
Abstract
Grafting is an important way to improve crop growth, stress resistance and yield. However, the molecular mechanism of grafting affecting flowering is not clear. In this study, roots of Paeonia broteri (PGPB) and Paeonia ostii (PGPO) were grafted with Paeonia suffruticosa ‘Luoyang Hong’ as the experimental material to measure transcriptome of the petals at a full blooming stage. A total of 41.84 Gb clean data were obtained; 80,390 unigenes and 439 differentially expressed genes (DEGs) were identified. Functional classification and enrichment of DEGs indicated that the differences in gene expression between PGPB and PGPO were mainly reflected in starch and sucrose metabolism, cell wall polysaccharides modification, redox activity, and signal transduction process. Differential expression of the hormone, protein kinase and ROS-related genes reflected the different degrees of body response of tree peony petals to different rootstocks. Besides, qRT-PCR analysis of expression profiles of the selected DEGs was in agreement with the results from RNA-seq analysis. This study investigated the molecular response of tree peony petals to grafting with two different rootstocks and laid the foundation for further study on the molecular mechanism of grafting to improve plant growth and development.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Grafting is an important cultivation method widely used in fruit crops, vegetable crops, ornamental plants and other species (Rouphael et al. 2010; Lee et al. 2010; Cabrera 2002) to improve the nutritional status of scion growth (Kumar et al. 2015), improve resistance (Colla et al. 2014; Kumar et al. 2015), promote flowering (Nienhuis and Rhodes 1977) and fruit set (Qaryouti et al. 2007). Tree peony (Paeonia suffruticosa Andr.) is a well-known ornamental plant with various flower types, rich colors and a deep flower culture in China, and grafting is also one of the main reproduction ways of ornamental tree peony (Li 1999). Excellent rootstocks can significantly improve the flowering ability of tree peony (Li 2016). But how the rootstock affects flowering, its internal mechanism is rarely known.
How did the rootstocks affect the growth and development of the scion? Plant vascular systems including xylem and phloem tissues establish links between aboveground and belowground tissues (Lough and Lucas 2006). Water, sugars, proteins, RNAs, hormones, etc. are circulated through the plant vascular systems, which function as transportation corridors between the rootstock and the scion (Spiegelman et al. 2013; Notaguchi et al. 2015). On the one hand, they are directly involved in life activities as metabolites, and on the other hand, they act as signals to indirectly regulate life activities (Martínez-Ballesta et al. 2010; Gaion and Carvalho 2017). After successful grafting, different rootstocks have different root vigor, which affects early shoot growth and leaf area development of kiwifruit (Clearwater et al. 2006). Studies have shown that by increasing the ability of the rootstock to absorb water (pay attention, nutrient uptake is intimately connected with water uptake), the growth of the scion can be increased, and the increase in the whole root system hydraulic conductance (Lsystem) may be due to an increase in hydraulic conductivity per unit surface area (Lpr) or an increase in the overall root surface area or a combination of both (Zhang et al. 2016). In addition, rootstock-induced changes in hormone levels, and/or signaling, such as cytokinin (CK), auxin (IAA), abscisic acid (ABA), reactive oxygen species (ROS), are obvious mechanisms for rootstock effects on scion growth (Gilroy et al. 2016; El-Showk et al. 2013; Aloni et al. 2010; Tworkoski and Fazio 2015).
The indexes of sugar metabolism, signal transduction, and antioxidant activity, etc. are important indicators to identify the advantages and disadvantages of grafting materials, and leaves and fruits are the parts that are of wide concern to researchers (Rouphael et al. 2010; Liu et al. 2011). Serving as the main ornamental organs and research objects, flowers also have important research value, especially for flower-viewing plants, because the condition of flower opening directly affects the growth and development of the whole plant (Jiménez et al. 2004; Van Doorn and Van Meeteren 2003; Yap et al. 2008). There were some studies on the flowering of tree peony using transcriptome. Transcriptome sequencing is important for digging up an intrinsic regulator of a physiological phenomenon. The transcriptome sequencing of tree peony focuses on petal development (Li et al. 2017), coloration mechanism (Gao et al. 2016), stress response (Zhang et al. 2017), flowering time (Hou et al. 2018), but there is little research on the molecular mechanism of petal metabolic activity affected by different rootstocks.
Prior to this article, our group has conducted a variety of studies on the physicochemical properties of two grafted tree peony. Paeonia suffruticosa ‘Luoyang Hong’ grafted on Paeonia broteri was used as the research object, because the plant height, crown width, leaf photosynthetic efficiency and flower continuous opening ability of it were significantly improved, and ‘Luoyang Hong’ grafted on Paeonia ostii was used as control (Li 2016). To further study the reason why the flowering status of P. suffruticosa ‘Luoyang Hong’ grafted on P. broteri was better than that of P. ostii, we carried out transcriptome sequencing research and determined the sugar content and antioxidant enzymes by using P. suffruticosa ‘Luoyang Hong’ petals grafted on these two different rootstocks. In this study, we used transcriptome sequencing to analyze tree peony petals on two different rootstocks and laid the foundation for the study of molecular mechanisms of plant grafting.
Materials and Methods
Plant Material
The test materials were purchased from the Zhengfeng Ecological Peony Garden in Henan Province. Roots of 3-year-old Paeonia broteri and Paeonia ostii seedlings were used as rootstocks, and the branches of 12-year-old Paeonia suffruticosa ‘Luoyang Hong’ were used as scions for grafting. P. suffruticosa ‘Luoyang Hong’ grafted on P. broteri was used as the research object, and ‘Luoyang Hong’ grafted on P. ostii was used as the control. The plants were cultivated for 5 years after grafting, and then, in 2014, ramets were planted in pots (pot size is 30 cm in upper diameter, 18 cm in lower diameter, and 25 cm in height) in the experimental field in the third area of Henan Agricultural University, and were routinely managed. At 9 a.m. on April 2017, the second day of flowers fully opening, the middle wheel petals were collected, three for each of P. suffruticosa ‘Luoyang Hong’ grafted on P. broteri (PGPB) and P. ostii (PGPO), 0.5 g per plant, then frozen in liquid nitrogen and placed in a refrigerator at − 80 °C until needed.
Petal RNA Extraction, Construction, and Sequencing of cDNA Libraries
RNA was extracted from six samples using a Total RNA Extraction Kit (Omega, Norcross, GA, USA). Construction of the cDNA library after quality testing of RNA samples was performed. After the cDNA library was tested, high-throughput sequencing was performed using the HiSeq 2500. The sequencing read was PE125.
Transcriptome Data Processing and Analysis
After raw data were filtered, the clean data were sequence assembled using Trinity software to obtain the unigene library of the species. BLAST software was used to compare the unigene sequence with the NR, Swiss-Prot, GO, COG, KOG, KEGG databases, using KOBAS 2.0 to obtain KEGG orthology results from unigenes in KEGG. After predicting the amino acid sequence of unigenes, HMMER software was used to compare with the Pfam database to obtain unigene’s annotation information. The reads obtained from the sequencing of each sample were compared with the unigene library using Bowtie, and the expression level was estimated by combining with RSEM. The fragments per kilobase of transcript per million mapped reads (FPKM) value is used to indicate the expression abundance of the corresponding unigene.
Differential Gene Expression Analysis
DESeq was used to analyze the differential expression between sample groups, and the Benjamini–Hochberg method was used to correct the p value obtained from the original hypothesis test. Finally, the corrected p value, false discovery rate (FDR), was used as a key indicator for screening DEGs. A FDR less than 0.01 and fold change (FC) of the two sample groups greater than or equal to 2 was used as the screening criteria for DEGs. Functional classification of DEGs was performed using MapMan (Thimm et al. 2004) and COG. Creating Mapping files are at http://www.plabipd.de/portal/mercator-sequence-annotation. Enrichment analysis of DEGs was annotated to the GO database using the topGO software. The enrichment factor was used to analyze the enrichment level of the DEGs in a certain pathway, and the Fisher’s exact test was used to calculate the significance of enrichment. The calculation formula for the enrichment factor is as follows:
Determination of Sugar Content and Antioxidant Enzyme Activity
The content of starch and total soluble sugar in petals was measured according to the method of Zou (1995), and determination of sucrose content used Zhang’s method (1990). Catalase (CAT), ascorbate peroxidase (APX), and peroxidase (POD) enzyme activities were determined by the method described in Wang (2006), Shen et al. (1996) and Li (2000), respectively. Three replicates were measured for each indicator, and the results were averaged. SPSS 20.0 was used to analyze the significance of differences in the values of the two sample groups.
Quantitative Real-Time PCR (qRT-PCR) Verification
Reverse transcription of RNA from two grafted peony petals was performed using the PrimeScript™ RT reagent Kit with gDNA Eraser (Perfect Real Time) (Takara, China). Specific primer design was performed at http://primer3.ut.ee/ (Table S1), and the ubiquitin gene was used as an internal reference (Liu et al. 2015). Genes were real-time quantified using SYBR® Premix Ex TaqTM II (Tli RNaseH Plus) (Takara, China) and the Applied Biosystems 7500 Fast Real-Time PCR System (Thermo Fisher, China). The cycling procedure was initiated at 94 °C for 5 min, followed by 35 cycles of 94 °C for 30 s, 57 °C for 30 s, and 72 °C for 1 min. The expression level of genes was calculated using the \({2^{ - \Delta \Delta {C_{\text{t}}}}}\) method. Each biological sample was examined in duplicate with three technical replicates.
Results
Transcriptome Sequencing Data and Functional Annotation of Unigenes
To explore the molecular mechanism of why the flowering status of P. suffruticosa ‘Luoyang Hong’ grafted on P. broteri was better than that of P. ostii, petals of P. suffruticosa ‘Luoyang Hong’ grafted on P. broteri and P. ostii were obtained for an RNA-seq analysis with three biological replicates. After quality control, a total of 41.84 Gb Clean Data was obtained from the six samples. Clean reads per sample ranged from 21,167,609 to 25,560,648 and the base number per sample ranged from 6,286,271,304 to 7,579,391,162. The percentage of GC content in each sample was 44.72% or more, and the percentage of Q30 bases was 95.50% or more (Table 1). A total of 80,390 unigenes were obtained after sequence assembly. Sequence alignments were performed between clean data of each sample and assembled transcripts or the unigene library. Mapped reads per sample ranged from 16,966,461 to 20,499,499. The percentage of the mapped ratio of each sample was 79.23% or more (Table 1). Total length of unigenes was 62,347,267 nt, with a mean of 775.56 nt. N50 of unigenes was 1249 nt (Fig. 1).

Unigene length distribution. N50 of 1249 nt was designated with a red vertical line. (Color figure online)
To understand the gene expression status of tree peony petals at the full blooming stage, functional characterizations of the sequencing results of petals grafted on two different rootstocks were performed. By selecting the BLAST parameters E-value no more than 1e−5 and HMMER parameters E value no more than 1e−10, the 30,421 unigenes were annotated into eight public databases out of 80,390 unigenes, accounting for 37.84% (Table 2). The number of unigenes annotated to the NR database (28,744, 35.76%) is the largest, followed by EggNOG (27,048, 33.65%), and the least is COG (8914, 11.09%).
Exploration of DEGs
Based on the RPKM of six samples, 439 DEGs were obtained; 98 were up-regulated, 341 were down-regulated (Fig. 2a), and 380 DEGs of them had annotated information. Hierarchical clustering of DEGs results in two major categories, one category contains 381 (86.79%) DEGs, and the overall expression of these genes is low, and the difference between the two sample groups was not significant. The other category contains 58 (13.21%) DEGs, the expression of these genes is relatively high, and the difference is larger in the two sample groups (Fig. 2b). According to the result of DEGs annotation, 43 DEGs had annotated information in 58 DEGs, and these DEGs were more involved in carbohydrate metabolism, cell wall modification, signaling and so on.

Statistical analysis and hierarchical clustering of DEGs. a Volcano plot of 439 DEGs. Each point in the figure represents a gene, the abscissa represents the logarithm of the multiple of the expression of a gene in the two samples, and the ordinate represents the negative logarithm of the false discovery rate. b Hierarchical clustering of 439 DEGs. Different columns in the figure represent different samples, and different lines represent different genes. The color represents the base 2 logarithms of the FPKM of the genes in the sample
Functional Classification and Enrichment of DEGs
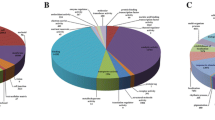
For the preliminary classification of 439 DEGs, the DEGs were first classified using the MapMan tool (Fig. 3). Among the 23 functional classifications of the DEGs, the top five are “secondary metabolism”, “lipid metabolism”, “transport”, “cell wall” and “hormone metabolism”(p value ≤ 0.05). Combined with 30,421 unigene functional annotation results, 380 out of 439 DEGs have annotation information. And then, through the COG database, all DEGs were classified into 18 categories (Fig. 4). In addition to the “general function prediction only” categories, the number of DEGs in “transcription” (22, 10.84%) and “replication, recombination and repair” (21, 10.34%) was greatest, and the “carbohydrate transport and metabolism” (20, 9.85%), “signal transduction mechanism” (20, 9.85%) and “secondary metabolite biosynthesis, transport, and metabolism” (20, 9.85%) followed. Enrichment analysis of DEGs annotated to the GO database indicated that under the first-level node, MF, the nodes with DEGs among the top ten nodes that were significantly enriched were “peroxidase activity”, “nucleic acid binding”, “chitin binding” and “ATPase activity, coupled to transmembrane movement of substances”, similarly, under CC the notes were “chloroplast thylakoid membrane”, “extracellular region”, under BP the notes were “plant-type cell wall modification” (Fig. S1). The results of the KEGG pathway enrichment analysis of DEGs indicated that the level of enrichment of DEGs in “starch and sucrose metabolism” and “pentose and glucuronate interconversions” pathway was significant and reliable (Fig. 5) (Fig. S2).

Function distribution of the significant DEGs. Numbers in the figure indicate the number of DEGs in each functional category

COG function classification of DEGs. Numbers in the figure indicate the number and percentage of DEGs in each functional category

DEGs KEGG pathway enrichment scatter plot. Each figure in the figure represents a KEGG path, and the path name is shown in the legend on the right. The abscissa is the enrichment factor, which represents the ratio of the proportion of genes annotated to a pathway in a differential gene to the proportion of genes in all genes annotated to that pathway. The ordinate is log10 (Q value), where Q value is the p value after the multiple hypothesis test correction. The first 19 pathways with the most reliable (that is, the smallest Q) enrichment were selected for the results
Screening of DEGs
Based on the functional classification and enrichment of DEGs, we can find that the expression levels of sugar transport and metabolism-related genes (starch, sucrose and cell wall polysaccharides, etc.) are significantly different in the petals of PGPB and PGPO, accompanied by differential expression of redox- and signaling-related genes.
The carbon source required for petal metabolism is transported from roots and leaves through the symplast and apoplast pathways. In the process of sucrose unloading through the apoplast pathway in the petal organ, CWINV1, INV1, SWEET5 and STP14 were down-regulated in PGPB compared with that of PGPO, and during starch degradation, BAM2, PHSL1, PHSH were up-regulated in PGPB compared with PGPO, whereas BAM3 had lower expression levels in PGPB compared with PGPO (Fig. 6a). Starch, total soluble sugar and sucrose contents in petals of two grafted tree peonies at a full flowering stage were determined. The starch content in petals of PGPB (44.30 mg/g) was significantly higher than that of PGPO (31.19 mg/g) (p < 0.05). The total soluble sugar content in petals of PGPB (325.77 mg/g) was significantly lower than that of PGPO (374.27 mg/g) (p < 0.05). And the sucrose content in petals of PGPB (44.76 mg/g) was significantly higher than that of PGPO (30.40 mg/g) (p < 0.05) (Fig. 6b). The difference in sugar content reflects the preference of two grafted tree peony for regulating sugar storage and utilization during the same period.

Screening of DEGs and determination of related physiological indicators. a Heatmap of the DEGs assigned to starch, sucrose and cell wall polysaccharides metabolism and redox and signal transduction pathways in the PGPB and PGPO. The FPKM values of the unigenes were log2 transformed. Red and blue indicate up- and down-regulated transcripts, respectively. b Various sugar content in petals of two grafted tree peonies. c CAT, APX and POD activity in petals of two grafted tree peonies. “*” and “**” indicate the significant differences and extremely significant differences between PGPB and PGPO at P < 0.05 and P < 0.01. The vertical bars represent the standard errors, n = 3
The metabolic activities of cell wall polysaccharides such as pectin and xyloglucan affect the cell homeostasis of petals by changing the ductility and permeability of cell walls (O’Donoghue and Sutherland 2012). Among the 21 DEGs involved in cell wall polysaccharide modification, 16 (76.19%) were directly involved in the decomposition of pectin, the glue in the cell wall, especially PME and PGLR were down-regulated in PGPB compared with PGPO (Fig. 6a). The decomposition of pectin can affect cell wall homeostasis on the one hand, and the oligogalacturonides (OGs), a decomposition product, can induce other reactions in combination with signaling factors (Ferrari et al. 2013).
Plant feels the natural environment all the time and can change their physiology and development in response to environmental factors (Ruberti et al. 2012). Protein kinases play an important role in the signal response process, especially receptor-like kinases (RLKs) play a key role in sensing external signals and activating a series of body reactions (Osakabe et al. 2013). In addition, RLKs have been known to have a major role in integrating environmental and plant hormone signaling (Diévart and Clark 2004). Among the 42 DEGs screened for signal transduction, there were 24 protein kinase-related DEGs and 18 hormone-related genes (Fig. 6a). Compared with PGPO, most of the protein kinase-related genes were down-regulated in PGPB (22, 91.67%), with only two up-regulated expressions (c57931.graph_c0 and c77169.graph_c0). Eighteen hormone-related DEGs were mainly divided into 4 categories: abscisic acid (synthesis-degradation: c60473.graph_c0, c82044.graph_c0; signal transduction: c66489.graph_c0, c77707.graph_c0), ethylene (synthesis-degradation: c67732.graph_c0; signal transduction: c67532.graph_c0, c70393.graph_c0, c72078.graph_c0), auxin (synthesis-degradation: c59738.graph_c0, c76099.graph_c0, c77195.graph_c0; signal transduction: c84099.graph_c0, c84099.graph_c1) and brassinosteroid (synthesis-degradation: c51816.graph_c0, c52056.graph_c0, c66214.graph_c0, c80431.graph_c0; signal transduction: c69403.graph_c0). The four hormone synthesis-degradation-related genes had both up-regulated and down-regulated expressions, whereas signal transduction-related genes were down-regulated in PGPB compared with PGPO, except c66489.graph_c0.
Environmental changes often cause plant redox imbalance, resulting in a rapid generation of oxidative stress resulting from the high production of reactive oxygen species (ROS), and the ROS is usually scavenged by enzymatic and non-enzymatic components (Blokhina et al. 2003). There were 8 DEGs involved in the redox reaction (Fig. 6a), in which APX and POD were up-regulated in PGPB compared with PGPO, which is consistent with the APX and POD activity in petals of PGPB being extremely significantly higher than that of PGPO (p < 0.01) (Fig. 6c), and no significant difference was found in the expression levels of CAT in PGPB and PGPO. The remaining 6 DEGs (c73943.graph_c0, c65195.graph_c0, c56353.graph_c0, c60267.graph_c0, c69471.graph_c0, c70653.graph_c0) involved in redox reduction were down-regulated in PGPB compared with PGPO.
Quantitative Real-Time PCR Validation of DEGs
To validate the transcriptome sequencing results, 12 DEGs were selected for qRT-PCR analysis. Details of these genes are listed in Table S1, and qRT-PCR results are presented in Fig. 7. According to the unigene annotation information, these 12 DEGs are mainly involved in pectin decomposition (c70889.graph_c0, c80163.graph_c0, c83387.graph_c0), anthocyanin metabolism (c67593.graph_c0, c75643.graph_c0), hormone metabolism (c59738.graph_c0, c76593.graph_c0, c67732.graph_c0) and antioxidant activity (c83515.graph_c0, c63288.graph_c0, c83513.graph_c0, c82527.graph_c0). According to the results of qRT-PCR, c67593.graph_c0, c75643.graph_c0 and c59738.graph_c0 were up-regulated in PGPB compared with PGPO, and the remaining 9 DEGs were down-regulated in PGPB. Overall, 12 genes showed similar expression patterns in PGPB and PGPO RNA-seq data compared with qRT-PCR results.

qRT-PCR results of 12 DEGs in PGPB and PGPO. Error bars represent standard errors from three technical replicates
Discussion
Grafting is one of the effective ways to improve the state of growth, stress resistance and yield of plants. After grafting, the rootstock regulates growth by transferring metabolites and signal substances to the aboveground part. In the literature, we found that the reported experimental studies on grafting were mainly focused on leaves and fruits, whereas the study of petals of grafted plants was rare (Rouphael et al. 2010; Lee et al. 2010; Schwarz et al. 2010; Liu et al. 2011; Kumar et al. 2015; Notaguchi et al. 2015; Gaion and Carvalho 2017). In this study, the transcription of tree peony petals on two different rootstocks was studied by means of transcriptome sequencing. Transcriptome results yielded 41.84 Gb clean data, and 80,390 unigenes were obtained after sequence assembly. Based on the RPKM of PGPB and PGPO, 439 DEGs were obtained, 98 were up-regulated, and 341 were down-regulated. In theory, a rootstock has no robust effect on scion growth only when it should hold up under different environmental conditions, and even different scions (at least relative other rootstocks) (Zhang et al. 2016). The number of DEGs in this study is much less than the previous measurements of our group (Li et al. 2017), indicating that only the difference in the rootstock does not differ from the growth period, causing a large number of DEGs in the tree peony petals. According to the functional classification and enrichment of DEGs, the gene expression differences of tree peony petals at a full blooming stage on two different rootstocks mainly focused on carbohydrate transport and metabolism, signal transduction and redox.
Sugar, serving as the junction between an energy substance and a signal in a complex metabolic network, is the basal metabolite of plant life activities, and sugar transport and metabolism research is an important part of studying rootstock-scion interactions (Martínez-Ballesta et al. 2010; Ruan et al. 2010; Bolouri-Moghaddam et al. 2010). The sugar source of petals is mainly from roots and leaves, and it is consumed as a substrate for respiration, or it is temporarily stored in the form of such sugar polymers as starch and cellulose (Ruan et al. 2010). The different sink strength influences source-sink relationships in plants and sugar allocation (Wardlaw 2010). During flowering, petal cells rapidly grow and differentiate to give them a stronger pool strength for nutrients from roots and leaves. Studies have shown that sucrose is the main form of long-distance transport of carbohydrates between plant sources and sinks (Liu et al. 2012). In this study, the sucrose content in petals of PGPB was significantly higher than that of PGPO, indicating that the roots and leaves of the PGPB provide more sucrose for the petals (Li 2016). Meanwhile, by screening the petal transcriptome data of PGPB and PGPO at a full flowering stage, it was found that the invertase gene (CWINV1, INV1) was down-regulated in PGPB in DEGs. Invertase is one of the most important enzymes to degrade sucrose (Ruan et al. 2010), so, down-regulated expression of CWINV1 and INV1 in PGPB may contribute to the preservation of sucrose. The content of starch in PGPB petals is significantly higher than that of PGPO, and β-amylase (BAM) or starch phosphorylase may play a regulatory role in it. In Arabidopsis, BAM2 was reported to have extremely low catalytic activity, and BAM3 played important roles in hydrolyzing starch to maltose in mesophyll cell chloroplasts (Li et al. 2009; Monroe et al. 2014; Fulton et al. 2008). Subsequently, maltose was degraded through the sequential actions of disproportionating enzyme (DPE2) and cytosolic starch phosphorylase to produce Glc-1P (Lu et al. 2006). In this study, BAM3 was the down-regulated in PGPB compared with that of PGPO, PHSL1 and PHSH were the opposite. This indicates that the rate of decomposition of starch into maltose in PGPB was low and the rate at which maltose continues to decompose into Glc-1P was high. And the increase in Glc-1P content reverses the inhibition of starch degradation. In addition, the up-regulation of ENPP in PGPB might increase the downstream product Glc-1P of ENPP enzyme, whereas the increase in Glc-1P will inhibit the decomposition of starch in the reverse direction.
In addition, cell wall modification is an important aspect of plant acclimation to environmental change (Sasidharan et al. 2011). As a glue for cell walls, pectin, and its dynamic changes affect the homeostasis of the cell wall (Caffall and Mohnen 2009). In previous studies, pectinesterase (PME) and polygalacturonase (PG) were considered key enzymes in the breakdown of pectin (Lionetti et al. 2007; Almeida and Huber 2007). Herein, the enrichment of metabolic pathways by DEGs using KEGG showed that cell wall pectin decomposition was significantly enriched, and up-regulation of PME, PGLR in PGPO petals suggests that the cell wall pectin in the PGPO petals is continuously decomposed. The action of PG degrades polygalacturonan into oligogalacturonides (OGs), which then bind to the OGs receptor, wall-associated receptor kinase (WAKs), thereby activating a series of receptor-like kinases (RLKs) to regulate the expression of stress-related genes (Ferrari et al. 2013; Kohorn and Kohorn 2012). In this study, the up-regulation of WAKLB, WAKLI, RLKs and other genes in PGPO suggests that the stress response induced by pectin decomposition has occurred. Therefore, in contrast, the stable cell wall state due to less decomposition of pectin may be the reason for the longer growth of PGPB petals.
Grafted plants complete rootstock-scion interactions through signal substances such as hormones and reactive oxygen species (ROS) (Aloni et al. 2010; Gilroy et al. 2016). There are many kinds of hormones in plants, such as gibberellin (GA), auxin (IAA), cytokinin (CK), brassinosteroid (BR), abscisic acid (ABA) and ethylene (ET). The ultimate function of each hormone is achieved through complex interactions and feedback regulation networks (Vanstraelen and Benková 2012). The mechanisms of hormone response to plant biotic and abiotic stress have been extensively studied. NILR1 (response to nematodes) (Mendy et al. 2017), ABA response-related genes (response to Agrobacterium) (Efetova et al. 2007) and AKR1 (response to Rhizobium) (Hur et al. 2009) play important roles in the process of biotic stress. At the same time, PLY and ERF act as the response element to ABA and ethylene, respectively, and play an important role in the stress process (Park et al. 2009; Bolt et al. 2017). In this study, differential expression of GEML4, PYL4, ERF5, EF105, ERF92, AKR1 and Y1743 indicates that certain stress factors may appear in grafted tree peony and exhibit different gene expression patterns in PGPB and PGPO. This stress factor may be derived from the root zone limitation. The process of plant redox reaction is accompanied by the production of ROS, which can act as signal-regulating substances, but also cause oxidative damage to plant tissues (Gilroy et al. 2016; Schippers et al. 2016; Mittler et al. 2011). Enzymatic antioxidants (APX, POD, etc.), as endogenous protective mechanisms, work in a complex cooperative network to reduce the cytotoxic effects of ROS in plant cells (Blokhina et al. 2003). In this study, APX and POD were up-regulated in PGPB compared to PGPO, which was positively associated with a higher activity of APX and POD in PGPB. This indicated that during the full blooming stage, the petals of tree peony grafted on P. broteri had a stronger ability to scavenge ROS to maintain the balance of the redox metabolism. Some transcriptome analyses suggested that the RLKs can be considered as key regulators in growth and developmental processes as well as in various environmental stress responses (Marshall et al. 2012; Osakabe et al. 2013; Cui et al. 2018). Pitorre et al. have shown that receptor-like kinase (RLK7), proline-rich extensin-like receptor kinase (PERK4) and cysteine-rich receptor-like kinase (CRK36) interact with ROS and ABA, respectively, to regulate signaling pathways to adapt to environmental changes (Tanaka et al. 2012; Pitorre et al. 2010; Bai et al. 2009). In this study, the up-regulated expression of protein kinase genes in PGPO compared with PGPB, such as RLK, PERK5, CRK10, CRK25, etc. related to stress response, indicates that P. ostii had a negative impact on the grafted tree peony petals, while its cell wall pectin degradation releases OGs, which triggers up-regulation of WAKs and may also trigger the expression of downstream stress-related genes.
In summary, we conducted a comprehensive analysis of tree peony petals on different rootstocks using RNA-seq. There were hundreds of DEGs mainly involved in starch and sucrose metabolism, cell wall polysaccharide modification and the signal transduction process involving hormones, ROS and protein kinases. This study provides a comprehensive insight into how different rootstocks affect the metabolism of petals at molecular levels and lays the foundation for further study on the molecular mechanism of grafting to improve plant growth and development.
References
Almeida DFP, Huber DJ (2007) Polygalacturonase-mediated dissolution and depolymerization of pectins in solutions mimicking the pH and mineral composition of tomato fruit apoplast. Plant Sci 172:1087–1094. https://doi.org/10.1016/j.plantsci.2007.03.008
Aloni B, Cohen R, Karni L, Aktas H, Edelstein M (2010) Hormonal signaling in rootstock-scion interactions. Sci Hortic 127:119–126. https://doi.org/10.1016/j.scienta.2010.09.003
Bai L, Zhang G, Zhou Y, Zhang Z, Wang W, Du Y, Wu Z, Song CP (2009) Plasma membrane-associated proline-rich extensin-like receptor kinase 4, a novel regulator of Ca2+ signalling, is required for abscisic acid responses in Arabidopsis thaliana. Plant J 60:314–327. https://doi.org/10.4161/psb.4.11.9739
Blokhina O, Virolainen E, Fagerstedt KV (2003) Antioxidants, oxidative damage and oxygen deprivation stress: a review. Ann Bot 91:179–194. https://doi.org/10.1093/aob/mcf118
Bolouri-Moghaddam MR, Le Roy K, Xiang L, Rolland F, Van den Ende W (2010) Sugar signalling and antioxidant network connections in plant cells. FEBS J 277:2022–2037. https://doi.org/10.1111/j.1742-4658.2010.07633.x
Bolt S, Zuther E, Zintl S, Hincha DK, Schmülling T (2017) ERF105 is a transcription factor gene of Arabidopsis thaliana required for freezing tolerance and cold acclimation. Plant Cell Environ 40:108–120. https://doi.org/10.1111/pce.12838
Cabrera RI (2002) Rose yield, dry matter partitioning and nutrient status responses to rootstock selection. Sci Hortic 95:75–83. https://doi.org/10.1016/S0304-4238(02)00020-1
Caffall KH, Mohnen D (2009) The structure, function, and biosynthesis of plant cell wall pectic polysaccharides. Carbohydr Res 344:1879–1900. https://doi.org/10.1016/j.carres.2009.05.021
Clearwater MJ, Seleznyova AN, Thorp TG, Blattmann P, Barnett AM, Lowe RG, Austin PT (2006) Vigor-controlling rootstocks affect early shoot growth and leaf area development of kiwifruit. Tree Physiol 26:505–515. https://doi.org/10.1093/treephys/26.4.505
Colla G, Fiorillo A, Cardarelli M, Rouphael Y (2014) Grafting to improve abiotic stress tolerance of fruit vegetables. Sci Hortic 1041:119–126. https://doi.org/10.1080/17421772.2014.961536
Cui JH, Ren GZ, Qiao HY, Xiang XD, Huang LS, Chang JH (2018) Comparative transcriptome analysis of seedling stage of two sorghum cultivars under salt stress. J Plant Growth Regul 37:986–998. https://doi.org/10.1007/s00344-018-9796-9
Diévart A, Clark SE (2004) LRR-containing receptors regulating plant development and defense. Development 131:251–261. https://doi.org/10.1242/dev.00998
Efetova M, Zeier J, Riederer M, Lee CW, Stingl N, Mueller M, Hartung W, Hedrich R, Deeken R (2007) A central role of abscisic acid in drought stress protection of Agrobacterium-induced tumors on Arabidopsis. Plant Physiol 145:853–862. https://doi.org/10.1104/pp.107.104851
El-Showk S, Ruonala R, Helariutta Y (2013) Crossing paths: cytokinin signalling and crosstalk. Development 140:1373–1383. https://doi.org/10.1242/dev.086371
Ferrari S, Savatin DV, Sicilia F, Gramegna G, Cervone F, Lorenzo GD (2013) Oligogalacturonides: plant damage-associated molecular patterns and regulators of growth and development. Front Plant Sci 4:49. https://doi.org/10.3389/fpls.2013.00049
Fulton DC, Stettler M, Mettler T, Vaughan CK, Li J, Francisco P, Gil M, Reinhold H, Eicke S, Messerli G, Dorken G, Halliday K, Smith AM, Smith SM, Zeeman SC (2008) Beta-AMYLASE4, a noncatalytic protein required for starch breakdown, acts upstream of three active beta-amylases in Arabidopsis chloroplasts. Plant Cell 20:1040–1058. https://doi.org/10.1105/tpc.107.056507
Gaion LA, Carvalho RF (2017) Long-distance signaling: what grafting has revealed? J Plant Growth Regul 37:694–704. https://doi.org/10.1007/s00344-017-9759-6
Gao LX, Yang HX, Liu HF, Yang J, Hu YH (2016) Extensive transcriptome changes underlying the flower color intensity variation in Paeonia ostii. Front Plant Sci 6:1205. https://doi.org/10.3389/fpls.2015.01205
Gilroy S, Białasek M, Suzuki N, Górecka M, Devireddy AR, Karpiński S, Mittler R (2016) ROS, calcium and electric signals: key mediators of rapid systemic signaling in plants. Plant Physiol 171:1606–1615. https://doi.org/10.1104/pp.16.00434
Hou XG, Guo Q, Wei WQ, Guo LL, Lguo DL, Zhang L (2018) Screening of genes related to early and late flowering in tree peony based on bulked segregant RNA sequencing and verification by quantitative real-time PCR. Molecules 23:689. https://doi.org/10.3390/molecules23030689
Hur YS, Shin KH, Kim S, Nam KH, Lee MS, Chun JY, Cheon CI (2009) Overexpression of GmAKR1, a stress-induced aldo/keto reductase from soybean, retards nodule development. Mol Cells 27:217–223. https://doi.org/10.1007/s10059-009-0027-x
Jiménez S, Garín A, Gogorcena Y, Betrán J, Moreno M (2004) Flower and foliar analysis for prognosis of sweet cherry nutrition: influence of different rootstocks. J Plant Nutr 27:701–712. https://doi.org/10.1081/PLN-120030376
Kohorn BD, Kohorn SL (2012) The cell wall-associated kinases, WAKs, as pectin receptors. Front Plant Sci 3:88. https://doi.org/10.3389/fpls.2012.00088
Kumar P, Lucini L, Rouphael Y, Cardarelli M, Kalunke RM, Colla G (2015) Insight into the role of grafting and arbuscular mycorrhiza on cadmium stress tolerance in tomato. Front Plant Sci 6:477. https://doi.org/10.3389/fpls.2015.00477
Lee JM, Kubota C, Tsao SJ, Bie Z, Echevarria PH, Morra L, Oda M (2010) Current status of vegetable grafting: Diffusion, grafting techniques, automation. Sci Hortic 127:93–105. https://doi.org/10.1016/j.scienta.2010.08.003
Li JY (1999) Chinese tree peony and herbaceous peony. China Forestry Press, Beijing
Li HS (2000) Principles and techniques of plant physiological and biochemical experiments. Higher Education Press, Beijing
Li Y (2016) The phloem sucrose transporting mechanism and key genes mining on Paeonia suffruticosa. Dissertation, Henan Agricultural University
Li J, Francisco P, Zhou W, Edner C, Steup M, Ritte G, Bond CS, Smith SM (2009) Catalytically-inactive beta-amylase BAM4 required for starch breakdown in Arabidopsis leaves is a starch-binding-protein. Arch Biochem Biophys 489:92–98. https://doi.org/10.1016/j.abb.2009.07.024
Li Y, Lu JX, Chang YH, Tang WL, Yang QS (2017) Comparative analysis of tree peony petal development by transcriptome sequencing. Acta Physiol Plant 39:216. https://doi.org/10.1007/s11738-017-2520-8
Lionetti V, Raiola A, Camardella L, Giovane A, Obel N, Pauly M, Favaron F, Cervone F, Bellincampi D (2007) Overexpression of pectin methylesterase inhibitors in Arabidopsis restricts fungal infection by Botrytis cinerea. Plant Physiol 143:1871–1880. https://doi.org/10.1104/pp.106.090803
Liu YF, Qi HY, Bai CM, Qi MF, Xu CQ, Hao JH, Li Y, Li TL (2011) Grafting helps improve photosynthesis and carbohydrate metabolism in leaves of muskmelon. Int J Biol Sci 7:1161–1170. https://doi.org/10.7150/ijbs.7.1161
Liu DD, Chao WM, Turgeon R (2012) Transport of sucrose, not hexose, in the phloem. J Exp Bot 63:4315. https://doi.org/10.1093/jxb/ers127
Liu CJ, Wang SL, Xue JQ, Zhu FY, Ren XX, Li MY, Zhang XX (2015) Molecular cloning of Ubiquitin protein gene and study on this gene as reference gene in tree peony. Acta Hortic Sin 42:1983–1992. https://doi.org/10.16420/j.issn.0513-353x.2015-0180
Lough TJ, Lucas WJ (2006) Integrative plant biology: role of phloem long-distance macromolecular trafficking. Annu Rev Plant Biol 57:203–232. https://doi.org/10.1146/annurev.arplant.56.032604.144145
Lu Y, Steichen JM, Yao J, Sharkey TD (2006) The role of cytosolic alpha-glucan phosphorylase in maltose metabolism and the comparison of amylomaltase in Arabidopsis and Escherichia coli. Plant Physiol 142:878–889. https://doi.org/10.1104/pp.106.086850
Marshall A, Aalen RB, Audenaert D, Beeckman T, Broadley MR, Butenko MA, Caño-Delgado AI, de Vries S, Dresselhaus T, Felix G, Graham NS, Foulkes J, Granier C, Greb T, Grossniklaus U, Hammond JP, Heidstra R, Hodgman C, Hothorn M, Inzé D, Ostergaard L, Russinova E, Simon R, Skirycz A, Stahl Y, Zipfel C, De Smet I (2012) Tackling drought stress: receptor-like kinases present new approaches. Plant Cell 24:2262–2278. https://doi.org/10.1105/tpc.112.096677
Martínez-Ballesta M, Alcaraz-López C, Muries B, Mota-Cadenas C, Carvajal M (2010) Physiological aspects of rootstock-scion interactions. Sci Hortic 127:112–118. https://doi.org/10.1016/j.scienta.2010.08.002
Mendy B, Wang’ombe MW, Radakovic ZS, Holbein J, Ilyas M, Chopra D, Holton N, Zipfel C, Grundler FM, Siddique S (2017) Arabidopsis leucine-rich repeat receptor-like kinase NILR1 is required for induction of innate immunity to parasitic nematodes. PLoS Pathog 13:e1006284. https://doi.org/10.1371/journal.ppat.1006284
Mittler R, Vanderauwera S, Suzuki N, Miller G, Tognetti VB, Vandepoele K, Gollery M, Shulaev V, Van Breusegem F (2011) ROS signaling: the new wave? Trends Plant Sci 16:300–309. https://doi.org/10.1016/j.tplants.2011.03.007
Monroe JD, Storm AR, Badley EM, Lehman MD, Platt SM, Saunders LK, Schmitz JM, Torres CE (2014) β-Amylase1 and β-amylase3 are plastidic starch hydrolases in Arabidopsis that seem to be adapted for different thermal, pH, and stress conditions. Plant Physiol 166:1748–1763. https://doi.org/10.1104/pp.114.246421
Nienhuis J, Rhodes AM (1977) Interspecific grafting to enhance flowering in wild species of Cucurbita. HortScience 12:458–459
Notaguchi M, Higashiyama T, Suzuki T (2015) Identification of mRNAs that move over long distances using an RNA-Seq analysis of Arabidopsis/Nicotiana benthamiana heterografts. Plant Cell Physiol 56:311–321. https://doi.org/10.1093/pcp/pcu210
O’Donoghue EM, Sutherland PW (2012) Cell wall polysaccharide distribution in Sandersonia aurantiaca flowers using immuno-detection. Protoplasma 249:843–849. https://doi.org/10.1007/s00709-011-0307-0
Osakabe Y, Yamaguchi-Shinozaki K, Shinozaki K, Tran LS (2013) Sensing the environment: key roles of membrane-localized kinases in plant perception and response to abiotic stress. J Exp Bot 64:445–458. https://doi.org/10.1093/jxb/ers354
Park SY, Fung P, Nishimura N, Jensen DR, Fujii H, Zhao Y, Lumba S, Santiago J, Rodrigues A, Chow TF, Alfred SE, Bonetta D, Finkelstein R, Provart NJ, Desveaux D, Rodriguez PL, McCourt P, Zhu JK, Schroeder JI, Volkman BF, Cutler SR (2009) Abscisic acid inhibits type 2C protein phosphatases via the PYR/PYL family of START proteins. Science 324:1068–1071. https://doi.org/10.1126/science.1173041
Pitorre D, Llauro C, Jobet E, Guilleminot J, Brizard JP, Delseny M, Lasserre E (2010) RLK7, a leucine-rich repeat receptor-like kinase, is required for proper germination speed and tolerance to oxidative stress in Arabidopsis thaliana. Planta 232:1339–1353. https://doi.org/10.1007/s00425-010-1260-4
Qaryouti MM, Qawasmi W, Hamdan H, Edwan M (2007) Tomato fruit yield and quality as affected by grafting and growing system. Acta Hortic. https://doi.org/10.17660/ActaHortic.2007.741.22
Rouphael Y, Schwarz D, Krumbein A, Colla G (2010) Impact of grafting on product quality of fruit vegetables. Sci Hortic 127:172–179. https://doi.org/10.1016/j.scienta.2010.09.001
Ruan YL, Jin Y, Yang YJ, Li GJ, Boyer JS (2010) Sugar input, metabolism, and signaling mediated by invertase: roles in development, yield potential, and response to drought and heat. Mol Plant 03:942–955. https://doi.org/10.1093/mp/ssq044
Ruberti I, Sessa G, Ciolfi A, Possenti M, Carabelli M, Morelli G (2012) Plant adaptation to dynamically changing environment: the shade avoidance response. Biotechnol Adv 30:1047–1058. https://doi.org/10.1016/j.biotechadv.2011.08.014
Sasidharan R, Voesenek L, Pierik R (2011) Cell wall modifying proteins mediate plant acclimatization to biotic and abiotic stresses. Crit Rev Plant Sci 30:548–562. https://doi.org/10.1080/07352689.2011.615706
Schippers JH, Foyer CH, van Dongen JT (2016) Redox regulation in shoot growth, SAM maintenance and flowering. Curr Opin Plant Biol 29:121–128. https://doi.org/10.1016/j.pbi.2015.11.009
Schwarz D, Rouphael Y, Colla G, Venema JH (2010) Grafting as a tool to improve tolerance of vegetables to abiotic stresses: thermal stress, water stress and organic pollutants. Sci Hortic 127:162–171. https://doi.org/10.1016/j.scienta.2010.09.016
Shen WB, Xu LL, Yang MB, Zeng RX (1996) Study on determination of ASP activity. Plant Physiol Commun. https://doi.org/10.13592/j.cnki.ppj.1996.03.016
Spiegelman Z, Golan G, Wolf S (2013) Don’t kill the messenger: long-distance trafficking of mRNA molecules. Plant Sci 213:1–8. https://doi.org/10.1016/j.plantsci.2013.08.011
Tanaka H, Osakabe Y, Katsura S, Mizuno S, Maruyama K, Kusakabe K, Mizoi J, Shinozaki K, Yamaguchi-Shinozaki K (2012) Abiotic stress-inducible receptor-like kinases negatively control ABA signaling in Arabidopsis. Plant J 70:599–613. https://doi.org/10.1111/j.1365-313X.2012.04901.x
Thimm O, Bläsing O, Gibon Y, Nagel A, Meyer S, Krüger P, Selbig J, Müller LA, Rhee SY, Stitt M (2004) MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J 37:914–939. https://doi.org/10.1111/j.1365-313X.2004.02016.x
Tworkoski T, Fazio G (2015) Effects of size-controlling apple rootstocks on growth, abscisic acid, and hydraulic conductivity of scion of different vigor. Int J Fruit Sci 15:369–381. https://doi.org/10.1080/15538362.2015.1009973
Van Doorn WG, Van Meeteren U (2003) Flower opening and closure: a review. J Exp Bot 54:1801–1812. https://doi.org/10.1093/jxb/erg213
Vanstraelen M, Benková E (2012) Hormonal interactions in the regulation of plant development. Annu Rev Cell Dev Biol 28:463–487. https://doi.org/10.1146/annurev-cellbio-101011-155741
Wang XK (2006) Principles and techniques of plant physiological biochemical experiment. Higher Education Press, Beijing
Wardlaw IF (2010) Tansley Review No. 27. The control of carbon partitioning in plants. New Phytol 116:341–381. https://doi.org/10.1111/j.1469-8137.1990.tb00524.x
Yap Y, Loh C, Ong B (2008) Regulation of flower development in Dendrobium crumenatum by changes in carbohydrate contents, water status and cell wall metabolism. Sci Hortic 119:59–66. https://doi.org/10.1016/j.scienta.2008.06.029
Zhang ZL (1990) Plant physiology experiment guide. Higher Education Press, Beijing
Zhang L, Marguerit E, Rossdeutsch L, Ollat N, Gambetta GA (2016) The influence of grapevine rootstocks on scion growth and drought resistance. Theor Exp Plant Physiol 28:143–157. https://doi.org/10.1007/s40626-016-0070-x
Zhang JP, Li DQ, Shi XH, Zhang D, Qiu S, Wei JF, Zhang J, Zhou JH, Zhu KY, Xia YP (2017) Mining and expression analysis of candidate genes involved in regulating the chilling requirement fulfillment of Paeonia lactiflora ‘Hang Baishao’. BMC Plant Biol 17:262. https://doi.org/10.1186/s12870-017-1205-1
Zou Q (1995) Plant physiological and biochemical experiment guidance. China Agricultural Press, Beijing
Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (31600568) and the Henan Agricultural University Doctoral Fund Project (30500565).
Author information
Authors and Affiliations
Contributions
Yan Li designed the experiment; Yihong Chang performed bioinformatics analysis on transcriptome data and wrote the manuscript; Jiuxing Lu helped in the experiments and data analysis; Rui Wang and Dan He performed the determination of physiological indicators and qRT-PCR. Qiusheng Yang confirmed the research content, controlled the progress of the test and the quality of the manuscript writing. Yonghua Li reviewed and edited the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Li, Y., Chang, Y., Lu, J. et al. Comparative Transcriptome Analysis of Tree Peony Petals on Two Different Rootstocks. J Plant Growth Regul 38, 1287–1299 (2019). https://doi.org/10.1007/s00344-019-09933-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00344-019-09933-w