
Abstract
Wolfram syndrome (OMIM 222300) is a rare autosomal recessive disease with a devastating array of symptoms, including diabetes mellitus, optic nerve atrophy, diabetes insipidus, hearing loss, and neurological dysfunction. The discovery of the causative gene, WFS1, has propelled research on this disease. However, a comprehensive understanding of the function of WFS1 remains unknown, making the development of effective treatment a pressing challenge. To bridge these knowledge gaps, disease models for Wolfram syndrome are indispensable, and understanding the characteristics of each model is critical. This review will provide a summary of the current knowledge regarding WFS1 function and offer a comprehensive overview of established disease models for Wolfram syndrome, covering animal models such as mice, rats, flies, and zebrafish, along with induced pluripotent stem cell (iPSC)-derived human cellular models. These models replicate key aspects of Wolfram syndrome, contributing to a deeper understanding of its pathogenesis and providing a platform for discovering potential therapeutic approaches.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Wolfram Syndrome (OMIM 222300) is a rare autosomal recessive disease characterized by several symptoms, including diabetes mellitus, optic nerve atrophy, diabetes insipidus, hearing loss, and neurological dysfunction. Two types of Wolfram syndrome have been identified: type 1 and type 2, caused by pathogenic variants of WFS1 and CISD2, respectively. Most Wolfram syndrome cases are classified as type 1, so we will refer to them as “Wolfram syndrome” throughout this review. Since the discovery of WFS1 as the causative gene of Wolfram syndrome, several animal and cellular models have been established. These disease models are indispensable tools in the research on Wolfram syndrome. However, a comprehensive summary of the characteristics and phenotypes of these experimental models is lacking. Therefore, this review aims to fill this gap by providing an overview of the animal and cellular models of Wolfram syndrome established to date. In this review, we first discuss (1) the clinical symptoms of Wolfram syndrome, (2) the function of WFS1 and the pathophysiology caused by its abnormalities, and (3) the characteristics of disease models of Wolfram syndrome.
Clinical features of Wolfram syndrome and the discovery of WFS1
This section describes the clinical features of Wolfram syndrome and the disease concept associated with the causative gene WFS1.
Major symptoms of Wolfram syndrome
The first cases of Wolfram syndrome were reported by Wolfram and Wagener (1938). The report described four siblings who developed juvenile-onset diabetes mellitus and optic nerve atrophy. Subsequently, these patients also developed hearing loss and neurogenic bladder (Paley and Tunbridge 1956). In 1956, Paley and Tunbridge reported two additional cases of diabetes and optic nerve atrophy, suggesting a hereditary component (Paley and Tunbridge 1956). In 1977, a comprehensive review of 91 cases by Cremers et al. (1977) led to the official designation of this condition as “Wolfram syndrome.” Since then, numerous reports on the clinical symptoms of patients have emerged. The first nationwide survey was conducted and reported by Barrett et al. (1995), revealing a prevalence of Wolfram syndrome at 1 in 770,000. Typically, patients with Wolfram syndrome develop diabetes mellitus at a median age of 6 years and optic atrophy around 11 years. Diabetes insipidus and sensorineural hearing loss typically manifest in the second decade of life, while renal tract abnormalities and neurological complications such as cerebellar ataxia appear by the fourth decade. Neurological and urinary tract manifestations are the most common causes of morbidity and mortality (Kinsley et al. 1995). Wolfram syndrome is characterized by a reduced size of intracranial volume, particularly in the brainstem and cerebellum, leading to balance impairment (Hershey et al. 2012; Pickett et al. 2012). However, the severity of these symptoms can vary among individuals with Wolfram syndrome (De Franco et al. 2017).
Discovery of WFS1
Wolfram syndrome was previously believed to be a mitochondrial disease due to the similarities in symptoms and the presence of mitochondrial DNA deletions in some patients (Bu and Rotter 1993; Bundey et al. 1993; Rötig et al. 1993; Vora and Lilleyman 1993). However, subsequent cases demonstrated the absence of mitochondrial DNA abnormalities, and the inheritance pattern suggested an abnormality in the nuclear genome of patients with Wolfram syndrome. Linkage analysis suggested the presence of a causative gene for Wolfram syndrome on chromosome 4p16 (Polymeropoulos et al. 1994). In 1998, Inoue et al., followed soon after by Strom et al., identified the WFS1 gene (Inoue et al. 1998; Strom et al. 1998) (Fig. 1). Since the discovery of WFS1, the understanding of the pathogenesis of Wolfram syndrome has remarkably advanced. Approximately 200 WFS1 variants associated with Wolfram syndrome have been reported, mostly found in exon 8 of WFS1 (Smith et al. 2004). WFS1 encodes the protein WFS1, also known as wolframin, which is an endoplasmic reticulum (ER)-membrane protein with multiple transmembrane domains comprising 890 amino acids (Strom et al. 1998; Takeda et al. 2001). WFS1 is expressed ubiquitously in various tissues, with the highest levels observed in the pancreas, heart, and brain, particularly in the hippocampus, amygdaloid area, and olfactory tubercle (Hofmann et al. 2003; Takeda et al. 2001). Recent studies have reported predominant localization of WFS1 in the ER membrane, particularly in the mitochondria-associated ER membranes, which serve as contact sites between the ER and mitochondria (Angebault et al. 2018; La Morgia et al. 2020).

The structure of WFS1. WFS1 consists of eight exons, with the largest one being exon 8. The location of reported variants is depicted in different colors. The image has been adapted from Clinvar (https://www.ncbi.nlm.nih.gov/clinvar/), and the information was accessed on July 23, 2023
Wolfram syndrome and WFS1-related disorders
Wolfram syndrome, a rare disorder caused by recessive WFS1 variants, is now recognized to exhibit a phenotypic spectrum. Recent findings suggest that its frequency may be higher in certain races and populations than previously assumed (Bansal et al. 2018; De Franco et al. 2017; Li et al. 2020; Marchand et al. 2021). Moreover, there are related disorders caused by a dominant inheritance form of WFS1, known as “WFS1-related disorders” or “Wolfram-like syndrome.” These disorders manifest with one or more symptoms observed in Wolfram syndrome. The severity and symptoms of WFS1-related disorders can vary, ranging from mild diabetes mellitus, hearing loss, congenital cataracts, and optic atrophy developing independently to severe neonatal cases that exhibit all these symptoms (Kobayashi et al. 2018; Mets et al. 2010; Morikawa et al. 2017). It has also been reported that specific heterozygous WFS1 variants can lead to nonsyndromic low-frequency sensorineural hearing loss (Bespalova et al. 2001; Cryns et al. 2002; Young et al. 2001).
WFS1 functions and their anomalies
Some WFS1 variants have also been implicated in other types of diabetes. Several large-scale genome-wide association studies have reported that several single nucleotide polymorphisms (SNPs) in WFS1 are associated with an increased genetic risk for type 2 diabetes mellitus (T2DM), irrespective of racial differences (Cheurfa et al. 2011; Elek et al. 2015; Heni et al. 2010; Lee et al. 2008; Long et al. 2012; Lyssenko et al. 2008; Sandhu et al. 2007; Sparsø et al. 2008; van Hoek et al. 2008; Westermark et al. 2010). However, a meta-analysis has shown that two specific WFS1 SNPs, rs734312 and rs10010131, have a significant protective effect against the risk of developing T2DM (Cheng et al. 2013). Accordingly, understanding the pathogenesis of Wolfram syndrome and WFS1-related disorders may provide valuable insights into the pathogenesis of other types of diabetes, including T2DM. In this section, we describe the known physiological functions of WFS1 and the pathological conditions that arise due to its abnormalities.
Interaction between WFS1 and Ca2+ channels or pumps
The ER serves as an intracellular Ca2+ store, and WFS1 is expressed on the ER membrane. Therefore, numerous studies have explored the relationship between WFS1 and intracellular Ca2+ homeostasis (La Morgia et al. 2020; Nguyen et al. 2020; Osman et al. 2003). The first study to demonstrate a connection between WFS1 and intracellular Ca2+ was reported by Osman et al. (2003), who showed elevated levels of intracellular Ca2+ in Xenopus oocytes overexpressing WFS1.
Takei et al. first revealed the relationship between Ca2+ concentration in the ER and the expression level of WFS1 (Takei et al. 2006). They found that WFS1 regulates ER Ca2+ storage and cytosolic Ca2+ homeostasis by increasing the Ca2+ uptake and store-operated Ca2+ entry (SOCE). This regulation involves the modulation of Ca2+ pumps and channels, such as sarcoendoplasmic reticulum ATPase (SERCA) and inositol 1,4,5-trisphosphate receptor (IP3R), localized on the ER membrane (Kunnappallil and Hasan 2022). WFS1 deficiency results in upregulated SERCA expression, leading to increased Ca2+ pumping into the ER (Zatyka et al. 2015) (Fig. 2A). Additionally, WFS1 forms a complex with neuronal calcium sensor 1 (NCS1) and IP3R to facilitate Ca2+ transfer between the ER and mitochondria. WFS1 deficiency reduces the expression level of NCS1, followed by impaired ER-mitochondrial contact and subsequent Ca2+ uptake into the mitochondria (Angebault et al. 2018) (Fig. 2B). Therefore, therapeutic approaches targeting intracellular Ca2+ signaling have been explored as potential treatments for Wolfram syndrome (Abreu et al. 2021; Akiyama et al. 2009; Clark et al. 2017; Crouzier et al. 2022a; Lu et al. 2014; Nguyen et al. 2020). Apart from its interactions with Ca2+ pumps and channels, WFS1 also interacts with the Na+/K+ ATPase beta-1 subunit, the V1A subunit of the H1 ATPase, and the voltage-dependent anion channel isoform 1 (VDAC1) (Gharanei et al. 2013; Zatyka et al. 2008; Zatyka et al. 2023). In WFS1-depleted cells, the H1 ATPase V1A subunit is degraded more rapidly (Gharanei et al. 2013).

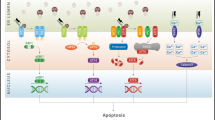
Multiple functions of WFS1. A WFS1 regulates Ca2+ uptake in the ER by modulating SERCA activity. WFS1 dysfunction increases ER Ca2+ levels and SOCE. Increased Ca2+ concentration activates Calpain-2, leading to cell death. B WFS1 activates IP3R through NCS1 and stimulates Ca2+ release from the ER. WFS1 dysfunction impairs IP3R activity, followed by decreased Ca2+ release from the ER and Ca2+ uptake into the mitochondria. C WFS1 stabilizes HRD1, an E3 ubiquitin ligase, and degrades ATF6. WFS1 abnormality induces the hyperactivation of ATF6. D WFS1 plays a vital role in maintaining the pH within secretory granules. WFS1 dysfunction leads to impaired granule acidification and insulin exocytosis. E The C-terminal of WFS1 binds to vesicular cargo proteins. The N-terminal of WFS1 is recognized by the protein transport protein SEC24, a component of coat protein complex II (COPII). WFS1 dysfunction disrupts the generation of mature COPII vesicles and hinders intercellular trafficking from the endoplasmic reticulum (ER) to the Golgi complex. ATF6 activating transcription factor 6, HRD1 HMG-CoA reductase degradation 1 homolog, SERCA sarcoendoplasmic reticulum ATPase, SOCE store-operated Ca2+ entry, IP3R 1,4,5-trisphosphate receptor, NCS1 neuronal calcium sensor 1, GRP75 glucose-regulated protein 75, VDAC1 voltage-dependent anion channel 1, MCU mitochondrial Ca2+ uniporter. (Created with BioRender.com)
WFS1 as a component of unfolded protein response
The link between WFS1 and ER stress was first described by Ueda et al. (2005). They demonstrated that the WFS1 expression level increased in response to ER stress. The gene expression levels of Wfs1 are considered ER stress markers (Lipson et al. 2006) due to the presence of a conserved sequence in the promoter region similar to the ER stress response element (Kakiuchi et al. 2006). ER stress-induced WFS1 upregulation requires the activation of inositol requiring 1 and PKR-like endoplasmic reticulum kinase, which are key regulators of the unfolded protein response (UPR) (Fonseca et al. 2005). Another UPR regulator, activating transcription factor (ATF) 6β, binds to the Wfs1 gene promoter and induces both gene and protein expression (Odisho et al. 2015).
WFS1 has additional roles in the regulation of ER stress and protein degradation. It stabilizes the E3 ubiquitin ligase HRD1 and regulates the degradation of ATF6α by facilitating ATF6 transport to the proteasome (Fonseca et al. 2010) (Fig. 2C). This means WFS1 acts as a UPR regulator; its deficiency in pancreatic β-cells induces pathogenic ER stress, leading to impaired cell cycle and accelerated cell apoptosis (Yamada et al. 2006). However, considering their close interaction, both ER stress and intracellular Ca2+ homeostasis may be involved in the pathogenesis of Wolfram syndrome. In fact, WFS1 deficiency induces ER stress, resulting in IP3R dysfunction and disturbed cytosolic Ca2+ homeostasis, which subsequently affects mitochondrial dynamics (Blackstone et al. 2016). This leads to inhibited mitochondrial fusion and trafficking, as well as augmented mitophagy, contributing to delayed neuronal development. ER stress is recognized for disrupting intracellular Ca2+ homeostasis and inducing cellular inflammation. The concept of “sterilized inflammation,” which refers to non-infectious inflammation triggered by pathogenic ER stress (Lerner et al. 2012; Oslowski et al. 2012), has recently emerged. In line with these observations, ER stress-induced sterilized inflammation caused by WFS1 dysfunction accelerates disease progression in Wolfram syndrome (Morikawa et al. 2022; Panfili et al. 2021).
WFS1 function in intracellular trafficking
WFS1 localization extends beyond the ER to include secretory granules (Hatanaka et al. 2011). In WFS1-deficient pancreatic β-cells, insulin secretory granule acidification becomes impaired, leading to impaired insulin exocytosis (Hatanaka et al. 2011) (Fig. 2D). The acidification of insulin granule is necessary for efficient proinsulin processing because endopeptidase PC1/3 and PC2 activate with a low pH optimum. While the precise interaction between WFS1 expressed on the insulin granule membrane and impaired insulin granule acidification has not been elucidated, it is speculated that WFS1 regulates the activities of V-type H+-ATPase and CLC-3Cl− channels expressed on the insulin granule membrane (Hatanaka et al. 2011). More recently, it was demonstrated that WFS1 serves as a vesicular cargo protein involved in intracellular trafficking (Wang et al. 2021). Specifically, the C-terminus of WFS1 directly binds to transported proteins within the ER lumen, while the N-terminus of WFS1 interacts with SEC24, a subunit of the coat protein complex II (COPII) located in the cytoplasm. This interaction allows for vesicular transport from the ER to the Golgi (Fig. 2E). This function of WFS1 has been shown to be necessary for the transportation of proteins such as proinsulin from the ER to the Golgi in pancreatic β-cells (Wang et al. 2021).
Experimental models of Wolfram syndrome
Experimental models are indispensable tools for conducting research to understand the pathogenesis of Wolfram syndrome and to develop therapeutic strategies. Over the years, several disease models have been established to study this condition. In the 2000s, Wfs1 knockout mice were generated as an early animal model. Subsequently, in the 2010s, disease models were extended to include rats, flies, and zebrafish. Recently, a Wfs1 pathogenic variant knock-in mouse model has also been reported. In this section, we summarize the phenotypic characteristics observed in these Wolfram syndrome animal models and also include information on induced pluripotent stem cell (iPSC)-derived cellular models (Table 1).
Animal models of Wolfram syndrome
The first animal model, Wolfram syndrome, was established by Ishihara et al. (2004). The whole-body Wfs1-deficient B6 background mice exhibited decreased pancreatic insulin content at 2 weeks old, impaired glucose homeostasis, and decreased insulin secretion around 17 weeks old. The diabetic phenotype in these mice resulted from ER stress-induced pancreatic β-cell death and was accelerated by crossing with agouti lethal yellow mice, a model that develops obesity, insulin resistance, and pancreatic β-cell hyperplasia (Akiyama et al. 2009). Similar to Wolfram syndrome patients, these mice also exhibited symptoms of central diabetes insipidus due to arginine vasopressin (AVP) deficiency (Kurimoto et al. 2021), impaired retinal functions (Bonnet Wersinger et al. 2014), and psychiatric symptoms observed in behavioral studies (Kato et al. 2008). In addition to the B6 strain, another whole-body Wfs1 knockout mouse model on the 129S6 background was developed (Kõks et al. 2009; Luuk et al. 2008). These 129S6 mice, in which LacZ replaces exon 8 of Wfs1, showed an earlier onset of diabetes compared to the B6 strain (Abreu et al. 2020). Interestingly, males were found to be at higher risk of developing diabetes than females (Noormets et al. 2011). The phenotype of this 129S6-background mouse model is well studied, and besides the diabetic phenotype, this mouse model recapitulates many of the symptoms observed in patients with Wolfram syndrome (Blackstone et al. 2016; Ivask et al. 2021; Luuk et al. 2009; Noormets et al. 2009, 2014; Richard et al. 2023; Visnapuu et al. 2013a, b; Waszczykowska et al. 2020). The only Wfs1-conditional knockout mouse model was reported by Riggs et al. These B6 background mice, with specifically knocked out Wfs1 in pancreatic β-cells, develop glucose intolerance and insulin deficiency by 12 weeks of age (Riggs et al. 2005). Most recently, a Wfs1 E864K knock-in mouse model was reported (Richard et al. 2023). These Wfs1 E864K knock-in mice develop an early onset of severe vestibular dysfunction besides hearing loss. Human patients with Wolfram syndrome carrying the WFS1 p.E864K variant develop low-frequency sensorineural hearing loss, optic atrophy, and impaired glucose tolerance. However, no vestibular neuropathy has been reported. These facts suggest that the role of WFS1 in the human vestibular system may not be as crucial as in rodents.
Besides the mouse model, the Wfs1-deficient rat is a well-established animal model for Wolfram syndrome (Plaas et al. 2017). This model involves whole-body Wfs1 knockout in Sprague–Dawley background rats, which show impaired glucose homeostasis starting at 3 months of age. At 15 months of age, these rats exhibit retinal gliosis, optic nerve atrophy, and reduced medullary volume. Although the onset age of each symptom is older than that in the mouse models, the larger body size of rats makes it advantageous for conducting therapeutic interventions and sample collection. Recently, the drosophila and zebrafish models with WFS1 homolog knockout have been reported as additional models for Wolfram syndrome. Optical transparency of the zebrafish embryo enables the observation of neural development in vivo. Drosophila has a short generation time and a small body size, making it possible to test many potential therapeutic molecules. Using these Wolfram syndrome zebrafish and Drosophila models, phenotypes caused by WFS1 deficiency, including neurodegeneration, visual dysfunction, and mitochondrial dysfunction, have been well studied (Cairns et al. 2021; Crouzier et al. 2022b; Kunnappallil and Hasan 2022; Sakakibara et al. 2018).
The pursuit of novel therapeutic approaches to combat Wolfram syndrome is actively underway. One promising strategy involves repurposing existing diabetic medications for the treatment of Wolfram syndrome, such as GLP1 receptor agonists (Panfili et al. 2023). Notably, exenatide and dulaglutide have been reported to improve glucose tolerance in Wolfram syndrome mouse models (Gorgogietas et al. 2023; Kondo et al. 2018; Sedman et al. 2016). Similarly, another GLP-1 receptor agonist, liraglutide, was found to prevent the development of glucose intolerance, reduce neuroinflammation, protect against retinal ganglion cell death, and delay the progression of hearing and vision loss in Wfs1 mutant rat models (Jagomäe et al. 2021; Seppa et al. 2019, 2021; Toots et al. 2018).
iPSC-derived models of Wolfram syndrome
The first successfully developed insulin-producing cells from patients with Wolfram syndrome were reported by Shang et al. (2014). In their study, the authors generated insulin-producing cells from skin fibroblast-derived iPSCs of Wolfram syndrome patients. These patient-derived insulin-producing cells showed lower insulin content and reduced tolerance to ER stress compared to controls. This cellular model provided crucial insights into ER stress-related diseases derived from human patients. Maxwell et al. improved the differential protocol for iPSC-derived β-cells (SC-β-cells) and used CRISPR/Cas9 techniques to explore the potential of gene therapy and personalized cell therapy for Wolfram syndrome (Maxwell et al. 2020). In their study, they successfully restored insulin secretion in patient-derived SC-β-cells through the correction of WFS1. Transplanting the gene-edited SC-β-cells into diabetic mice resulted in improved blood glucose profile. In addition, they found that SC-β-cells differentiated from Wolfram syndrome patients with mild WFS1 variants retained their insulin secretory capacity (Kitamura et al. 2022). This indicates that these SC-β-cell models exhibit phenotypes consistent with the symptoms observed in patients. Using these SC-β-cell models, a combination treatment of 4-phenylbutyric acid and tauroursodeoxycholic acid has been demonstrated to effectively address the diabetic phenotype in Wolfram syndrome patients with the WFS1 p.R558C variant (Kitamura et al. 2022). Besides pancreatic β-cells, neural cells have also been developed as a disease model from Wolfram syndrome patient iPSCs. Impaired neurite outgrowth was observed in neurons derived from Wolfram syndrome patients, and this morphological change was prevented by treatment with valproic acid (Pourtoy-Brasselet et al. 2021). Valproic acid has been reported to reduce ER stress and act as a histone deacetylase inhibitor for neurons. It is not clear how valproic acid promotes neurite outgrowth in the neuronal cell model of Wolfram syndrome. However, valproic acid may exert neuroprotective effects by reducing ER stress and through pleiotropic mechanisms. Zatyka et al. demonstrated that the interaction between WFS1 and VDAC1 is essential for mitochondrial function and dynamics in iPSC-derived neural cells (Zatyka et al. 2023). These models could prove useful for studying the pathogenesis of brainstem atrophy and neurodegeneration observed in Wolfram syndrome.
Future expectations
Numerous clinical and basic studies have significantly contributed to understanding the function of WFS1 and advancing toward a cure for Wolfram syndrome. In future, further research is expected to uncover the unknown pathogenesis, particularly regarding the development of optic nerve atrophy and neurological symptoms associated with WFS1 dysfunction. Furthermore, the development of therapeutic agents against Wolfram syndrome necessitates the use of patient-derived disease models. In addition to pancreatic β-cells and neural cells, the development of retinal ganglion, inner ear, and AVP-producing neural cells is important. These patient-derived cellular models can serve as invaluable tools for developing preclinical and personalized therapeutic reagents against visual impairment, hearing loss, and central diabetes insipidus in Wolfram syndrome. Research on Wolfram syndrome, including the development of various disease models, continues to serve as a prototype for studying ER stress-related diseases.
Data availability
Data sharing not applicable – no new data generated.
References
Abreu D, Asada R, Revilla JMP, Lavagnino Z, Kries K, Piston DW, Urano F (2020) Wolfram syndrome 1 gene regulates pathways maintaining beta-cell health and survival. Lab Invest 100:849–862. https://doi.org/10.1038/s41374-020-0408-5
Abreu D, Stone SI, Pearson TS, Bucelli RC, Simpson AN, Hurst S, Brown CM, Kries K, Onwumere C, Gu H, Hoekel J, Tychsen L, Van Stavern GP, White NH, Marshall BA, Hershey T, Urano F (2021) A phase Ib/IIa clinical trial of dantrolene sodium in patients with Wolfram syndrome. JCI Insight 6(15):e145188. https://doi.org/10.1172/jci.insight.145188
Akiyama M, Hatanaka M, Ohta Y, Ueda K, Yanai A, Uehara Y, Tanabe K, Tsuru M, Miyazaki M, Saeki S, Saito T, Shinoda K, Oka Y, Tanizawa Y (2009) Increased insulin demand promotes while pioglitazone prevents pancreatic beta cell apoptosis in Wfs1 knockout mice. Diabetologia 52:653–663. https://doi.org/10.1007/s00125-009-1270-6
Angebault C, Fauconnier J, Patergnani S, Rieusset J, Danese A, Affortit CA, Jagodzinska J, Mégy C, Quiles M, Cazevieille C, Korchagina J, Bonnet-Wersinger D, Milea D, Hamel C, Pinton P, Thiry M, Lacampagne A, Delprat B, Delettre C (2018) ER-mitochondria cross-talk is regulated by the Ca(2+) sensor NCS1 and is impaired in Wolfram syndrome. Sci Signal 11:eaaq1380. https://doi.org/10.1126/scisignal.aaq1380
Bansal V, Boehm BO, Darvasi A (2018) Identification of a missense variant in the WFS1 gene that causes a mild form of Wolfram syndrome and is associated with risk for type 2 diabetes in Ashkenazi Jewish individuals. Diabetologia 61:2180–2188. https://doi.org/10.1007/s00125-018-4690-3
Barrett TG, Bundey SE, Macleod AF (1995) Neurodegeneration and diabetes: UK nationwide study of Wolfram (DIDMOAD) syndrome. Lancet 346:1458–1463. https://doi.org/10.1016/s0140-6736(95)92473-6
Bespalova IN, Van Camp G, Bom SJ, Brown DJ, Cryns K, DeWan AT, Erson AE, Flothmann K, Kunst HP, Kurnool P, Sivakumaran TA, Cremers CW, Leal SM, Burmeister M, Lesperance MM (2001) Mutations in the Wolfram syndrome 1 gene (WFS1) are a common cause of low frequency sensorineural hearing loss. Hum Mol Genet 10:2501–2508. https://doi.org/10.1093/hmg/10.22.2501
Blackstone C, Cagalinec M, Liiv M, Hodurova Z, Hickey MA, Vaarmann A, Mandel M, Zeb A, Choubey V, Kuum M, Safiulina D, Vasar E, Veksler V, Kaasik A (2016) Role of mitochondrial dynamics in neuronal development: mechanism for Wolfram syndrome. PLOS Biol 14:e1002511
Bonnet Wersinger D, Benkafadar N, Jagodzinska J, Hamel C, Tanizawa Y, Lenaers G, Delettre C (2014) Impairment of visual function and retinal ER stress activation in Wfs1-deficient mice. PLoS ONE 9:e97222. https://doi.org/10.1371/journal.pone.0097222
Bu X, Rotter JI (1993) Wolfram syndrome: a mitochondrial-mediated disorder? Lancet 342:598–600. https://doi.org/10.1016/0140-6736(93)91416-j
Bundey S, Fielder A, Poulton K (1993) Wolfram syndrome: mitochondrial disorder. Lancet 342:1059–1060. https://doi.org/10.1016/0140-6736(93)92918-J
Cairns G, Burté F, Price R, O’Connor E, Toms M, Mishra R, Moosajee M, Pyle A, Sayer JA, Yu-Wai-Man P (2021) A mutant wfs1 zebrafish model of Wolfram syndrome manifesting visual dysfunction and developmental delay. Sci Rep 11:20491. https://doi.org/10.1038/s41598-021-99781-0
Cheng S, Wu Y, Wu W, Zhang D (2013) Association of rs734312 and rs10010131 polymorphisms in WFS1 gene with type 2 diabetes mellitus: a meta-analysis. Endocr J 60:441–447. https://doi.org/10.1507/endocrj.EJ12-0325
Cheurfa N, Brenner GM, Reis AF, Dubois-Laforgue D, Roussel R, Tichet J, Lantieri O, Balkau B, Fumeron F, Timsit J, Marre M, Velho G (2011) Decreased insulin secretion and increased risk of type 2 diabetes associated with allelic variations of the WFS1 gene: the Data from Epidemiological Study on the Insulin Resistance Syndrome (DESIR) prospective study. Diabetologia 54:554–562. https://doi.org/10.1007/s00125-010-1989-0
Clark AL, Kanekura K, Lavagnino Z, Spears LD, Abreu D, Mahadevan J, Yagi T, Semenkovich CF, Piston DW, Urano F (2017) Targeting cellular calcium homeostasis to prevent cytokine-mediated beta cell death. Sci Rep 7:5611. https://doi.org/10.1038/s41598-017-05935-4
Cremers CW, Wijdeveld PG, Pinckers AJ (1977) Juvenile diabetes mellitus, optic atrophy, hearing loss, diabetes insipidus, atonia of the urinary tract and bladder, and other abnormalities (Wolfram syndrome). A review of 88 cases from the literature with personal observations on 3 new patients. Acta Paediatr Scand Suppl 264:1–16. https://doi.org/10.1111/j.1651-2227.1977.tb15069.x
Crouzier L, Danese A, Yasui Y, Richard EM, Liévens JC, Patergnani S, Couly S, Diez C, Denus M, Cubedo N, Rossel M, Thiry M, Su TP, Pinton P, Maurice T, Delprat B (2022a) Activation of the sigma-1 receptor chaperone alleviates symptoms of Wolfram syndrome in preclinical models. Sci Transl Med 14:eabh3763. https://doi.org/10.1126/scitranslmed.abh3763
Crouzier L, Richard EM, Diez C, Alzaeem H, Denus M, Cubedo N, Delaunay T, Glendenning E, Baxendale S, Liévens JC, Whitfield TT, Maurice T, Delprat B (2022b) Morphological, behavioral and cellular analyses revealed different phenotypes in Wolfram syndrome wfs1a and wfs1b zebrafish mutant lines. Hum Mol Genet 31:2711–2727. https://doi.org/10.1093/hmg/ddac065
Cryns K, Pfister M, Pennings RJ, Bom SJ, Flothmann K, Caethoven G, Kremer H, Schatteman I, Köln KA, Tóth T, Kupka S, Blin N, Nürnberg P, Thiele H, van de Heyning PH, Reardon W, Stephens D, Cremers CW, Smith RJ, Van Camp G (2002) Mutations in the WFS1 gene that cause low-frequency sensorineural hearing loss are small non-inactivating mutations. Hum Genet 110:389–394. https://doi.org/10.1007/s00439-002-0719-1
De Franco E, Flanagan SE, Yagi T, Abreu D, Mahadevan J, Johnson MB, Jones G, Acosta F, Mulaudzi M, Lek N, Oh V, Petz O, Caswell R, Ellard S, Urano F, Hattersley AT (2017) Dominant ER stress-inducing WFS1 mutations underlie a genetic syndrome of neonatal/infancy-onset diabetes, congenital sensorineural deafness, and congenital cataracts. Diabetes 66:2044–2053. https://doi.org/10.2337/db16-1296
Elek Z, Németh N, Nagy G, Németh H, Somogyi A, Hosszufalusi N, Sasvári-Székely M, Rónai Z (2015) Micro-RNA binding site polymorphisms in the WFS1 gene are risk factors of diabetes mellitus. PLoS ONE 10:e0139519. https://doi.org/10.1371/journal.pone.0139519
Fonseca SG, Fukuma M, Lipson KL, Nguyen LX, Allen JR, Oka Y, Urano F (2005) WFS1 is a novel component of the unfolded protein response and maintains homeostasis of the endoplasmic reticulum in pancreatic beta-cells. J Biol Chem 280:39609–39615. https://doi.org/10.1074/jbc.M507426200
Fonseca SG, Ishigaki S, Oslowski CM, Lu S, Lipson KL, Ghosh R, Hayashi E, Ishihara H, Oka Y, Permutt MA, Urano F (2010) Wolfram syndrome 1 gene negatively regulates ER stress signaling in rodent and human cells. J Clin Invest 120:744–755. https://doi.org/10.1172/JCI39678
Gharanei S, Zatyka M, Astuti D, Fenton J, Sik A, Nagy Z, Barrett TG (2013) Vacuolar-type H+-ATPase V1A subunit is a molecular partner of Wolfram syndrome 1 (WFS1) protein, which regulates its expression and stability. Hum Mol Genet 22:203–217. https://doi.org/10.1093/hmg/dds400
Gorgogietas V, Rajaei B, Heeyoung C, Santacreu BJ, Marín-Cañas S, Salpea P, Sawatani T, Musuaya A, Arroyo MN, Moreno-Castro C, Benabdallah K, Demarez C, Toivonen S, Cosentino C, Pachera N, Lytrivi M, Cai Y, Carnel L, Brown C, Urano F, Marchetti P, Gilon P, Eizirik DL, Cnop M, Igoillo-Esteve M (2023) GLP-1R agonists demonstrate potential to treat Wolfram syndrome in human preclinical models. Diabetologia 66:1306–1321. https://doi.org/10.1007/s00125-023-05905-8
Hatanaka M, Tanabe K, Yanai A, Ohta Y, Kondo M, Akiyama M, Shinoda K, Oka Y, Tanizawa Y (2011) Wolfram syndrome 1 gene (WFS1) product localizes to secretory granules and determines granule acidification in pancreatic beta-cells. Hum Mol Genet 20:1274–1284. https://doi.org/10.1093/hmg/ddq568
Heni M, Ketterer C, Thamer C, Herzberg-Schäfer SA, Guthoff M, Stefan N, Machicao F, Staiger H, Fritsche A, Häring HU (2010) Glycemia determines the effect of type 2 diabetes risk genes on insulin secretion. Diabetes 59:3247–3252. https://doi.org/10.2337/db10-0674
Hershey T, Lugar HM, Shimony JS, Rutlin J, Koller JM, Perantie DC, Paciorkowski AR, Eisenstein SA, Permutt MA, Washington University Wolfram Study Group (2012) Early brain vulnerability in Wolfram syndrome. PLoS ONE 7:e40604. https://doi.org/10.1371/journal.pone.0040604
Hofmann S, Philbrook C, Gerbitz KD, Bauer MF (2003) Wolfram syndrome: structural and functional analyses of mutant and wild-type wolframin, the WFS1 gene product. Hum Mol Genet 12:2003–2012. https://doi.org/10.1093/hmg/ddg214
Inoue H, Tanizawa Y, Wasson J, Behn P, Kalidas K, Bernal-Mizrachi E, Mueckler M, Marshall H, Donis-Keller H, Crock P, Rogers D, Mikuni M, Kumashiro H, Higashi K, Sobue G, Oka Y, Permutt MA (1998) A gene encoding a transmembrane protein is mutated in patients with diabetes mellitus and optic atrophy (Wolfram syndrome). Nat Genet 20:143–148. https://doi.org/10.1038/2441
Ishihara H, Takeda S, Tamura A, Takahashi R, Yamaguchi S, Takei D, Yamada T, Inoue H, Soga H, Katagiri H, Tanizawa Y, Oka Y (2004) Disruption of the WFS1 gene in mice causes progressive beta-cell loss and impaired stimulus-secretion coupling in insulin secretion. Hum Mol Genet 13:1159–1170. https://doi.org/10.1093/hmg/ddh125
Ivask M, Volke V, Raasmaja A, Kõks S (2021) High-fat diet associated sensitization to metabolic stress in Wfs1 heterozygous mice. Mol Genet Metab 134:203–211. https://doi.org/10.1016/j.ymgme.2021.07.002
Jagomäe T, Seppa K, Reimets R, Pastak M, Plaas M, Hickey MA, Kukker KG, Moons L, De Groef L, Vasar E, Kaasik A, Terasmaa A, Plaas M (2021) Early intervention and lifelong treatment with GLP1 receptor agonist liraglutide in a Wolfram syndrome rat model with an emphasis on visual neurodegeneration, sensorineural hearing loss and diabetic phenotype. Cells 10:3193. https://doi.org/10.3390/cells10113193
Kakiuchi C, Ishiwata M, Hayashi A, Kato T (2006) XBP1 induces WFS1 through an endoplasmic reticulum stress response element-like motif in SH-SY5Y cells. J Neurochem 97:545–555. https://doi.org/10.1111/j.1471-4159.2006.03772.x
Kato T, Ishiwata M, Yamada K, Kasahara T, Kakiuchi C, Iwamoto K, Kawamura K, Ishihara H, Oka Y (2008) Behavioral and gene expression analyses of Wfs1 knockout mice as a possible animal model of mood disorder. Neurosci Res 61:143–158. https://doi.org/10.1016/j.neures.2008.02.002
Kinsley BT, Swift M, Dumont RH, Swift RG (1995) Morbidity and mortality in the Wolfram syndrome. Diabetes Care 18:1566–1570. https://doi.org/10.2337/diacare.18.12.1566
Kitamura RA, Maxwell KG, Ye W, Kries K, Brown CM, Augsornworawat P, Hirsch Y, Johansson MM, Weiden T, Ekstein J, Cohen J, Klee J, Leslie K, Simeonov A, Henderson MJ, Millman JR, Urano F (2022) Multidimensional analysis and therapeutic development using patient iPSC-derived disease models of Wolfram syndrome. JCI Insight 7:e156549. https://doi.org/10.1172/jci.insight.156549
Kobayashi M, Miyagawa M, Nishio SY, Moteki H, Fujikawa T, Ohyama K, Sakaguchi H, Miyanohara I, Sugaya A, Naito Y, Morita SY, Kanda Y, Takahashi M, Ishikawa K, Nagano Y, Tono T, Oshikawa C, Kihara C, Takahashi H, Noguchi Y, Usami SI (2018) WFS1 mutation screening in a large series of Japanese hearing loss patients: massively parallel DNA sequencing-based analysis. PLoS ONE 13:e0193359. https://doi.org/10.1371/journal.pone.0193359
Kõks S, Soomets U, Paya-Cano JL, Fernandes C, Luuk H, Plaas M, Terasmaa A, Tillmann V, Noormets K, Vasar E, Schalkwyk LC (2009) Wfs1 gene deletion causes growth retardation in mice and interferes with the growth hormone pathway. Physiol Genomics 37:249–259. https://doi.org/10.1152/physiolgenomics.90407.2008
Kondo M, Tanabe K, Amo-Shiinoki K, Hatanaka M, Morii T, Takahashi H, Seino S, Yamada Y, Tanizawa Y (2018) Activation of GLP-1 receptor signalling alleviates cellular stresses and improves beta cell function in a mouse model of Wolfram syndrome. Diabetologia 61:2189–2201. https://doi.org/10.1007/s00125-018-4679-y
Kunnappallil RS, Hasan G (2022) A Drosophila Wolfram Syndrome 1 (WFS1) homologue synergises with the intracellular Ca2+ release channel, IP3R to affect mitochondrial morphology and function. https://doi.org/10.1101/2022.11.10.515972
Kurimoto J, Takagi H, Miyata T, Hodai Y, Kawaguchi Y, Hagiwara D, Suga H, Kobayashi T, Sugiyama M, Onoue T, Ito Y, Iwama S, Banno R, Tanabe K, Tanizawa Y, Arima H (2021) Deficiency of WFS1 leads to the impairment of AVP secretion under dehydration in male mice. Pituitary 24:582–588. https://doi.org/10.1007/s11102-021-01135-6
La Morgia C, Maresca A, Amore G, Gramegna LL, Carbonelli M, Scimonelli E, Danese A, Patergnani S, Caporali L, Tagliavini F, Del Dotto V, Capristo M, Sadun F, Barboni P, Savini G, Evangelisti S, Bianchini C, Valentino ML, Liguori R, Tonon C, Giorgi C, Pinton P, Lodi R, Carelli V (2020) Calcium mishandling in absence of primary mitochondrial dysfunction drives cellular pathology in Wolfram syndrome. Sci Rep 10:4785. https://doi.org/10.1038/s41598-020-61735-3
Lee YH, Kang ES, Kim SH, Han SJ, Kim CH, Kim HJ, Ahn CW, Cha BS, Nam M, Nam CM, Lee HC (2008) Association between polymorphisms in SLC30A8, HHEX, CDKN2A/B, IGF2BP2, FTO, WFS1, CDKAL1, KCNQ1 and type 2 diabetes in the Korean population. J Hum Genet 53:991–998. https://doi.org/10.1007/s10038-008-0341-8
Lerner AG, Upton JP, Praveen PV, Ghosh R, Nakagawa Y, Igbaria A, Shen S, Nguyen V, Backes BJ, Heiman M, Heintz N, Greengard P, Hui S, Tang Q, Trusina A, Oakes SA, Papa FR (2012) IRE1alpha induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab 16:250–264. https://doi.org/10.1016/j.cmet.2012.07.007
Li M, Wang S, Xu K, Chen Y, Fu Q, Gu Y, Shi Y, Zhang M, Sun M, Chen H, Han X, Li Y, Tang Z, Cai L, Li Z, Shi Y, Yang T, Polychronakos C (2020) High prevalence of a monogenic cause in Han Chinese diagnosed with Type 1 diabetes, partly driven by nonsyndromic recessive WFS1 mutations. Diabetes 69:121–126. https://doi.org/10.2337/db19-0510
Lipson KL, Fonseca SG, Ishigaki S, Nguyen LX, Foss E, Bortell R, Rossini AA, Urano F (2006) Regulation of insulin biosynthesis in pancreatic beta cells by an endoplasmic reticulum-resident protein kinase IRE1. Cell Metab 4:245–254. https://doi.org/10.1016/j.cmet.2006.07.007
Long J, Edwards T, Signorello LB, Cai Q, Zheng W, Shu XO, Blot WJ (2012) Evaluation of genome-wide association study-identified type 2 diabetes loci in African Americans. Am J Epidemiol 176:995–1001. https://doi.org/10.1093/aje/kws176
Lu S, Kanekura K, Hara T, Mahadevan J, Spears LD, Oslowski CM, Martinez R, Yamazaki-Inoue M, Toyoda M, Neilson A, Blanner P, Brown CM, Semenkovich CF, Marshall BA, Hershey T, Umezawa A, Greer PA, Urano F (2014) A calcium-dependent protease as a potential therapeutic target for Wolfram syndrome. Proc Natl Acad Sci U S A 111:E5292–E5301. https://doi.org/10.1073/pnas.1421055111
Luuk H, Koks S, Plaas M, Hannibal J, Rehfeld JF, Vasar E (2008) Distribution of Wfs1 protein in the central nervous system of the mouse and its relation to clinical symptoms of the Wolfram syndrome. J Comp Neurol 509:642–660. https://doi.org/10.1002/cne.21777
Luuk H, Plaas M, Raud S, Innos J, Sütt S, Lasner H, Abramov U, Kurrikoff K, Kõks S, Vasar E (2009) Wfs1-deficient mice display impaired behavioural adaptation in stressful environment. Behav Brain Res 198:334–345. https://doi.org/10.1016/j.bbr.2008.11.007
Lyssenko V, Jonsson A, Almgren P, Pulizzi N, Isomaa B, Tuomi T, Berglund G, Altshuler D, Nilsson P, Groop L (2008) Clinical risk factors, DNA variants, and the development of type 2 diabetes. N Engl J Med 359:2220–2232. https://doi.org/10.1056/NEJMoa0801869
Marchand L, Li M, Leblicq C, Rafique I, Alarcon-Martinez T, Lange C, Rendon L, Tam E, Courville-Le Bouyonnec A, Polychronakos C (2021) Monogenic causes in the Type 1 diabetes genetics consortium cohort: low genetic risk for autoimmunity in case selection. J Clin Endocrinol Metab 106:1804–1810. https://doi.org/10.1210/clinem/dgab056
Maxwell KG, Augsornworawat P, Velazco-Cruz L, Kim MH, Asada R, Hogrebe NJ, Morikawa S, Urano F, Millman JR (2020) Gene-edited human stem cell-derived beta cells from a patient with monogenic diabetes reverse preexisting diabetes in mice. Sci Transl Med 12:eaax9106. https://doi.org/10.1126/scitranslmed.aax9106
Mets RB, Emery SB, Lesperance MM, Mets MB (2010) Congenital cataracts in two siblings with Wolfram syndrome. Ophthalmic Genet 31:227–229. https://doi.org/10.3109/13816810.2010.516056
Morikawa S, Tajima T, Nakamura A, Ishizu K, Ariga T (2017) A novel heterozygous mutation of the WFS1 gene leading to constitutive endoplasmic reticulum stress is the cause of Wolfram syndrome. Pediatr Diabetes 18:934–941. https://doi.org/10.1111/pedi.12513
Morikawa S, Blacher L, Onwumere C, Urano F (2022) Loss of function of WFS1 causes ER stress-mediated inflammation in pancreatic beta-cells. Front Endocrinol (lausanne) 13:849204. https://doi.org/10.3389/fendo.2022.849204
Nguyen LD, Fischer TT, Abreu D, Arroyo A, Urano F, Ehrlich BE (2020) Calpain inhibitor and ibudilast rescue beta cell functions in a cellular model of Wolfram syndrome. Proc Natl Acad Sci U S A 117:17389–17398. https://doi.org/10.1073/pnas.2007136117
Noormets K, Kõks S, Kavak A, Arend A, Aunapuu M, Keldrimaa A, Vasar E, Tillmann V (2009) Male mice with deleted Wolframin (Wfs1) gene have reduced fertility. Reprod Biol Endocrinol 7:82. https://doi.org/10.1186/1477-7827-7-82
Noormets K, Kõks S, Muldmaa M, Mauring L, Vasar E, Tillmann V (2011) Sex differences in the development of diabetes in mice with deleted wolframin (Wfs1) gene. Exp Clin Endocrinol Diabetes 119:271–275. https://doi.org/10.1055/s-0030-1265163
Noormets K, Kõks S, Ivask M, Aunapuu M, Arend A, Vasar E, Tillmann V (2014) Energy metabolism and thyroid function of mice with deleted wolframin (Wfs1) gene. Exp Clin Endocrinol Diabetes 122:281–286. https://doi.org/10.1055/s-0034-1372582
Odisho T, Zhang L, Volchuk A (2015) ATF6beta regulates the Wfs1 gene and has a cell survival role in the ER stress response in pancreatic beta-cells. Exp Cell Res 330:111–122. https://doi.org/10.1016/j.yexcr.2014.10.007
Oslowski CM, Hara T, O’Sullivan-Murphy B, Kanekura K, Lu S, Hara M, Ishigaki S, Zhu LJ, Hayashi E, Hui ST, Greiner D, Kaufman RJ, Bortell R, Urano F (2012) Thioredoxin-interacting protein mediates ER stress-induced beta cell death through initiation of the inflammasome. Cell Metab 16:265–273. https://doi.org/10.1016/j.cmet.2012.07.005
Osman AA, Saito M, Makepeace C, Permutt MA, Schlesinger P, Mueckler M (2003) Wolframin expression induces novel ion channel activity in endoplasmic reticulum membranes and increases intracellular calcium. J Biol Chem 278:52755–52762. https://doi.org/10.1074/jbc.M310331200
Paley RG, Tunbridge RE (1956) Primary optic atrophy in diabetes mellitus. Diabetes 5:295–296. https://doi.org/10.2337/diab.5.4.295
Panfili E, Mondanelli G, Orabona C, Belladonna ML, Gargaro M, Fallarino F, Orecchini E, Prontera P, Proietti E, Frontino G, Tirelli E, Iacono A, Vacca C, Puccetti P, Grohmann U, Esposito S, Pallotta MT (2021) Novel mutations in the WFS1 gene are associated with Wolfram syndrome and systemic inflammation. Hum Mol Genet 30:265–276. https://doi.org/10.1093/hmg/ddab040
Panfili E, Frontino G, Pallotta MT (2023) GLP-1 receptor agonists as promising disease-modifying agents in WFS1 spectrum disorder. Front Clin Diabetes Healthc 4:1171091. https://doi.org/10.3389/fcdhc.2023.1171091
Pickett KA, Duncan RP, Paciorkowski AR, Permutt MA, Marshall B, Hershey T, Earhart GM, Washington University Wolfram Study Group (2012) Balance impairment in individuals with Wolfram syndrome. Gait Posture 36:619–624. https://doi.org/10.1016/j.gaitpost.2012.06.008
Plaas M, Seppa K, Reimets R, Jagomäe T, Toots M, Koppel T, Vallisoo T, Nigul M, Heinla I, Meier R, Kaasik A, Piirsoo A, Hickey MA, Terasmaa A, Vasar E (2017) Wfs1-deficient rats develop primary symptoms of Wolfram syndrome: insulin-dependent diabetes, optic nerve atrophy and medullary degeneration. Sci Rep 7:10220. https://doi.org/10.1038/s41598-017-09392-x
Polymeropoulos MH, Swift RG, Swift M (1994) Linkage of the gene for Wolfram syndrome to markers on the short arm of chromosome 4. Nat Genet 8:95–97. https://doi.org/10.1038/ng0994-95
Pourtoy-Brasselet S, Sciauvaud A, Boza-Moran MG, Cailleret M, Jarrige M, Polvèche H, Polentes J, Chevet E, Martinat C, Peschanski M, Aubry L (2021) Human iPSC-derived neurons reveal early developmental alteration of neurite outgrowth in the late-occurring neurodegenerative Wolfram syndrome. Am J Hum Genet 108:2171–2185. https://doi.org/10.1016/j.ajhg.2021.10.001
Richard EM, Brun E, Korchagina J, Crouzier L, Affortit C, Alves S, Cazevieille C, Mausset-Bonnefont AL, Lenoir M, Puel JL, Maurice T, Thiry M, Wang J, Delprat B (2023) Wfs 1(E864K) knock-in mice illuminate the fundamental role of Wfs1 in endocochlear potential production. Cell Death Dis 14:387. https://doi.org/10.1038/s41419-023-05912-y
Riggs AC, Bernal-Mizrachi E, Ohsugi M, Wasson J, Fatrai S, Welling C, Murray J, Schmidt RE, Herrera PL, Permutt MA (2005) Mice conditionally lacking the Wolfram gene in pancreatic islet beta cells exhibit diabetes as a result of enhanced endoplasmic reticulum stress and apoptosis. Diabetologia 48:2313–2321. https://doi.org/10.1007/s00125-005-1947-4
Rötig A, Cormier V, Chatelain P, Francois R, Saudubray JM, Rustin P, Munnich A (1993) Deletion of mitochondrial DNA in a case of early-onset diabetes mellitus, optic atrophy, and deafness (Wolfram syndrome, MIM 222300). J Clin Invest 91:1095–1098. https://doi.org/10.1172/JCI116267
Sakakibara Y, Sekiya M, Fujisaki N, Quan X, Iijima KM (2018) Knockdown of wfs1, a fly homolog of Wolfram syndrome 1, in the nervous system increases susceptibility to age- and stress-induced neuronal dysfunction and degeneration in Drosophila. PLOS Genet 14:e1007196. https://doi.org/10.1371/journal.pgen.1007196
Sandhu MS, Weedon MN, Fawcett KA, Wasson J, Debenham SL, Daly A, Lango H, Frayling TM, Neumann RJ, Sherva R, Blech I, Pharoah PD, Palmer CN, Kimber C, Tavendale R, Morris AD, McCarthy MI, Walker M, Hitman G, Glaser B, Permutt MA, Hattersley AT, Wareham NJ, Barroso I (2007) Common variants in WFS1 confer risk of type 2 diabetes. Nat Genet 39:951–953. https://doi.org/10.1038/ng2067
Sedman T, Rünkorg K, Krass M, Luuk H, Plaas M, Vasar E, Volke V (2016) Exenatide is an effective antihyperglycaemic agent in a mouse model of Wolfram syndrome 1. J Diabetes Res 2016:9239530. https://doi.org/10.1155/2016/9239530
Seppa K, Toots M, Reimets R, Jagomae T, Koppel T, Pallase M, Hasselholt S, Krogsbaek Mikkelsen M, Randel Nyengaard J, Vasar E, Terasmaa A, Plaas M (2019) GLP-1 receptor agonist liraglutide has a neuroprotective effect on an aged rat model of Wolfram syndrome. Sci Rep 31:15742
Seppa K, Jagomäe T, Kukker KG, Reimets R, Pastak M, Vasar E, Terasmaa A, Plaas M (2021) Liraglutide, 7,8-DHF and their co-treatment prevents loss of vision and cognitive decline in a Wolfram syndrome rat model. Sci Rep 11:2275. https://doi.org/10.1038/s41598-021-81768-6
Shang L, Hua H, Foo K, Martinez H, Watanabe K, Zimmer M, Kahler DJ, Freeby M, Chung W, LeDuc C, Goland R, Leibel RL, Egli D (2014) β-Cell dysfunction due to increased ER stress in a stem cell model of Wolfram syndrome. Diabetes 63:923–933. https://doi.org/10.2337/db13-0717
Smith CJ, Crock PA, King BR, Meldrum CJ, Scott RJ (2004) Phenotype-genotype correlations in a series of wolfram syndrome families. Diabetes Care 27:2003–2009. https://doi.org/10.2337/diacare.27.8.2003
Sparsø T, Andersen G, Albrechtsen A, Jørgensen T, Borch-Johnsen K, Sandbaek A, Lauritzen T, Wasson J, Permutt MA, Glaser B, Madsbad S, Pedersen O, Hansen T (2008) Impact of polymorphisms in WFS1 on prediabetic phenotypes in a population-based sample of middle-aged people with normal and abnormal glucose regulation. Diabetologia 51:1646–1652. https://doi.org/10.1007/s00125-008-1064-2
Strom TM, Hörtnagel K, Hofmann S, Gekeler F, Scharfe C, Rabl W, Gerbitz KD, Meitinger T (1998) Diabetes insipidus, diabetes mellitus, optic atrophy and deafness (DIDMOAD) caused by mutations in a novel gene (wolframin) coding for a predicted transmembrane protein. Hum Mol Genet 7:2021–2028. https://doi.org/10.1093/hmg/7.13.2021
Takeda K, Inoue H, Tanizawa Y, Matsuzaki Y, Oba J, Watanabe Y, Shinoda K, Oka Y (2001) WFS1 (Wolfram syndrome 1) gene product: predominant subcellular localization to endoplasmic reticulum in cultured cells and neuronal expression in rat brain. Hum Mol Genet 10:477–484. https://doi.org/10.1093/hmg/10.5.477
Takei D, Ishihara H, Yamaguchi S, Yamada T, Tamura A, Katagiri H, Maruyama Y, Oka Y (2006) WFS1 protein modulates the free Ca(2+) concentration in the endoplasmic reticulum. FEBS Lett 580:5635–5640. https://doi.org/10.1016/j.febslet.2006.09.007
Toots M, Seppa K, Jagomäe T, Koppel T, Pallase M, Heinla I, Terasmaa A, Plaas M, Vasar E (2018) Preventive treatment with liraglutide protects against development of glucose intolerance in a rat model of Wolfram syndrome. Sci Rep 8:10183. https://doi.org/10.1038/s41598-018-28314-z
Ueda K, Kawano J, Takeda K, Yujiri T, Tanabe K, Anno T, Akiyama M, Nozaki J, Yoshinaga T, Koizumi A, Shinoda K, Oka Y, Tanizawa Y (2005) Endoplasmic reticulum stress induces Wfs1 gene expression in pancreatic beta-cells via transcriptional activation. Eur J Endocrinol 153:167–176. https://doi.org/10.1530/eje.1.01945
van Hoek M, Dehghan A, Witteman JC, van Duijn CM, Uitterlinden AG, Oostra BA, Hofman A, Sijbrands EJ, Janssens AC (2008) Predicting type 2 diabetes based on polymorphisms from genome-wide association studies: a population-based study. Diabetes 57:3122–3128. https://doi.org/10.2337/db08-0425
Visnapuu T, Plaas M, Reimets R, Raud S, Terasmaa A, Kõks S, Sütt S, Luuk H, Hundahl CA, Eskla KL, Altpere A, Alttoa A, Harro J, Vasar E (2013a) Evidence for impaired function of dopaminergic system in Wfs1-deficient mice. Behav Brain Res 244:90–99. https://doi.org/10.1016/j.bbr.2013.01.046
Visnapuu T, Raud S, Loomets M, Reimets R, Sütt S, Luuk H, Plaas M, Kõks S, Volke V, Alttoa A, Harro J, Vasar E (2013b) Wfs1-deficient mice display altered function of serotonergic system and increased behavioral response to antidepressants. Front Neurosci 7:132. https://doi.org/10.3389/fnins.2013.00132
Vora AJ, Lilleyman JS (1993) Wolfram syndrome: mitochondrial disorder. Lancet 342:1059. https://doi.org/10.1016/0140-6736(93)92918-J
Wang L, Liu H, Zhang X, Song E, Wang Y, Xu T, Li Z (2021) WFS1 functions in ER export of vesicular cargo proteins in pancreatic beta-cells. Nat Commun 12:6996. https://doi.org/10.1038/s41467-021-27344-y
Waszczykowska A, Zmysłowska A, Braun M, Ivask M, Koks S, Jurowski P, Młynarski W (2020) Multiple retinal anomalies in Wfs1-deficient mice. Diagnostics (basel) 10:607. https://doi.org/10.3390/diagnostics10090607
Westermark P, Neuman RJ, Wasson J, Atzmon G, Wainstein J, Yerushalmi Y, Cohen J, Barzilai N, Blech I, Glaser B, Permutt MA (2010) Gene-gene interactions lead to higher risk for development of Type 2 diabetes in an Ashkenazi Jewish population. PLoS ONE 5:e9903
Wolfram DJ, Wagener HP (1938) Diabetes mellitus and simple optic atrophy among siblings: report of four cases. Mayo Clin Proc 13:715–718
Yamada T, Ishihara H, Tamura A, Takahashi R, Yamaguchi S, Takei D, Tokita A, Satake C, Tashiro F, Katagiri H, Aburatani H, Miyazaki J, Oka Y (2006) WFS1-deficiency increases endoplasmic reticulum stress, impairs cell cycle progression and triggers the apoptotic pathway specifically in pancreatic beta-cells. Hum Mol Genet 15:1600–1609. https://doi.org/10.1093/hmg/ddl081
Young TL, Ives E, Lynch E, Person R, Snook S, MacLaren L, Cater T, Griffin A, Fernandez B, Lee MK, King MC (2001) Non-syndromic progressive hearing loss DFNA38 is caused by heterozygous missense mutation in the Wolfram syndrome gene WFS1. Hum Mol Genet 10:2509–2514. https://doi.org/10.1093/hmg/10.22.2509
Zatyka M, Ricketts C, da Silva XG, Minton J, Fenton S, Hofmann-Thiel S, Rutter GA, Barrett TG (2008) Sodium-potassium ATPase 1 subunit is a molecular partner of Wolframin, an endoplasmic reticulum protein involved in ER stress. Hum Mol Genet 17:190–200. https://doi.org/10.1093/hmg/ddm296
Zatyka M, Da Silva XG, Bellomo EA, Leadbeater W, Astuti D, Smith J, Michelangeli F, Rutter GA, Barrett TG (2015) Sarco(endo)plasmic reticulum ATPase is a molecular partner of Wolfram syndrome 1 protein, which negatively regulates its expression. Hum Mol Genet 24:814–827. https://doi.org/10.1093/hmg/ddu499
Zatyka M, Rosenstock TR, Sun C, Palhegyi AM, Hughes GW, Lara-Reyna S, Astuti D, di Maio A, Sciauvaud A, Korsgen ME, Stanulovic V, Kocak G, Rak M, Pourtoy-Brasselet S, Winter K, Varga T, Jarrige M, Polvèche H, Correia J, Frickel EM, Hoogenkamp M, Ward DG, Aubry L, Barrett T, Sarkar S (2023) Depletion of WFS1 compromises mitochondrial function in hiPSC-derived neuronal models of Wolfram syndrome. Stem Cell Rep 18:1090–1106. https://doi.org/10.1016/j.stemcr.2023.04.002
Acknowledgements
SM thanks Dr. Fumihiko Urano (Washington University School of Medicine in St. Louis), Dr. Toshihiro Tajima (Department of Pediatrics, Jichi Medical University), and Dr. Atsushi Manabe (Department of Pediatrics, Hokkaido University Graduate School of Medicine) for their mentorship and Sachiko Nakayama, Yui Sakurai, Ryoko Miyoshi, Kaoru Yokoki, and Miki Kaiho (Department of Pediatrics, Hokkaido University) for their invaluable secretarial assistance.
Funding
SM received funding from The Japanese Society for Pediatric Endocrinology Future Development Grant, which is supported by Novo Nordisk Pharma Ltd. KT received funding from Grants-in-Aid from the Ministry of Education, Culture, Sports, Science and Technology, Japan (23K08011).
Author information
Authors and Affiliations
Contributions
SM performed the literature search and wrote the draft. SM, KT, NK, NH, and AK critically reviewed the manuscript. All authors approved the final version of the manuscript to be published.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflict of interest to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Morikawa, S., Tanabe, K., Kaneko, N. et al. Comprehensive overview of disease models for Wolfram syndrome: toward effective treatments. Mamm Genome 35, 1–12 (2024). https://doi.org/10.1007/s00335-023-10028-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00335-023-10028-x