression and recurrence. Given the abundance of TAMs, DAMs, and MDSCs within the TME, the potential for successful targeted immunotherapies directed at innate immunity is substantial. Other efforts combating GSC enrichment and vascular pathology represent mechanisms to resensitize these tumors to standard of care interventions and could enhance the efficacy of our current clinical options. With better understanding of contributing mechanisms, future combination therapies have potential for improving patient outcomes.
References
Cancer Genome Atlas Research Network (2008) Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455:1061–1068. https://doi.org/10.1038/nature07385
Åberg M, Eriksson O, Siegbahn A (2015) Tissue factor noncoagulant signaling: mechanisms and implications for cell migration and apoptosis. Semin Thromb Hemostat 41:691–699
Achyut BR, Angara K, Jain M, Borin TF, Rashid MH, Iskander ASM et al (2017) Canonical NFκB signaling in myeloid cells is required for the glioblastoma growth. Sci Rep. https://doi.org/10.1038/s41598-017-14079-4
An Z, Knobbe-Thomsen CB, Wan X, Fan QW, Reifenberger G, Weiss WA (2018) EGFR cooperates with EGFRvIII to recruit macrophages in glioblastoma. Cancer Res 78:6785–6794. https://doi.org/10.1158/0008-5472.Can-17-3551
Anfray C, Ummarino A, Andón FT, Allavena P (2019) Current strategies to target tumor-associated-macrophages to improve anti-tumor immune responses. Cells 1:46. https://doi.org/10.3390/cells9010046
Babelova A, Moreth K, Tsalastra-Greul W, Zeng-Brouwers J, Eickelberg O, Young MF et al (2009) Biglycan, a danger signal that activates the NLRP3 inflammasome via toll-like and P2X receptors. J Biol Chem 284:24035–24048. https://doi.org/10.1074/jbc.m109.014266
Bar EE, Lin A, Mahairaki V, Matsui W, Eberhart CG (2010) Hypoxia increases the expression of stem-cell markers and promotes clonogenicity in glioblastoma neurospheres. Am J Pathol 177:1491–1502. https://doi.org/10.2353/ajpath.2010.091021
Barrett JA, Cai H, Miao J, Khare PD, Gonzalez P, Dalsing-Hernandez J et al (2018) Regulated intratumoral expression of IL-12 using a RheoSwitch Therapeutic System® (RTS®) gene switch as gene therapy for the treatment of glioma. Cancer Gene Ther 25:106–116. https://doi.org/10.1038/s41417-018-0019-0
Bartoschek M, Pietras K (2018) PDGF family function and prognostic value in tumor biology. Biochem Biophys Res Commun 503:984–990. https://doi.org/10.1016/j.bbrc.2018.06.106
Bassi R, Giussani P, Anelli V, Colleoni T, Pedrazzi M, Patrone M et al (2008) HMGB1 as an autocrine stimulus in human T98G glioblastoma cells: role in cell growth and migration. J Neurooncol 87:23–33. https://doi.org/10.1007/s11060-007-9488-y
Basu S (2000) Necrotic but not apoptotic cell death releases heat shock proteins, which deliver a partial maturation signal to dendritic cells and activate the NF-kappaB pathway. Int Immunol 12:1539–1546. https://doi.org/10.1093/intimm/12.11.1539
Basu S, Binder RJ, Ramalingam T, Srivastava PK (2001) CD91 is a common receptor for heat shock proteins gp96, hsp90, hsp70, and Calreticulin. Immunity 14:303–313. https://doi.org/10.1016/s1074-7613(01)00111-x
Bennett JP, Keeney PM, Brohawn DG (2019) RNA sequencing reveals small and variable contributions of infectious agents to transcriptomes of postmortem nervous tissues from amyotrophic lateral sclerosis, Alzheimer’s disease and Parkinson’s disease subjects, and increased expression of genes from D. Front Neurosci 13:235. https://doi.org/10.3389/fnins.2019.00235
Bernier LP, Bohlen CJ, York EM, Choi HB, Kamyabi A, Dissing-Olesen L et al (2019) Nanoscale surveillance of the brain by microglia via cAMP-regulated filopodia. Cell Rep 27:2895–2908. https://doi.org/10.1016/j.celrep.2019.05.010
Bhaduri A, Di Lullo E, Jung D, Muller S, Crouch EE, Espinosa CS et al (2020) Outer radial glia-like cancer stem cells contribute to heterogeneity of glioblastoma. Cell Stem Cell 26:48–63. https://doi.org/10.1016/j.stem.2019.11.015
Boulakirba S, Pfeifer A, Mhaidly R, Obba S, Goulard M, Schmitt T et al (2018) IL-34 and CSF-1 display an equivalent macrophage differentiation ability but a different polarization potential. Sci Rep 8:256. https://doi.org/10.1038/s41598-017-18433-4
Bours MJL, Swennen ELR, Di Virgilio F, Cronstein BN, Dagnelie PC (2006) Adenosine 5′-triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation. Pharmacol Ther 112:358–404. https://doi.org/10.1016/j.pharmthera.2005.04.013
Brat DJ, Castellano-Sanchez AA, Hunter SB, Pecot M, Cohen C, Hammond EH et al (2004) Pseudopalisades in glioblastoma are hypoxic, express extracellular matrix proteases, and are formed by an actively migrating cell population. Cancer Res 64:920–927. https://doi.org/10.1158/0008-5472.can-03-2073
Brat DJ, Prayson RA, Ryken TC, Olson JJ (2008) Diagnosis of malignant glioma: role of neuropathology. J Neurooncol 89:287–311. https://doi.org/10.1007/s11060-008-9618-1
Brat DJ, Van Meir EG (2004) Vaso-occlusive and prothrombotic mechanisms associated with tumor hypoxia, necrosis, and accelerated growth in glioblastoma. Lab Invest 84:397–405. https://doi.org/10.1038/labinvest.3700070
Braun M, Vaibhav K, Saad NM, Fatima S, Vender JR, Baban B et al (2017) White matter damage after traumatic brain injury: A role for damage associated molecular patterns. Biochim Biophys Acta Mol Basis Dis 1863:2614–2626. https://doi.org/10.1016/j.bbadis.2017.05.020
Brennan Cameron W, Verhaak Roel GW, McKenna A, Campos B, Noushmehr H et al (2013) The somatic genomic landscape of glioblastoma. Cell 155:462–477. https://doi.org/10.1016/j.cell.2013.09.034
Caiazzo A, Ramis-Conde I (2015) Multiscale modelling of palisade formation in gliobastoma multiforme. J Theor Biol 383:145–156. https://doi.org/10.1016/j.jtbi.2015.07.021
Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B et al (2007) A perivascular niche for brain tumor stem cells. Cancer Cell 11:69–82. https://doi.org/10.1016/j.ccr.2006.11.020
Cameron R, McKenna A, Campos B, Noushmehr H, Sofie R, Zheng S et al (2013) The somatic genomic landscape of glioblastoma. Cell 155:462–477. https://doi.org/10.1016/j.cell.2013.09.034
Caponegro MD, Oh K, Madeira MM, Radin D, Sterge N, Tayyab M et al (2021) A distinct microglial subset at the tumor–stroma interface of glioma. Glia 69:1767–1781. https://doi.org/10.1002/glia.23991
Capper D, Jones DTW, Sill M, Hovestadt V, Schrimpf D, Sturm D et al (2018) DNA methylation-based classification of central nervous system tumours. Nature 555:469–474. https://doi.org/10.1038/nature26000
Caruso R, Parisi A, Bonanno A, Paparo D, Quattrocchi E, Branca G et al (2012) Histologic coagulative tumour necrosis as a prognostic indicator of aggressiveness in renal, lung, thyroid and colorectal carcinomas: a brief review. Oncol Lett 3:16–18. https://doi.org/10.3892/ol.2011.420
Catalano M, D’Alessandro G, Trettel F, Limatola C (2020) Role of infiltrating microglia/macrophages in glioma. Adv Exp Med Biol 1202:281–298. https://doi.org/10.1007/978-3-030-30651-9_14
Catar R, Moll G, Hosp I, Simon M, Luecht C, Zhao H et al (2021) Transcriptional regulation of thrombin-induced endothelial VEGF induction and proangiogenic response. Cells 10:910. https://doi.org/10.3390/cells10040910
Cayrol C, Girard JP (2009) The IL-1-like cytokine IL-33 is inactivated after maturation by caspase-1. Proc Natl Acad Sci 106:9021–9026. https://doi.org/10.1073/pnas.0812690106
Ceccarelli M, Barthel FP, Malta TM, Sabedot TS, Salama SR, Murray BA et al (2016) Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell 164:550–563. https://doi.org/10.1016/j.cell.2015.12.028
Chandrasekar N, Mohanam S, Gujrati M, Olivero WC, Dinh DH, Rao JS (2003) Downregulation of uPA inhibits migration and PI3k/Akt signaling in glioblastoma cells. Oncogene 22:392–400. https://doi.org/10.1038/sj.onc.1206164
Chang AL, Miska J, Wainwright DA, Dey M, Rivetta CV, Yu D et al (2016) CCL2 produced by the glioma microenvironment is essential for the recruitment of regulatory T cells and myeloid-derived suppressor cells. Can Res 76:5671–5682. https://doi.org/10.1158/0008-5472.can-16-0144
Chang WH, Lai AG (2019) Transcriptional landscape of DNA repair genes underpins a pan-cancer prognostic signature associated with cell cycle dysregulation and tumor hypoxia. DNA Repair 78:142–153. https://doi.org/10.1016/j.dnarep.2019.04.008
Chen C-J, Kono H, Golenbock D, Reed G, Akira S, Rock KL (2007) Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat Med 13:851–856. https://doi.org/10.1038/nm1603
Chen J-WE, Pedron S, Shyu P, Hu Y, Sarkaria JN, Harley BAC (2018) Influence of hyaluronic acid transitions in tumor microenvironment on glioblastoma malignancy and invasive behavior. Front Mater. https://doi.org/10.3389/fmats.2018.00039
Chen Z, Feng X, Herting CJ, Garcia VA, Nie K, Pong WW et al (2017) Cellular and molecular identity of tumor-associated macrophages in glioblastoma. Can Res 77:2266–2278. https://doi.org/10.1158/0008-5472.can-16-2310
Chen Z, Hambardzumyan D (2018) Immune microenvironment in glioblastoma subtypes. Front Immunol 9:1004. https://doi.org/10.3389/fimmu.2018.01004
Chen Z, Herting CJ, Ross JL, Gabanic B, Puigdelloses Vallcorba M et al (2020) Genetic driver mutations introduced in identical cell-of-origin in murine glioblastoma reveal distinct immune landscapes but similar response to checkpoint blockade. Glia 68:2148–2166. https://doi.org/10.1002/glia.23883
Chen Z, Ross JL, Hambardzumyan D (2019) Intravital 2-photon imaging reveals distinct morphology and infiltrative properties of glioblastoma-associated macrophages. Proc Natl Acad Sci 116:14254–14259. https://doi.org/10.1073/pnas.1902366116
Cheng P, Corzo CA, Luetteke N, Yu B, Nagaraj S, Bui MM et al (2008) Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J Exp Med 205:2235–2249. https://doi.org/10.1084/jem.20080132
Cheng W, Ren X, Zhang C, Cai J, Liu Y, Han S et al (2016) Bioinformatic profiling identifies an immune-related risk signature for glioblastoma. Neurology 86:2226–2234. https://doi.org/10.1212/wnl.0000000000002770
Choi BD, Fecci PE, Sampson JH (2012) Regulatory T cells move in when gliomas say “I DO.” Clin Cancer Res 18:6086–6088. https://doi.org/10.1158/1078-0432.ccr-12-2801
Clarke RH, Moosa S, Anzivino M, Wang Y, Floyd DH, Purow BW et al (2014) Sustained radiosensitization of hypoxic glioma cells after oxygen pretreatment in an animal model of glioblastoma and in vitro models of tumor hypoxia. PLoS ONE 9:e111199. https://doi.org/10.1371/journal.pone.0111199
Corrigan F, Mander KA, Leonard AV, Vink R (2016) Neurogenic inflammation after traumatic brain injury and its potentiation of classical inflammation. J Neuroinflamm 13:264. https://doi.org/10.1186/s12974-016-0738-9
Couto M, Coelho-Santos V, Santos L, Fontes-Ribeiro C, Silva AP, Gomes CMF (2019) The interplay between glioblastoma and microglia cells leads to endothelial cell monolayer dysfunction via the interleukin-6-induced JAK2/STAT3 pathway. J Cell Physiol 234:19750–19760. https://doi.org/10.1002/jcp.28575
Cummings TJ, Hulette CM, Bigner SH, Riggins GJ, McLendon RE (2000) HAM56-immunoreactive macrophages in untreated infiltrating gliomas. Arch Pathol Lab Med 125:637–641
Dai Z, Zhu MM, Peng Y, Machireddy N, Evans CE, Machado R et al (2018) Therapeutic targeting of vascular remodeling and right heart failure in PAH with HIF-2α inhibitor. Am J Respir Crit Care Med 198:1423–1434. https://doi.org/10.1164/rccm.201710-2079oc
Deczkowska A, Keren-Shaul H, Weiner A, Colonna M, Schwartz M, Amit I (2018) Disease-associated microglia: a universal immune sensor of neurodegeneration. Cell 173:1073–1081. https://doi.org/10.1016/j.cell.2018.05.003
Deryugina EI, Soroceanu L, Strongin AY (2002) Up-Regulation of vascular endothelial growth factor by membrane-type 1 matrix metalloproteinase stimulates human glioma xenograft growth and angiogenesis. Can Res 62:580–588
Deutsch MB, Panageas KS, Lassman AB, DeAngelis LM (2013) Steroid management in newly diagnosed glioblastoma. J Neurooncol 113:111–116. https://doi.org/10.1007/s11060-013-1096-4
Dirkse A, Golebiewska A, Buder T, Nazarov PV, Muller A, Poovathingal S et al (2019) Stem cell-associated heterogeneity in Glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment. Nat Commun 10:1787. https://doi.org/10.1038/s41467-019-09853-z
Doucette T, Rao G, Rao A, Shen L, Aldape K, Wei J et al (2013) Immune heterogeneity of glioblastoma subtypes: extrapolation from the cancer genome atlas. Cancer Immunol Res 1:112–122. https://doi.org/10.1158/2326-6066.cir-13-0028
Du W, Bos PD (2020) Tracing bone marrow-derived microglia in brain metastatic tumors. Methods Enzymol. https://doi.org/10.1016/bs.mie.2019.08.017
Dubinski DJ, Hasselblatt M, Schneider-Hohendorf T, Bogdahn U, Stummer W, Wiendl H et al (2015) CD4+ T effector memory cell dysfunction is associated with the accumulation of granulocytic myeloid-derived suppressor cells in glioblastoma patients. Neuro Oncol 18:807–818. https://doi.org/10.1093/neuonc/nov280
Echizenya S, Ishii Y, Kitazawa S, Tanaka T, Matsuda S, Watanabe E et al (2019) Discovery of a new pyrimidine synthesis inhibitor eradicating glioblastoma-initiating cells. Neuro Oncol 22(2):29–239. https://doi.org/10.1093/neuonc/noz170
Edwin NC, Khoury MN, Sohal D, McCrae KR, Ahluwalia MS, Khorana AA (2016) Recurrent venous thromboembolism in glioblastoma. Thromb Res 137:184–188. https://doi.org/10.1016/j.thromres.2015.11.027
Eigenbrod T, Park J-H, Harder J, Iwakura Y, Núñez G (2008) Cutting edge: critical role for mesothelial cells in necrosis-induced inflammation through the recognition of IL-1α released from dying cells. J Immunol 181:8194–8198. https://doi.org/10.4049/jimmunol.181.12.8194
Elste AP, Petersen I (2010) Expression of proteinase-activated receptor 1–4 (PAR 1–4) in human cancer. J Mol Histol 41:89–99. https://doi.org/10.1007/s10735-010-9274-6
Feng X, Szulzewsky F, Yerevanian A, Chen Z, Heinzmann D, Rasmussen RD et al (2015) Loss of CX3CR1 increases accumulation of inflammatory monocytes and promotes gliomagenesis. Oncotarget 6:15077–15094. https://doi.org/10.18632/oncotarget.3730
Feng X, Zhang H, Meng L, Song H, Zhou Q, Qu C et al (2020) Hypoxia-induced acetylation of PAK1 enhances autophagy and promotes brain tumorigenesis via phosphorylating ATG5. Autophagy 17(3):723–742. https://doi.org/10.1080/15548627.2020.1731266
Filatova A, Acker T, Garvalov BK (2013) The cancer stem cell niche(s): the crosstalk between glioma stem cells and their microenvironment. Biochim Biophys Acta 1830:2496–2508. https://doi.org/10.1016/j.bbagen.2012.10.008
Fu Y, Wang D, Wang H, Cai M, Li C, Zhang X et al (2019) TSPO deficiency induces mitochondrial dysfunction, leading to hypoxia, angiogenesis and a growth-promoting metabolic shift towards glycolysis in glioblastoma. Neuro Oncol 22(2):240–252. https://doi.org/10.1093/neuonc/noz183
Galstyan A, Markman JL, Shatalova ES, Chiechi A, Korman AJ, Patil R et al (2019) Blood–brain barrier permeable nano immunoconjugates induce local immune responses for glioma therapy. Nat Commun 10:3850. https://doi.org/10.1038/s41467-019-11719-3
Gao Y, Liu B, Feng L, Sun B, He S, Yang Y et al (2019) Targeting JUN, CEBPB, and HDAC3: a novel strategy to overcome drug resistance in hypoxic glioblastoma. Front Oncol 9:33. https://doi.org/10.3389/fonc.2019.00033
Garcia-Barros M, Paris F, Cordon-Cardo C, Lyden D, Rafii S, Haimovitz-Friedman A et al (2003) Tumor response to radiotherapy regulated by endothelial cell apoptosis. Science 300:1155–1159. https://doi.org/10.1126/science.1082504
Geirsdottir L, David E, Keren-Shaul H, Weiner A, Bohlen SC, Neuber J et al (2019) Cross-species single-cell analysis reveals divergence of the primate microglia program. Cell 179:1609-1622 e1616. https://doi.org/10.1016/j.cell.2019.11.010
Gil-Bernabé AM, Ferjancic S, Tlalka M, Zhao L, Allen PD, Im JH et al (2012) Recruitment of monocytes/macrophages by tissue factor-mediated coagulation is essential for metastatic cell survival and premetastatic niche establishment in mice. Blood 119:3164–3175. https://doi.org/10.1182/blood-2011-08-376426
Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S et al (2010) Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330:841–845. https://doi.org/10.1126/science.1194637
Gofrit S, Shavit-Stein E (2019) The neuro-glial coagulonome: the thrombin receptor and coagulation pathways as major players in neurological diseases. Neural Regen Res 14:2043. https://doi.org/10.4103/1673-5374.262568
Graham CH, Forsdike J, Fitzgerald CJ, Macdonald-Goodfellow S (1999) Hypoxia-mediated stimulation of carcinoma cell invasiveness via upregulation of urokinase receptor expression. Int J Cancer 80:617–623. https://doi.org/10.1002/(sici)1097-0215(19990209)80:4%3c617::aid-ijc22%3e3.0.co;2-c
Greenhalgh AD, Brough D, Robinson EM, Girard S, Rothwell NJ, Allan SM (2012) Interleukin-1 receptor antagonist is beneficial after subarachnoid haemorrhage in rat by blocking haem-driven inflammatory pathology. Dis Model Mech 5:823–833. https://doi.org/10.1242/dmm.008557
Grégoire H, Roncali L, Rousseau A, Chérel M, Delneste Y, Jeannin P et al (2020) Targeting tumor associated macrophages to overcome conventional treatment resistance in glioblastoma. Front Pharmacol. https://doi.org/10.3389/fphar.2020.00368
Griess B, Mir S, Datta K, Teoh-Fitzgerald M (2020) Scavenging reactive oxygen species selectively inhibits M2 macrophage polarization and their pro-tumorigenic function in part, via Stat3 suppression. Free Radical Biol Med 147:48–60. https://doi.org/10.1016/j.freeradbiomed.2019.12.018
Gularyan SK, Gulin AA, Anufrieva KS, Shender V, Shakhparonov MI, Bastola S et al (2020) Investigation of inter- and intra-tumoral heterogeneity of glioblastoma using TOF-SIMS. Mol Cell Proteomics mcp.RA120.00198. https://doi.org/10.1074/mcp.ra120.001986
Haage V, Semtner M, Vidal RO, Hernandez DP, Pong WW, Chen Z et al (2019) Comprehensive gene expression meta-analysis identifies signature genes that distinguish microglia from peripheral monocytes/macrophages in health and glioma. Acta Neuropathol Commun. https://doi.org/10.1186/s40478-019-0665-y
Hambardzumyan D, Becher OJ, Rosenblum MK, Pandolfi PP, Manova-Todorova K, Holland EC (2008) PI3K pathway regulates survival of cancer stem cells residing in the perivascular niche following radiation in medulloblastoma in vivo. Genes Dev 22:436–448. https://doi.org/10.1101/gad.1627008
Hambardzumyan D, Bergers G (2015) Glioblastoma: defining tumor niches. Trends Cancer 1:252–265. https://doi.org/10.1016/j.trecan.2015.10.009
Hambardzumyan D, Gutmann DH, Kettenmann H (2016) The role of microglia and macrophages in glioma maintenance and progression. Nat Neurosci 19:20–27. https://doi.org/10.1038/nn.4185
Hambardzumyan D, Squatrito M, Carbajal E, Holland EC (2008) Glioma formation, cancer stem cells, and Akt signaling. Stem Cell Rev 4:203–210. https://doi.org/10.1007/s12015-008-9021-5
Han D, Yu T, Dong N, Wang B, Sun F, Jiang D (2019) Napabucasin, a novel STAT3 inhibitor suppresses proliferation, invasion and stemness of glioblastoma cells. J Exp Clin Cancer Res. https://doi.org/10.1186/s13046-019-1289-6
Hanisch U-K, Kettenmann H (2007) Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci 10:1387–1394. https://doi.org/10.1038/nn1997
Heddleston JM, Wu Q, Rivera M, Minhas S, Lathia JD, Sloan AE et al (2012) Hypoxia-induced mixed-lineage leukemia 1 regulates glioma stem cell tumorigenic potential. Cell Death Differ 19:428–439. https://doi.org/10.1038/cdd.2011.109
Heimberger AB, Abou-Ghazal M, Reina-Ortiz C, Yang DS, Sun W, Qiao W et al (2008) Incidence and prognostic impact of FoxP3+ regulatory T cells in human gliomas. Clin Cancer Res 14:5166–5172. https://doi.org/10.1158/1078-0432.ccr-08-0320
Henze A-T, Mazzone M (2016) The impact of hypoxia on tumor-associated macrophages. J Clin Investig 126:3672–3679. https://doi.org/10.1172/jci84427
Hernandez C, Huebener P, Schwabe RF (2016) Damage-associated molecular patterns in cancer: a double-edged sword. Oncogene 35:5931–5941. https://doi.org/10.1038/onc.2016.104
Herting CJ, Chen Z, Maximov V, Duffy A, Szulzewsky F, Shayakhmetov DM et al (2019) Tumour-associated macrophage-derived interleukin-1 mediates glioblastoma-associated cerebral oedema. Brain. https://doi.org/10.1093/brain/awz331
Hira VVV, Aderetti DA, Van Noorden CJF (2018) Glioma stem cell niches in human glioblastoma are periarteriolar. J Histochem Cytochem. https://doi.org/10.1369/0022155417752676
Hoffmann C, Mao X, Brown-Clay J, Moreau F, Al Absi A, Wurzer H et al (2018) Hypoxia promotes breast cancer cell invasion through HIF-1α-mediated up-regulation of the invadopodial actin bundling protein CSRP2. Sci Rep 8:10191. https://doi.org/10.1038/s41598-018-28637-x
Hofmann MA, Drury S, Fu C, Qu W, Taguchi A, Lu Y et al (1999) RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell 97:889–901. https://doi.org/10.1016/s0092-8674(00)80801-6
Holla FK, Postma TJ, Blankenstein MA, Van Mierlo TJM, Vos MJ, Sizoo EM et al (2016) Prognostic value of the S100B protein in newly diagnosed and recurrent glioma patients: a serial analysis. J Neurooncol 129:525–532. https://doi.org/10.1007/s11060-016-2204-z
Homma T, Fukushima T, Vaccarella S, Yonekawa Y, Di Patre PL, Franceschi S et al (2006) Correlation among pathology, genotype, and patient outcomes in glioblastoma. J Neuropathol Exp Neurol 65:846–854. https://doi.org/10.1097/01.jnen.0000235118.75182.94
Hong B, Muili K, Bolyard C, Russell L, Lee TJ, Banasavadi-Siddegowda Y et al (2019) Suppression of HMGB1 released in the glioblastoma tumor microenvironment reduces tumoral edema. Mol Therapy Oncolytics 12:93–102. https://doi.org/10.1016/j.omto.2018.11.005
Hope C, Foulcer S, Jagodinsky J, Chen SX, Jensen JL, Patel S et al (2016) Immunoregulatory roles of versican proteolysis in the myeloma microenvironment. Blood 128:680–685. https://doi.org/10.1182/blood-2016-03-705780
Hori M, Mori H, Aoki S, Abe O, Masumoto T, Kunimatsu S et al (2010) Three-dimensional susceptibility-weighted imaging at 3 T using various image analysis methods in the estimation of grading intracranial gliomas. Magn Reson Imaging 28:594–598. https://doi.org/10.1016/j.mri.2010.01.002
Hsu SPC, Chen Y-C, Chiang H-C, Huang Y-C, Huang C-C, Wang H-E et al (2020) Rapamycin and hydroxychloroquine combination alters macrophage polarization and sensitizes glioblastoma to immune checkpoint inhibitors. J Neurooncol 146:417–426. https://doi.org/10.1007/s11060-019-03360-3
Hu FA, Dzaye OD, Hahn A, Yu Y, Scavetta RJ, Dittmar G et al (2015) Glioma-derived versican promotes tumor expansion via glioma-associated microglial/macrophages Toll-like receptor 2 signaling. Neuro Oncol 17:200–210. https://doi.org/10.1093/neuonc/nou324
Hu S, Wu G, Zheng J, Liu X, Zhang Y (2019) Astrocytic thrombin-evoked VEGF release is dependent on p44/42 MAPKs and PAR1. Biochem Biophys Res Commun 509:585–589. https://doi.org/10.1016/j.bbrc.2018.12.168
Huang M, Zhang D, Wu JY, Xing K, Yeo E, Li C et al (2020) Wnt-mediated endothelial transformation into mesenchymal stem cell-like cells induces chemoresistance in glioblastoma. Sci Transl Med 12:eaay522. https://doi.org/10.1126/scitranslmed.aay7522
Hubert P, Roncarati P, Demoulin S, Pilard C, Ancion M, Reynders C et al (2021) Extracellular HMGB1 blockade inhibits tumor growth through profoundly remodeling immune microenvironment and enhances checkpoint inhibitor-based immunotherapy. J Immunother Cancer 9:e001966. https://doi.org/10.1136/jitc-2020-001966
Inukai M, Hara A, Yasui Y, Kumabe T, Matsumoto T, Saegusa M (2015) Hypoxia-mediated cancer stem cells in pseudopalisades with activation of hypoxia-inducible factor-1alpha/Akt axis in glioblastoma. Hum Pathol 46:1496–1505. https://doi.org/10.1016/j.humpath.2015.06.008
Irtenkauf SM, Sobiechowski S, Hasselbach LA, Nelson KK, Transou AD, Carlton ET et al (2017) Optimization of glioblastoma mouse orthotopic xenograft models for translational research. Comp Med 67:300–314
Ishikawa E, Miyazaki T, Takano S, Akutsu H (2021) Anti-angiogenic and macrophage-based therapeutic strategies for glioma immunotherapy. Brain Tumor Pathol. https://doi.org/10.1007/s10014-021-00402-5
Iyer SS, Pulskens WP, Sadler JJ, Butter LM, Teske GJ, Ulland TK et al (2009) Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc Natl Acad Sci 106:20388–20393. https://doi.org/10.1073/pnas.0908698106
Jackson CM, Choi J, Lim M (2019) Mechanisms of immunotherapy resistance: lessons from glioblastoma. Nat Immunol. https://doi.org/10.1038/s41590-019-0433-y
Jacob F, Salinas RD, Zhang DY, Nguyen PTT, Schnoll JG, Wong SZH et al (2020) A patient-derived glioblastoma organoid model and biobank recapitulates inter- and intra-tumoral heterogeneity. Cell 180:188-204e122. https://doi.org/10.1016/j.cell.2019.11.036
Jahchan NS, Mujal AM, Pollack JL, Binnewies M, Sriram V, Reyno L et al (2019) Tuning the tumor myeloid microenvironment to fight cancer. Front Immunol. https://doi.org/10.3389/fimmu.2019.01611
Jayaprakash P, Dong H, Zou M, Bhatia A, O’Brien K, Chen M et al (2015) Hsp90 and Hsp90 together operate a hypoxia and nutrient paucity stress-response mechanism during wound healing. J Cell Sci 128:1475–1480. https://doi.org/10.1242/jcs.166363
Jayaraj RL, Azimullah S, Beiram R, Jalal FY, Rosenberg GA (2019) Neuroinflammation: friend and foe for ischemic stroke. J Neuroinflamm. https://doi.org/10.1186/s12974-019-1516-2
Jeanmougin M, Håvik AB, Cekaite L, Brandal P, Sveen A, Meling TR et al (2020) Improved prognostication of glioblastoma beyond molecular subtyping by transcriptional profiling of the tumor microenvironment. Mol Oncol 14:1016–1027. https://doi.org/10.1002/1878-0261.12668
Jiang D, Liang J, Fan J, Yu S, Chen S, Luo Y et al (2005) Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat Med 11:1173–1179. https://doi.org/10.1038/nm1315
Johnson GB, Brunn GJ, Kodaira Y, Platt JL (2002) Receptor-mediated monitoring of tissue well-being via detection of soluble heparan sulfate by toll-like receptor 4. J Immunol 168:5233–5239. https://doi.org/10.4049/jimmunol.168.10.5233
Jordan JT, Sun W, Hussain SF, DeAngulo G, Prabhu SS, Heimberger AB (2008) Preferential migration of regulatory T cells mediated by glioma-secreted chemokines can be blocked with chemotherapy. Cancer Immunol Immunother 57:123–131. https://doi.org/10.1007/s00262-007-0336-x
Joseph JV, Conroy S, Pavlov K, Sontakke P, Tomar T, Eggens-Meijer E et al (2015) Hypoxia enhances migration and invasion in glioblastoma by promoting a mesenchymal shift mediated by the HIF1α–ZEB1 axis. Cancer Lett 359:107–116. https://doi.org/10.1016/j.canlet.2015.01.010
Jung E, Osswald M, Ratliff M, Dogan H, Xie R, Weil S et al (2021) Tumor cell plasticity, heterogeneity, and resistance in crucial microenvironmental niches in glioma. Nat Commun. https://doi.org/10.1038/s41467-021-21117-3
Jung J, Zhang Y, Celiku O, Zhang W, Song H, Williams BJ, et al (2019) Mitochondrial NIX promotes tumor survival in the hypoxic niche of glioblastoma. Cancer Res canres.0198.0201. https://doi.org/10.1158/0008-5472.can-19-0198
Kaffes I, Szulzewsky F, Chen Z, Herting CJ, Gabanic B, Velázquez Vega JE et al (2019) Human mesenchymal glioblastomas are characterized by an increased immune cell presence compared to proneural and classical tumors. Oncoimmunology. https://doi.org/10.1080/2162402x.2019.1655360
Kawai N, Lin W, Cao W-D, Ogawa D, Miyake K, Haba R et al (2014) Correlation between 18F-fluoromisonidazole PET and expression of HIF-1α and VEGF in newly diagnosed and recurrent malignant gliomas. Eur J Nucl Med Mol Imaging 41:1870–1878. https://doi.org/10.1007/s00259-014-2776-9
Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK et al (2017) A unique microglia type associated with restricting development of alzheimer’s disease. Cell 169:1276-1290e1217. https://doi.org/10.1016/j.cell.2017.05.018
Kim S, Takahashi H, Lin W-W, Descargues P, Grivennikov S, Kim Y et al (2009) Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature 457:102–106. https://doi.org/10.1038/nature07623
Kitabayashi T, Dong Y, Furuta T, Sabit H, Jiapaer S, Zhang J et al (2019) Identification of GSK3β inhibitor kenpaullone as a temozolomide enhancer against glioblastoma. Sci Rep. https://doi.org/10.1038/s41598-019-46454-8
Kitange GJ, Carlson BL, Schroeder MA, Decker PA, Morlan BW, Wu W et al (2010) Expression of CD74 in high grade gliomas: a potential role in temozolomide resistance. J Neurooncol 100:177–186. https://doi.org/10.1007/s11060-010-0186-9
Kohm AP, Carpentier PA, Anger HA, Miller SD (2002) Cutting edge: CD4+CD25+ regulatory T cells suppress antigen-specific autoreactive immune responses and central nervous system inflammation during active experimental autoimmune encephalomyelitis. J Immunol 169:4712–4716. https://doi.org/10.4049/jimmunol.169.9.4712
Kono H, Karmarkar D, Iwakura Y, Rock KL (2010) Identification of the cellular sensor that stimulates the inflammatory response to sterile cell death. J Immunol 184:4470–4478. https://doi.org/10.4049/jimmunol.0902485
Krenzlin H, Lorenz V, Alessandri B (2017) The involvement of thrombin in the pathogenesis of glioblastoma. J Neurosci Res 95:2080–2085. https://doi.org/10.1002/jnr.24049
Krishnamachary B, Berg-Dixon S, Kelly B, Agani F, Feldser D, Ferreira G et al (2003) Regulation of colon carcinoma cell invasion by hypoxia-inducible factor 1. Can Res 63:1138–1143
Kumar V (2013) Adenosine as an endogenous immunoregulator in cancer pathogenesis: where to go? Purinergic Signal 9:145–165. https://doi.org/10.1007/s11302-012-9349-9
Kvisten M, Mikkelsen VE, Stensjoen AL, Solheim O, Van Der Want J, Torp SH (2019) Microglia and macrophages in human glioblastomas: a morphological and immunohistochemical study. Mol Clin Oncol 11:31–36. https://doi.org/10.3892/mco.2019.1856
Landry AP, Balas M, Alli S, Spears J, Zador Z (2020) Distinct regional ontogeny and activation of tumor associated macrophages in human glioblastoma. Sci Rep. https://doi.org/10.1038/s41598-020-76657-3
Lathia JD, Mack SC, Mulkearns-Hubert EE, Valentim CLL, Rich JN (2015) Cancer stem cells in glioblastoma. Genes Dev 29:1203–1217. https://doi.org/10.1101/gad.261982.115
Lee J, Jo DH, Kim JH, Cho CS, Han JE, Kim Y et al (2019) Development of a patient-derived xenograft model of glioblastoma via intravitreal injection in mice. Exp Mol Med 51:1–9. https://doi.org/10.1038/s12276-019-0241-3
Li J, Kaneda MM, Ma J, Li M, Shepard RM, Patel K et al (2021) PI3Kγ inhibition suppresses microglia/TAM accumulation in glioblastoma microenvironment to promote exceptional temozolomide response. Proc Natl Acad Sci 118:e2009290118. https://doi.org/10.1073/pnas.2009290118
Li Z, Bao S, Wu Q, Wang H, Eyler C, Sathornsumetee S et al (2009) Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell 15:501–513. https://doi.org/10.1016/j.ccr.2009.03.018
Li Z, Wang J, Zhang X, Liu P, Zhang X, Wang J et al (2020) Proinflammatory S100A8 induces PD-L1 expression in macrophages, mediating tumor immune escape. J Immunol. https://doi.org/10.4049/jimmunol.1900753
Li Z, Zhang J, Zheng H, Li C, Xiong J, Wang W et al (2019) Modulating lncRNA SNHG15/CDK6/miR-627 circuit by palbociclib, overcomes temozolomide resistance and reduces M2-polarization of glioma associated microglia in glioblastoma multiforme. J Exp Clin Cancer Res 38:380. https://doi.org/10.1186/s13046-019-1371-0
Liao Z, She C, Ma L, Sun Z, Li P, Zhang X et al (2019) KDELR2 promotes glioblastoma tumorigenesis targeted by HIF1a via mTOR signaling pathway. Cell Mol Neurobiol. https://doi.org/10.1007/s10571-019-00715-2
Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L et al (2017) Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541:481–487. https://doi.org/10.1038/nature21029
Liesz A, Suri-Payer E, Veltkamp C, Doerr H, Sommer C, Rivest S et al (2009) Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat Med 15:192–199. https://doi.org/10.1038/nm.1927
Lisi L, Ciotti GMP, Chiavari M, Pizzoferrato M, Mangiola A, Kalinin S et al (2019) Phospho-mTOR expression in human glioblastoma microglia-macrophage cells. Neurochem Int 129:104485. https://doi.org/10.1016/j.neuint.2019.104485
Liu S, Tang Y, Yuan X, Yuan D, Liu J, Li B et al (2018) Inhibition of Rb and mTOR signaling associates with synergistic anticancer effect of palbociclib and erlotinib in glioblastoma cells. Investig New Drugs. https://doi.org/10.1007/s10637-018-0575-z
Liu T, Ma W, Xu H, Huang M, Zhang D, He Z et al (2018) PDGF-mediated mesenchymal transformation renders endothelial resistance to anti-VEGF treatment in glioblastoma. Nat Commun. https://doi.org/10.1038/s41467-018-05982-z
Liu X, Chen J, Li W, Hang C, Dai Y (2020) Inhibition of casein kinase II by CX-4945, but not yes-associated protein (YAP) by verteporfin, enhances the antitumor efficacy of temozolomide in glioblastoma. Transl Oncol 13:70–78. https://doi.org/10.1016/j.tranon.2019.09.006
Lo Dico A, Valtorta S, Ottobrini L, Moresco RM (2019) Role of metformin and AKT axis modulation in the reversion of hypoxia induced TMZ-resistance in glioma cells. Front Oncol. https://doi.org/10.3389/fonc.2019.00463
Lohr J, Ratliff T, Huppertz A, Ge Y, Dictus C, Ahmadi R et al (2011) Effector T-cell infiltration positively impacts survival of glioblastoma patients and is impaired by tumor-derived TGF-β. Clin Cancer Res 17:4296–4308. https://doi.org/10.1158/1078-0432.Ccr-10-2557
Lowe KL, Navarro-Núñez L, Bénézech C, Nayar S, Kingston BL, Nieswandt B et al (2015) The expression of mouse CLEC-2 on leucocyte subsets varies according to their anatomical location and inflammatory state. Eur J Immunol 45:2484–2493. https://doi.org/10.1002/eji.201445314
Lucero R, Zappulli V, Sammarco A, Murillo OD, Cheah PS, Srinivasan S et al (2020) Glioma-derived miRNA-containing extracellular vesicles induce angiogenesis by reprogramming brain endothelial cells. Cell Rep 7:2065–2074. https://doi.org/10.1016/j.celrep.2020.01.073
Luo W, Yan D, Song Z, Zhu X, Liu X, Li X (2019) miR-126-3p sensitizes glioblastoma cells to temozolomide by inactivating Wnt/β-catenin signaling via targeting SOX2. Life Sci 226:98–106. https://doi.org/10.1016/j.lfs.2019.04.023
Ma YY, He XJ, Wang HJ, Xia YJ, Wang SL, Ye ZY et al (2011) Interaction of coagulation factors and tumor-associated macrophages mediates migration and invasion of gastric cancer. Cancer Sci 102:336–342. https://doi.org/10.1111/j.1349-7006.2010.01795.x
Magnus N, D’Asti E, Garnier D, Meehan B, Rak J (2013) Brain neoplasms and coagulation. Semin Thromb Hemost 39:881–895. https://doi.org/10.1055/s-0033-1357483
Majc B, Novak M, Jerala NK, Jewett A, Breznik B (2021) Immunotherapy of glioblastoma: current strategies and challenges in tumor model development. Cells 10:265. https://doi.org/10.3390/cells10020265
Marín-Hernández A, Gallardo-Pérez JC, Ralph SJ, Rodríguez-Enríquez S, Moreno-Sánchez R (2009) HIF-1alpha modulates energy metabolism in cancer cells by inducing over-expression of specific glycolytic isoforms. Mini Rev Med Chem 9:1084–1101. https://doi.org/10.2174/138955709788922610
Martinez-Lage M, Lynch TM, Bi Y, Cocito C, Way GP, Pal S et al (2019) Immune landscapes associated with different glioblastoma molecular subtypes. Acta Neuropathol Commun. https://doi.org/10.1186/s40478-019-0803-6
Mathys H, Adaikkan C, Gao F, Young JZ, Manet E, Hemberg M et al (2017) Temporal tracking of microglia activation in neurodegeneration at single-cell resolution. Cell Rep 21:366–380. https://doi.org/10.1016/j.celrep.2017.09.039
McCoy MG, Nyanyo D, Hung CK, Goerger JP, Zipfel W, Williams RM et al (2019) Endothelial cells promote 3D invasion of GBM by IL-8-dependent induction of cancer stem cell properties. Sci Rep. https://doi.org/10.1038/s41598-019-45535-y
McKelvey KJ, Hudson AL, Prasanna Kumar R, Wilmott JS, Attrill GH, Long GV et al (2020) Temporal and spatial modulation of the tumor and systemic immune response in the murine Gl261 glioma model. PLoS ONE 15:e0226444. https://doi.org/10.1371/journal.pone.0226444
Miroshnikova YA, Mouw JK, Barnes JM, Pickup MW, Kim Y, Lobo K et al (2016) Tissue mechanics promote IDH1-dependent HIF1α–tenascin C feedback to regulate glioblastoma aggression. Nat Cell Biol 18:1336–1345. https://doi.org/10.1038/ncb3429
Miska J, Lee-Chang C, Rashidi A, Muroski ME, Chang AL, Lopez-Rosas A et al (2019) HIF-1α is a metabolic switch between glycolytic-driven migration and oxidative phosphorylation-driven immunosuppression of Tregs in glioblastoma. Cell Rep 27:226-237.e224. https://doi.org/10.1016/j.celrep.2019.03.029
Mitsuka K, Kawataki T, Satoh E, Asahara T, Horikoshi T, Kinouchi H (2013) Expression of indoleamine 2,3-dioxygenase and correlation with pathological malignancy in gliomas. Neurosurgery 72:1031–1039. https://doi.org/10.1227/NEU.0b013e31828cf945
Mohanam S, Sawaya R, McCutcheon I, Ali-Osman F, Boyd D, Rao JS (1993) Modulation of in vitro invasion of human glioblastoma cells by urokinase-type plasminogen activator receptor antibody. Can Res 53:4143–4147
Mooney J, Bernstock JD, Ilyas A, Ibrahim A, Yamashita D, Markert JM et al (2019) Current approaches and challenges in the molecular therapeutic targeting of glioblastoma. World Neurosurg. https://doi.org/10.1016/j.wneu.2019.05.205
Mori T, Abe T, Wakabayashi Y, Hikawa T, Matsuo K, Yamada Y et al (2000) Up-regulation of urokinase-type plasminogen activator and its receptor correlates with enhanced invasion activity of human glioma cells mediated by transforming growth factor-alpha or basic fibroblast growth factor. J Neurooncol 46:115–123. https://doi.org/10.1023/a:1006339717748
Moussion C, Ortega N, Girard J-P (2008) The IL-1-like cytokine IL-33 Is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: a novel ‘alarmin’? PLoS ONE 3:e3331. https://doi.org/10.1371/journal.pone.0003331
Musah-Eroje A, Watson S (2019) Adaptive changes of glioblastoma cells following exposure to hypoxic (1% oxygen) tumour microenvironment. Int J Mol Sci 20:2091. https://doi.org/10.3390/ijms20092091
Neftel C, Laffy J, Filbin MG, Hara T, Shore ME, Rahme GJ et al (2019) An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell. https://doi.org/10.1016/j.cell.2019.06.024
Niechi I, Uribe-Ojeda A, Erices J, Torres Á, Uribe D, Rocha J et al (2019) Adenosine depletion as a new strategy to decrease glioblastoma stem-like cells aggressiveness. Cells. https://doi.org/10.3390/cells8111353
Nikolova T, Christmann M, Kaina B (2009) FEN1 is overexpressed in testis, lung and brain tumors. Anticancer Res 29:2453–2459
Nimmerjahn A (2005) Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308:1314–1318. https://doi.org/10.1126/science.1110647
Ochocka N, Segit P, Walentynowicz KA, Wojnicki K, Cyranowski S, Swatler J et al (2021) Single-cell RNA sequencing reveals functional heterogeneity of glioma-associated brain macrophages. Nat Commun. https://doi.org/10.1038/s41467-021-21407-w
Olar A, Aldape KD (2014) Using the molecular classification of glioblastoma to inform personalized treatment. J Pathol 232:165–177. https://doi.org/10.1002/path.4282
Olmez I, Zhang Y, Manigat L, Benamar M, Brenneman B, Nakano I, et al (2018) Combined c-Met/Trk inhibition overcomes resistance to CDK4/6 inhibitors in glioblastoma. Cancer Res canres.3124.3201. https://doi.org/10.1158/0008-5472.can-17-3124
Orzan F, Pagani F, Cominelli M, Triggiani L, Calza S, De Bacco F et al (2020) A simplified integrated molecular and immunohistochemistry-based algorithm allows high accuracy prediction of glioblastoma transcriptional subtypes. Lab Investig. https://doi.org/10.1038/s41374-020-0437-0
Ostrom QT, Gittleman H, Truitt G, Boscia A, Kruchko C, Barnholtz-Sloan JS (2018) CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2011–2015. Neuro Oncol 20:iv1–iv86. https://doi.org/10.1093/neuonc/noy131
Otvos B, Alban TJ, Grabowski MM, Bayik D, Mulkearns-Hubert EE, Radivoyevitch T et al (2021) Preclinical modeling of surgery and steroid therapy for glioblastoma reveals changes in immunophenotype that are associated with tumor growth and outcome. Clin Cancer Res 27:2038–2049. https://doi.org/10.1158/1078-0432.CCR-20-3262
Otvos B, Silver DJ, Mulkearns-Hubert EE, Alvarado AG, Turaga SM, Sorensen MD et al (2016) Cancer stem cell-secreted macrophage migration inhibitory factor stimulates myeloid derived suppressor cell function and facilitates glioblastoma immune evasion. Stem Cells 34:2026–2039. https://doi.org/10.1002/stem.2393
Ozawa Y, Yamamuro S, Sano E, Tatsuoka J, Hanashima Y, Yoshimura S et al (2020) Indoleamine 2,3-dioxygenase 1 is highly expressed in glioma stem cells. Biochem Biophys Res Commun. https://doi.org/10.1016/j.bbrc.2020.01.148
Pacioni S, D’Alessandris QG, Buccarelli M, Boe A, Martini M, Larocca LM et al (2019) Brain invasion along perivascular spaces by glioma cells: relationship with blood-brain barrier. Cancers 12:E18. https://doi.org/10.3390/cancers12010018
Pan YB, Wang S, Yang B, Jiang Z, Lenahan C, Wang J et al (2020) Transcriptome analyses reveal molecular mechanisms underlying phenotypic differences among transcriptional subtypes of glioblastoma. J Cell Mol Med 24:3901–3916. https://doi.org/10.1111/jcmm.14976
Park SJ, Kim H, Kim SH, Joe E-h, Jou I (2019) Epigenetic downregulation of STAT6 increases HIF-1α expression via mTOR/S6K/S6, leading to enhanced hypoxic viability of glioma cells. Acta Neuropathol Commun. https://doi.org/10.1186/s40478-019-0798-z
Peterziel H, Muller J, Danner A, Barbus S, Liu HK, Radlwimmer B et al (2012) Expression of podoplanin in human astrocytic brain tumors is controlled by the PI3K-AKT-AP-1 signaling pathway and promoter methylation. Neuro Oncol 14:426–439. https://doi.org/10.1093/neuonc/nos055
Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD et al (2006) Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 9:157–173. https://doi.org/10.1016/j.ccr.2006.02.019
Pietrobono D, Giacomelli C, Marchetti L, Martini C, Trincavelli ML (2020) High adenosine extracellular levels induce glioblastoma aggressive traits modulating the mesenchymal stromal cell secretome. Int J Mol Sci 21:7706. https://doi.org/10.3390/ijms21207706
Pinton L, Masetto E, Vettore M, Solito S, Magri S, D’Andolfi M et al (2019) The immune suppressive microenvironment of human gliomas depends on the accumulation of bone marrow-derived macrophages in the center of the lesion. J Immunother Cancer. https://doi.org/10.1186/s40425-019-0536-x
Pitter KL, Tamagno I, Alikhanyan K, Hosni-Ahmed A, Pattwell SS, Donnola S et al (2016) Corticosteroids compromise survival in glioblastoma. Brain 139:1458–1471. https://doi.org/10.1093/brain/aww046
Poh AR, Ernst M (2018) Targeting macrophages in cancer: from bench to bedside. Front Oncol. https://doi.org/10.3389/fonc.2018.00049
Pore N, Liu S, Haas-Kogan DA, O’Rourke DM, Maity A (2003) PTEN mutation and epidermal growth factor receptor activation regulate vascular endothelial growth factor (VEGF) mRNA expression in human glioblastoma cells by transactivating the proximal VEGF promoter. Can Res 63:236–241
Prins RM, Soto H, Konkankit V, Odesa SK, Eskin A, Yong WH et al (2011) Gene expression profile correlates with T-cell infiltration and relative survival in glioblastoma patients vaccinated with dendritic cell immunotherapy. Clin Cancer Res 17:1603–1615. https://doi.org/10.1158/1078-0432.ccr-10-2563
Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF et al (2013) CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med 19:1264–1272. https://doi.org/10.1038/nm.3337
Qiu W, Song S, Chen W, Zhang J, Yang H, Chen Y (2019) Hypoxia-induced EPHB2 promotes invasive potential of glioblastoma. Int J Clin Exp Pathol 12:539–548
Quail DF, Bowman RL, Akkari L, Quick ML, Schuhmacher AJ, Huse JT et al (2016) The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science 352:aad3018. https://doi.org/10.1126/science.aad3018
Quail DF, Joyce JA (2017) The microenvironmental landscape of brain tumors. Cancer Cell 31:326–341. https://doi.org/10.1016/j.ccell.2017.02.009
Quezada C, Garrido W, Oyarzún C, Fernández K, Segura R, Melo R et al (2013) 5′-ectonucleotidase mediates multiple-drug resistance in glioblastoma multiforme cells. J Cell Physiol 228:602–608. https://doi.org/10.1002/jcp.24168
Quintana FJ, Cohen IR (2005) Heat shock proteins as endogenous adjuvants in sterile and septic inflammation. J Immunol 175:2777–2782. https://doi.org/10.4049/jimmunol.175.5.2777
Rao G, Latha K, Ott M, Sabbagh A, Marisetty A, Ling X et al (2020) Anti–PD-1 induces M1 polarization in the glioma microenvironment and exerts therapeutic efficacy in the absence of CD8 cytotoxic T cells. Clin Cancer Res 26:4699–4712. https://doi.org/10.1158/1078-0432.ccr-19-4110
Rayes J, Lax S, Wichaiyo S, Watson SK, Di Y, Lombard S et al (2017) The podoplanin-CLEC-2 axis inhibits inflammation in sepsis. Nat Commun. https://doi.org/10.1038/s41467-017-02402-6
Reardon DA, Lassman AB, Schiff D, Yunus SA, Gerstner ER, Cloughesy TF et al (2018) Phase 2 and biomarker study of trebananib, an angiopoietin-blocking peptibody, with and without bevacizumab for patients with recurrent glioblastoma. Cancer 124:1438–1448. https://doi.org/10.1002/cncr.31172
Richards CH, Mohammed Z, Qayyum T, Horgan PG, McMillan DC (2011) The prognostic value of histological tumor necrosis in solid organ malignant disease: a systematic review. Future Oncol 7:1223–1235. https://doi.org/10.2217/fon.11.99
Richards RI, Robertson SA, O’Keefe LV, Fornarino D, Scott A, Lardelli M et al (2016) The enemy within: innate surveillance-mediated cell death, the common mechanism of neurodegenerative disease. Front Neurosci 10:193. https://doi.org/10.3389/fnins.2016.00193
Riedl J, Preusser M, Nazari PMS, Posch F, Panzer S, Marosi C et al (2017) Podoplanin expression in primary brain tumors induces platelet aggregation and increases risk of venous thromboembolism. Blood 129:1831–1839. https://doi.org/10.1182/blood-2016-06-720714
Roedig H, Nastase MV, Wygrecka M, Schaefer L (2019) Breaking down chronic inflammatory diseases: the role of biglycan in promoting a switch between inflammation and autophagy. FEBS J 286:2965–2979. https://doi.org/10.1111/febs.14791
Roh JS, Sohn DH (2018) Damage-associated molecular patterns in inflammatory diseases. Immune Netw 18:e27. https://doi.org/10.4110/in.2018.18.e27
Rong Y, Durden DL, Van Meir EG, Brat DJ (2006) ‘Pseudopalisading’ necrosis in glioblastoma: a familiar morphologic feature that links vascular pathology, hypoxia, and angiogenesis. J Neuropathol Exp Neurol 65:529–539. https://doi.org/10.1097/00005072-200606000-00001
Rong Y, Hu F, Huang R, Mackman N, Horowitz JM, Jensen RL et al (2006) Early growth response gene-1 regulates hypoxia-induced expression of tissue factor in glioblastoma multiforme through hypoxia-inducible factor-1-independent mechanisms. Can Res 66:7067–7074. https://doi.org/10.1158/0008-5472.can-06-0346
Rong Y, Post DE, Pieper RO, Durden DL, Van Meir EG, Brat DJ (2005) PTEN and hypoxia regulate tissue factor expression and plasma coagulation by glioblastoma. Can Res 65:1406–1413. https://doi.org/10.1158/0008-5472.can-04-3376
Rose M, Duhamel M, Aboulouard S, Kobeissy F, Le Rhun E, Desmons A et al (2020) The role of a proprotein convertase inhibitor in reactivation of tumor-associated macrophages and inhibition of glioma growth. Mol Therapy Oncolytics 17:31–46. https://doi.org/10.1016/j.omto.2020.03.005
Ross JL, Chen Z, Herting CJ, Grabovska Y, Szulzewsky F, Puigdelloses M et al (2020) Platelet-derived growth factor beta is a potent inflammatory driver in paediatric high-grade glioma. Brain. https://doi.org/10.1093/brain/awaa382
Ross JL, Cooper LAD, Kong J, Gutman D, Williams M, Tucker-Burden C et al (2017) 5-Aminolevulinic acid guided sampling of glioblastoma microenvironments identifies pro-survival signaling at infiltrative margins. Sci Rep. https://doi.org/10.1038/s41598-017-15849-w
Ruf W (2012) Tissue factor and cancer. Thromb Res 130:S84–S87. https://doi.org/10.1016/j.thromres.2012.08.285
Ruf W, Disse J, Carneiro-Lobo TC, Yokota N, Schaffner F (2011) Tissue factor and cell signalling in cancer progression and thrombosis. J Thromb Haemost 9:306–315. https://doi.org/10.1111/j.1538-7836.2011.04318.x
Ruf W, Rothmeier AS, Graf C (2016) Targeting clotting proteins in cancer therapy—progress and challenges. Thromb Res 140:S1–S7. https://doi.org/10.1016/s0049-3848(16)30090-1
Sabelstrom H, Petri R, Shchors K, Jandial R, Schmidt C, Sacheva R et al (2019) Driving neuronal differentiation through reversal of an ERK1/2-miR-124-SOX9 axis abrogates glioblastoma aggressiveness. Cell Rep 28:2064–2079. https://doi.org/10.1016/j.celrep.2019.07.071
Scaffidi P, Misteli T, Bianchi ME (2002) Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 418:191–195. https://doi.org/10.1038/nature00858
Schaefer L, Babelova A, Kiss E, Hausser H-J, Baliova M, Krzyzankova M et al (2005) The matrix component biglycan is proinflammatory and signals through toll-like receptors 4 and 2 in macrophages. J Clin Investig 115:2223–2233. https://doi.org/10.1172/jci23755
Scheibner KA, Lutz MA, Boodoo S, Fenton MJ, Powell JD, Horton MR (2006) Hyaluronan fragments act as an endogenous danger signal by engaging TLR2. J Immunol 177:1272–1281. https://doi.org/10.4049/jimmunol.177.2.1272
Schweickert PG, Yang Y, White EE, Cresswell GM, Elzey BD, Ratliff TL et al (2021) Thrombin-PAR1 signaling in pancreatic cancer promotes an immunosuppressive microenvironment. J Thromb Haemost 19:161–172. https://doi.org/10.1111/jth.15115
Scott IC, Majithiya JB, Sanden C, Thornton P, Sanders PN, Moore T et al (2018) Interleukin-33 is activated by allergen- and necrosis-associated proteolytic activities to regulate its alarmin activity during epithelial damage. Sci Rep. https://doi.org/10.1038/s41598-018-21589-2
Seidel S, Garvalov BK, Wirta V, Von Stechow L, Schänzer A, Meletis K et al (2010) A hypoxic niche regulates glioblastoma stem cells through hypoxia inducible factor 2α. Brain 133:983–995. https://doi.org/10.1093/brain/awq042
Seo Y-S, Ko IO, Park H, Jeong YJ, Park J-A, Kim KS et al (2019) Radiation-induced changes in tumor vessels and microenvironment contribute to therapeutic resistance in glioblastoma. Front Oncol. https://doi.org/10.3389/fonc.2019.01259
Sharma I, Singh A, Siraj F, Saxena S (2018) IL-8/CXCR1/2 signalling promotes tumor cell proliferation, invasion and vascular mimicry in glioblastoma. J Biomed Sci 25:62. https://doi.org/10.1186/s12929-018-0464-y
Shlomovitz I, Erlich Z, Speir M, Zargarian S, Baram N, Engler M et al (2019) Necroptosis directly induces the release of full-length biologically active IL-33 in vitro and in an inflammatory disease model. FEBS J 286:507–522. https://doi.org/10.1111/febs.14738
Silva EJ, Argyris P, Zou X, Ross KF, Herzberg MC (2014) S100A8/A9 regulates MMP-2 expression and invasion and migration by carcinoma cells. Int J Biochem Cell Biol 55:279–287
Sims GP, Rowe DC, Rietdijk ST, Herbst R, Coyle AJ (2010) HMGB1 and RAGE in inflammation and cancer. Annu Rev Immunol 28:367–388. https://doi.org/10.1146/annurev.immunol.021908.132603
Skaga E, Kulesskiy E, Fayzullin A, Sandberg CJ, Potdar S, Kyttälä A et al (2019) Intertumoral heterogeneity in patient-specific drug sensitivities in treatment-naïve glioblastoma. BMC Cancer. https://doi.org/10.1186/s12885-019-5861-4
Snyder B, Shell B, Cunningham JT, Cunningham RL (2017) Chronic intermittent hypoxia induces oxidative stress and inflammation in brain regions associated with early-stage neurodegeneration. Physiol Rep. https://doi.org/10.14814/phy2.13258
Soda Y, Marumoto T, Friedmann-Morvinski D, Soda M, Liu F, Michiue H et al (2011) Transdifferentiation of glioblastoma cells into vascular endothelial cells. Proc Natl Acad Sci USA 108:4274–4280. https://doi.org/10.1073/pnas.1016030108
Soeda A, Park M, Lee D, Mintz A, Androutsellis-Theotokis A, McKay RD et al (2009) Hypoxia promotes expansion of the CD133-positive glioma stem cells through activation of HIF-1α. 28. Oncogene 28:3949–3959. https://doi.org/10.1038/onc.2009.252
Sorelle ED, Yecies DW, Liba O, Bennett FC, Graef CM, Dutta R et al (2019) Spatiotemporal tracking of brain-tumor-associated myeloid cells in vivo through optical coherence tomography with plasmonic labeling and speckle modulation. ACS Nano 13:7985–7995. https://doi.org/10.1021/acsnano.9b02656
Spence AM, Muzi M, Swanson KR, O’Sullivan F, Rockhill JK, Rajendran JG et al (2008) Regional hypoxia in glioblastoma multiforme quantified with [18F]Fluoromisonidazole positron emission tomography before radiotherapy: correlation with time to progression and survival. Clin Cancer Res 14:2623–2630. https://doi.org/10.1158/1078-0432.ccr-07-4995
Srikrishna G (2012) S100A8 and S100A9: new insights into their roles in malignancy. J Innate Immun 4:31–40. https://doi.org/10.1159/000330095
Stichel D, Ebrahimi A, Reuss D, Schrimpf D, Ono T, Shirahata M et al (2018) Distribution of EGFR amplification, combined chromosome 7 gain and chromosome 10 loss, and TERT promoter mutation in brain tumors and their potential for the reclassification of IDHwt astrocytoma to glioblastoma. Acta Neuropathol 136:793–803. https://doi.org/10.1007/s00401-018-1905-0
Sun C, Dai X, Zhao D, Wang H, Rong X, Huang Q et al (2019) Mesenchymal stem cells promote glioma neovascularization in vivo by fusing with cancer stem cells. BMC Cancer. https://doi.org/10.1186/s12885-019-6460-0
Sun S, He M, Wang Y, Yang H, Al-Abed Y (2018) Folic acid derived-P5779 mimetics regulate DAMP-mediated inflammation through disruption of HMGB1:TLR4:MD-2 axes. PLoS ONE 13:e0193028. https://doi.org/10.1371/journal.pone.0193028
Sun X, Liu X, Xia M, Shao Y, Zhang XD (2019) Multicellular gene network analysis identifies a macrophage-related gene signature predictive of therapeutic response and prognosis of gliomas. J Transl Med. https://doi.org/10.1186/s12967-019-1908-1
Suzuki-Inoue K (2019) Platelets and cancer-associated thrombosis: focusing on the platelet activation receptor CLEC-2 and podoplanin. Blood 134:1912–1918
Tafani M, Di Vito M, Frati A, Pellegrini L, De Santis E, Sette G et al (2011) Pro-inflammatory gene expression in solid glioblastoma microenvironment and in hypoxic stem cells from human glioblastoma. J Neuroinflamm 8:32. https://doi.org/10.1186/1742-2094-8-32
Talasila KM, Røsland GV, Hagland HR, Eskilsson E, Flønes IH, Fritah S et al (2016) The angiogenic switch leads to a metabolic shift in human glioblastoma. Neuro Oncol. https://doi.org/10.1093/neuonc/now175
Tamura R, Miyoshi H, Sampetrean O, Shinozaki M, Morimoto Y, Iwasawa C et al (2019) Visualization of spatiotemporal dynamics of human glioma stem cell invasion. Mol Brain. https://doi.org/10.1186/s13041-019-0462-3
Tamura R, Tanaka T, Akasaki Y, Murayama Y, Yoshida K, Sasaki H (2020) The role of vascular endothelial growth factor in the hypoxic and immunosuppressive tumor microenvironment: perspectives for therapeutic implications. Med Oncol. https://doi.org/10.1007/s12032-019-1329-2
Tawil N, Bassawon R, Rak J (2019) Oncogenes and clotting factors: the emerging role of tumor cell genome and epigenome in cancer-associated thrombosis. Semin Thromb Hemost 45:373–384. https://doi.org/10.1055/s-0039-1687891
Taylor KR, Yamasaki K, Radek KA, Nardo AD, Goodarzi H, Golenbock D et al (2007) Recognition of hyaluronan released in sterile injury involves a unique receptor complex dependent on toll-like receptor 4, CD44, and MD-2. J Biol Chem 282:18265–18275. https://doi.org/10.1074/jbc.m606352200
Tehrani M, Friedman TM, Olson JJ, Brat DJ (2007) Intravascular thrombosis in central nervous system malignancies: a potential role in astrocytoma progression to glioblastoma. Brain Pathol 18:164–171. https://doi.org/10.1111/j.1750-3639.2007.00108.x
Terraneo L, Samaja M (2017) Comparative response of brain to chronic hypoxia and hyperoxia. Int J Mol Sci 18:1914. https://doi.org/10.3390/ijms18091914
Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang T-H et al (2018) The immune landscape of cancer. Immunity 48:812-830.e814. https://doi.org/10.1016/j.immuni.2018.03.023
Tian J, Avalos AM, Mao S-Y, Chen B, Senthil K, Wu H et al (2007) Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol 8:487–496. https://doi.org/10.1038/ni1457
Tirosh I, Suva ML (2020) Tackling the many facets of glioblastoma heterogeneity. Cell Stem Cell 26:303–304. https://doi.org/10.1016/j.stem.2020.02.005
Trousseau A (1865) Phlegmatia alba dolens. Clinique Médicale de L’hôtel-dieu de Paris 2nd edn. J.-B. Baillière et fils, City, pp 654–712
Tu Y, Xie P, Du X, Fan L, Bao Z, Sun G et al (2019) S100A11 functions as novel oncogene in glioblastoma via S100A11/ANXA2/NF-κB positive feedback loop. J Cell Mol Med. https://doi.org/10.1111/jcmm.14574
Unruh D, Horbinski C (2020) Beyond thrombosis: the impact of tissue factor signaling in cancer. J Hematol Oncol. https://doi.org/10.1186/s13045-020-00932-z
Unruh D, Mirkov S, Wray B, Drumm M, Lamano J, Li YD et al (2019) Methylation-dependent tissue factor suppression contributes to the reduced malignancy of IDH1-mutant gliomas. Clin Cancer Res 25:747–759. https://doi.org/10.1158/1078-0432.ccr-18-1222
Unruh D, Schwarze SR, Khoury L, Thomas C, Wu M, Chen L et al (2016) Mutant IDH1 and thrombosis in gliomas. Acta Neuropathol 132:917–930. https://doi.org/10.1007/s00401-016-1620-7
Uribe D, Torres Á, Rocha JD, Niechi I, Oyarzún C, Sobrevia L et al (2017) Multidrug resistance in glioblastoma stem-like cells: role of the hypoxic microenvironment and adenosine signaling. Doi. https://doi.org/10.1016/j.mam.2017.01.009
Vabulas RM (2001) Endocytosed HSP60s use toll-like receptor 2 (TLR2) and TLR4 to activate the toll/interleukin-1 receptor signaling pathway in innate immune cells. J Biol Chem 276:31332–31339. https://doi.org/10.1074/jbc.m103217200
Velásquez C, Mansouri S, Gutiérrez O, Mamatjan Y, Mollinedo P, Karimi S et al (2019) Hypoxia can induce migration of glioblastoma cells through a methylation-dependent control of ODZ1 gene expression. Front Oncol. https://doi.org/10.3389/fonc.2019.01036
Verhaak RGW, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD et al (2010) Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17:98–110. https://doi.org/10.1016/j.ccr.2009.12.020
Vincentelli C, Hwang SN, Holder CA, Brat DJ (2012) The use of neuroimaging to guide the histologic diagnosis of central nervous system lesions. Adv Anat Pathol 19:97–107. https://doi.org/10.1097/pap.0b013e318248b747
Wainwright DA, Balyasnikova IV, Chang AL, Ahmed AU, Moon KS, Auffinger B et al (2012) IDO expression in brain tumors increases the recruitment of regulatory T cells and negatively impacts survival. Clin Cancer Res 18:6110–6121. https://doi.org/10.1158/1078-0432.ccr-12-2130
Wang P, Yan Q, Liao B, Zhao L, Xiong S, Wang J et al (2020) The HIF1α/HIF2α-miR210-3p network regulates glioblastoma cell proliferation, dedifferentiation and chemoresistance through EGF under hypoxic conditions. Cell Death Dis. https://doi.org/10.1038/s41419-020-03150-0
Wang P, Zhao L, Gong S, Xiong S, Wang J, Zou D et al (2021) HIF1α/HIF2α–Sox2/Klf4 promotes the malignant progression of glioblastoma via the EGFR–PI3K/AKT signalling pathway with positive feedback under hypoxia. Cell Death Dis. https://doi.org/10.1038/s41419-021-03598-8
Wang Q, Hu B, Hu X, Kim H, Squatrito M, Scarpace L et al (2017) Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell 32:42-56e46. https://doi.org/10.1016/j.ccell.2017.06.003
Wang Z, Shi Y, Ying C, Jiang Y, Hu J (2021) Hypoxia-induced PLOD1 overexpression contributes to the malignant phenotype of glioblastoma via NF-κB signaling. Oncogene 40:1458–1475. https://doi.org/10.1038/s41388-020-01635-y
Wei Q, Singh O, Ekinci C, Gill J, Li M, Mamatjan Y et al (2021) TNFα secreted by glioma associated macrophages promotes endothelial activation and resistance against anti-angiogenic therapy. Acta Neuropathol Commun. https://doi.org/10.1186/s40478-021-01163-0
Weinstein JR, Koerner IP, Möller T (2010) Microglia in ischemic brain injury. Future Neurol 5:227–246. https://doi.org/10.2217/fnl.10.1
Wen Y-T, Wu AT, Bamodu OA, Wei L, Lin C-M, Yen Y et al (2019) A novel multi-target small molecule, LCC-09, inhibits stemness and therapy-resistant phenotypes of glioblastoma cells by increasing miR-34a and deregulating the DRD4/Akt/mTOR signaling axis. Cancers. https://doi.org/10.3390/cancers11101442
Wight TN, Kang I, Evanko SP, Harten IA, Chang MY, Pearce OMT et al (2020) Versican—a critical extracellular matrix regulator of immunity and inflammation. Front Immunol. https://doi.org/10.3389/fimmu.2020.00512
Wippold FJ 2nd, Lämmle M, Anatelli F, Lennerz J, Perry A (2006) Neuropathology for the neuroradiologist: palisades and pseudopalisades. AJNR Am J Neuroradiol 27:2037–2041
Wojtukiewicz MZ, Mysliwiec M, Matuszewska E, Sulkowski S, Zimnoch L, Politynska B et al (2021) Imbalance in coagulation/fibrinolysis inhibitors resulting in extravascular thrombin generation in gliomas of varying levels of malignancy. Biomolecules 11:663. https://doi.org/10.3390/biom11050663
Won WJ, Deshane JS, Leavenworth JW, Oliva CR, Griguer CE (2019) Metabolic and functional reprogramming of myeloid-derived suppressor cells and their therapeutic control in glioblastoma. Cell Stress 3:47–65. https://doi.org/10.15698/cst2019.02.176
Woroniecka KI, Rhodin KE, Chongsathidkiet P, Keith KA, Fecci PE (2018) T-cell dysfunction in glioblastoma: applying a new framework. Clin Cancer Res 24:3792–3802. https://doi.org/10.1158/1078-0432.ccr-18-0047
Wu T, Luo Q, Ouyang G (2015) Periostin: a potent chemotactic factor for recruiting tumor-associated macrophage. Protein Cell 6:235–237. https://doi.org/10.1007/s13238-015-0141-9
Xie L, Yang S-H (2015) Interaction of astrocytes and T cells in physiological and pathological conditions. Brain Res 1623:63–73. https://doi.org/10.1016/j.brainres.2015.03.026
Xiong N, Li J, Yuan H, Xu H, Zhao H (2020) Hypoxic cancer-secreted exosomal miR-182-5p promotes glioblastoma angiogenesis by targeting Kruppel-like factor 2 and 4. Mol Cancer Res. https://doi.org/10.1158/1541-7786.mcr-19-0725
Xu D, Young J, Song D, Esko JD (2011) Heparan sulfate is essential for high mobility group protein 1 (HMGB1) signaling by the receptor for advanced glycation end products (RAGE). J Biol Chem 286:41736–41744. https://doi.org/10.1074/jbc.m111.299685
Xu H-S, Qin X-L, Zong H-L, He X-G, Cao L (2017) Cancer stem cell markers in glioblastoma—an update. Eur Rev Med Pharmacol Sci 21:3207–3211
Xu K, Boas DA, Sakadzic S, LaManna JC (2017) Brain tissue PO2 measurement during normoxia and hypoxia using two-photon phosphorescence lifetime microscopy. Adv Exp Med Biol 977:149–153. https://doi.org/10.1007/978-3-319-55231-6_20
Xu L, Xiao H, Xu M, Zhou C, Yi L, Liang H (2011) Glioma-derived T cell immunoglobulin- and mucin domain-containing molecule-4 (TIM4) contributes to tumor tolerance. J Biol Chem 286:36694–36699. https://doi.org/10.1074/jbc.m111.292540
Yamaki T, Shibahra I, Matsuda K-I, Kanemura Y, Konta T, Kanamori M et al (2020) Relationships between recurrence patterns and subventricular zone involvement or CD133 expression in glioblastoma. J Neurooncol 146:489–499. https://doi.org/10.1007/s11060-019-03381-y
Yamasaki R, Lu H, Butovsky O, Ohno N, Rietsch AM, Cialic R et al (2014) Differential roles of microglia and monocytes in the inflamed central nervous system. J Exp Med 211:1533–1549. https://doi.org/10.1084/jem.20132477
Yang I, Tihan T, Han SJ, Wrensch MR, Wiencke J, Sughrue ME et al (2010) CD8+ T-cell infiltrate in newly diagnosed glioblastoma is associated with long-term survival. J Clin Neurosci 17:1381–1385. https://doi.org/10.1016/j.jocn.2010.03.031
Yi L, Xiao H, Xu M, Ye X, Hu J, Li F et al (2011) Glioma-initiating cells: a predominant role in microglia/macrophages tropism to glioma. J Neuroimmunol 232:75–82. https://doi.org/10.1016/j.jneuroim.2010.10.011
Yona S, Kim K-W, Wolf Y, Mildner A, Varol D, Breker M et al (2013) fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 38:79–91. https://doi.org/10.1016/j.immuni.2012.12.001
Yu J, Zhong B, Xiao Q, Du L, Hou Y, Sun H-S et al (2020) Induction of programmed necrosis: a novel anti-cancer strategy for natural compounds. Pharmacol Ther 214:107593. https://doi.org/10.1016/j.pharmthera.2020.107593
Yu M, Wang H, Ding A, Golenbock DT, Latz E, Czura CJ et al (2006) HMGB1 signals through toll-like receptor (TLR) 4 and TLR2. Shock 26:174–179. https://doi.org/10.1097/01.shk.0000225404.51320.82
Zagzag D, Zhong H, Scalzitti JM, Laughner E, Simons JW, Semenza GL (2000) Expression of hypoxia-inducible factor 1alpha in brain tumors: association with angiogenesis, invasion, and progression. Cancer 88:2606–2618
Zhang B, Chen Y, Shi X, Zhou M, Bao L, Hatanpaa KJ et al (2020) Regulation of branched-chain amino acid metabolism by hypoxia-inducible factor in glioblastoma. Cell Mol Life Sci. https://doi.org/10.1007/s00018-020-03483-1
Zhao L, Zhang J, Liu Z, Wang Y, Xuan S, Zhao P (2021) Comprehensive characterization of alternative mRNA splicing events in glioblastoma: implications for prognosis, molecular subtypes, and immune microenvironment remodeling. Front Oncol. https://doi.org/10.3389/fonc.2020.555632
Zheng Z-Q, Chen J-T, Zheng M-C, Yang L-J, Wang J-M, Liu Q-L et al (2020) Nestin +/CD31 + cells in the hypoxic perivascular niche regulate glioblastoma chemoresistance by upregulating JAG1 and DLL4. Neuro Oncol. https://doi.org/10.1093/neuonc/noaa265
Zhou W, Ke SQ, Huang Z, Flavahan W, Fang X, Paul J et al (2015) Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat Cell Biol 17:170–182. https://doi.org/10.1038/ncb3090
Acknowledgements
This work was supported by the National Institutes of Health (DJB; R01 CA214928, R01 CA247905, P50 CA221747, U01 CA217613 and U01 CA199288) and the Cancer Research Institute Irvington Fellowship (JLR). Illustrations were created by Andrea Charest, MS using Adobe Illustrator in collaboration with the authors.
Author information
Authors and Affiliations
Contributions
Conceptualization: DJB; literature search: SMM; writing—original draft preparation: SMM; writing—revision, review and editing: SMM, JLR, CLO, and DJB.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no potential conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Markwell, S.M., Ross, J.L., Olson, C.L. et al. Necrotic reshaping of the glioma microenvironment drives disease progression. Acta Neuropathol 143, 291–310 (2022). https://doi.org/10.1007/s00401-021-02401-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-021-02401-4