
Abstract
Aims/hypothesis
Loss of functional beta cells results in a gradual progression of insulin insufficiency in Wolfram syndrome caused by recessive WFS1 mutations. However, beta cell dysfunction in Wolfram syndrome has yet to be fully characterised, and there are also no specific treatment recommendations. In this study, we aimed to characterise beta cell secretory defects and to examine the potential effects of a glucagon-like peptide-1 (GLP-1) receptor agonist on diabetes in Wolfram syndrome.
Methods
Insulin secretory function was assessed by the pancreatic perfusion method in mice used as a model of Wolfram syndrome. In addition, granule dynamics in living beta cells were examined using total internal reflection fluorescence microscopy. Acute and chronic effects of exendin-4 (Ex-4) on glucose tolerance and insulin secretion were examined in young Wfs1−/− mice without hyperglycaemia. Molecular events associated with Ex-4 treatment were investigated using pancreatic sections and isolated islets. In addition, we retrospectively observed a woman with Wolfram syndrome who had been treated with liraglutide for 24 weeks.
Results
Treatment with liraglutide ameliorated our patient’s glycaemic control and resulted in a 20% reduction of daily insulin dose along with an off-drug elevation of fasting C-peptide immunoreactivity. Glucose-stimulated first-phase insulin secretion and potassium-stimulated insulin secretion decreased by 53% and 59%, respectively, in perfused pancreases of 10-week-old Wfs1−/− mice compared with wild-type (WT) mice. The number of insulin granule fusion events in the first phase decreased by 41% in Wfs1−/− beta cells compared with WT beta cells. Perfusion with Ex-4 increased insulin release in the first and second phases by 3.9-fold and 5.6-fold, respectively, in Wfs1−/− mice compared with perfusion with saline as a control. The physiological relevance of the effects of Ex-4 was shown by the fact that a single administration potentiated glucose-stimulated insulin secretion and improved glucose tolerance in Wfs1−/− mice. Four weeks of administration of Ex-4 resulted in an off-drug amelioration of glucose excursions after glucose loading in Wfs1−/− mice, with insulin secretory dynamics that were indistinguishable from those in WT mice, despite the fact that there was no alteration in beta cell mass. In association with the functional improvements, Ex-4 treatment reversed the increases in phosphorylated eukaryotic initiation factor (EIF2α) and thioredoxin interacting protein (TXNIP), and the decrease in phosphorylated AMP-activated kinase (AMPK), in the beta cells of the Wfs1−/− mice. Furthermore, Ex-4 treatment modulated the transcription of oxidative and endoplasmic reticulum stress-related markers in isolated islets, implying that it was able to mitigate the cellular stresses resulting from Wfs1 deficiency.
Conclusions/interpretation
Our study provides deeper insights into the pathophysiology of beta cell dysfunction caused by WFS1 deficiency and implies that activation of the GLP-1 receptor signal may alleviate insulin insufficiency and aid glycaemic control in Wolfram syndrome.
Similar content being viewed by others

Avoid common mistakes on your manuscript.

Introduction
Wolfram syndrome (OMIM 222300) is an autosomal recessive disorder characterised by a combination of juvenile-onset diabetes mellitus, optic atrophy, diabetes insipidus, deafness and neurodegeneration [1, 2]. Diabetes mellitus, the earliest manifestation, is followed by the onset of the other clinical features [1, 3, 4]. Autopsy analysis has revealed a selective loss of pancreatic beta cells in affected individuals, accounting for the insulin insufficiency [5]. Since genetic analysis has identified the major causative gene (WFS1) [6,7,8], the combination of known phenotypes and insights into the molecular genetics of WFS1 has allowed a better understanding of this complicated disease.
WFS1 encodes a 100 kDa protein (Wolfram syndrome 1 [WFS1]), which is localised in the endoplasmic reticulum (ER) [9] as well as in secretory granules in mouse pancreatic beta cells [10]. The disruption of this gene in mice (Wfs1−/−) predisposes beta cells to ER stress through perturbations of cellular Ca2+ homeostasis [11,12,13] and aberrant ER stress responses [14], accounting for the beta cell dysfunction and cellular loss seen. In addition, previous studies have demonstrated that WFS1 is required for maintaining the acidic environment of the insulin granules [10], suggesting that WFS1 contributes to insulin biosynthesis and vesicle exocytosis by producing a suitable pH of the insulin granules to maintain such cellular processes [15, 16]. Although the secretory defects in Wfs1−/− mice have yet to be fully characterised, they interact with the processes of beta cell failure.
As beta cells are the most susceptible tissues in Wolfram syndrome, diabetes mellitus in humans develops during childhood and is an important target for intervention. Although molecular and cellular experiments have focused on the underlying pathophysiology [14, 17, 18], research into interventions targeting the processes of beta cell failure and their clinical correlates is still in its infancy. Our present goal was to elucidate the therapeutic effects on insulin insufficiency in Wolfram syndrome of activating the glucagon-like peptide-1 (GLP-1) receptor signal. We also retrospectively observed the effects of liraglutide treatment on beta cell function and glycaemic control in an insulin-dependent woman with Wolfram syndrome. We further studied the alterations of insulin secretion at the systemic and cellular levels in Wfs1−/− mice and their modification by exendin-4 (Ex-4).
Methods
Clinical data collection
A 25-year-old Japanese woman was diagnosed with Wolfram syndrome based on findings of insulin-dependent diabetes mellitus, optic atrophy, hearing loss and diabetes insipidus. Her diabetes mellitus had been diagnosed at the age of 5 years, and this was followed by optic atrophy and other manifestations (see electronic supplementary material [ESM) Table 1). She had become blind at the age of 20. Her parents were consanguineous, and she had two siblings who had been diagnosed with Wolfram syndrome. She had been reported to harbour a homozygous 1515_1530del, p.508Y_L512 mutation in exon 8 of the WFS1 gene (ESM Table 1) [8]. She had started insulin therapy at the age of 15, and experienced fluctuations of blood glucose with recurrent episodes of hypoglycaemia. However, no diabetic microvascular complications had so far developed. She was being treated with subcutaneous injections of liraglutide at a dose of 0.9 mg once daily, which is the dosage approved in Japan, in combination with insulin therapy. Before this intervention, she had been admitted to hospital and her residual beta cell function assessed.
We retrospectively studied this participant’s clinical course of intervention with liraglutide. All clinical data were collected at the Akita University hospital. The difference (Δ) in C-peptide immunoreactivity (CPR) was assessed using the following formula: (CPR at 6 min after intravenous infusion of 1 mg of glucagon) − (basal CPR). The secretory units of islets in transplantation (SUIT) index and CPR index (CPI) were calculated as previously described [19]. The collection and use of retrospective clinical data were approved by the institutional review board of the Akita University hospital (approval number 723) and Yamaguchi University hospital (approval number H22-47-2), and appropriate written informed consent was obtained from the patient.
Animal studies
The Wfs1 knockout (Wfs1−/−) mice, with a C57BL/6J background, were obtained as described [12] and were housed with a free access to food and water in pathogen-free facilities with a 12 h light/dark cycle at the Animal Care Facility of Yamaguchi University Graduate School of Medicine, Ube, Japan. All experiments were performed on 5- to 12-week-old male Wfs1−/− mice and age-matched male littermate control WT mice. Animal care and experimental procedures were approved by the Animal Ethics Committee of Yamaguchi University Graduate School of Medicine (approval number 25-020) and carried out according to the Yamaguchi University Experimentation Regulations and the NIH guidelines. Randomisation and blinding were carried out for all experiments. The data analysis was performed with all experimental results obtained.
Perfusion experiment
Perfusion experiments were performed as previously described [20]. Briefly, we used 10-week-old male mice that had been fasted for 16 h. After induction of anaesthesia, the superior mesenteric and renal arteries were ligated, and the aorta was tied off just below the diaphragm. The perfusate (KRB HEPES [KRBH] buffer supplemented with 4.6% dextran and 0.25% BSA and infused with 95% O2/5% CO2) was infused into a catheter placed in the aorta and collected from the portal vein. The perfusion protocol began with the initial step (i.e. from 1 to 5 min; see Figs 1a,c and 2a,c,e) following a 30 min equilibration period using the same buffer. The flow rate of the perfusate was 1 ml/min. For Ex-4 treatment, perfusion with 10 nmol/l Ex-4 (Sigma-Aldrich, St Louis, MO, USA) was initiated 5 min before perfusion with 16.7 mmol/l glucose and continued for the duration of the observation period. For bafilomycin A1 (BafA1) treatment, the pancreas was perfused with KRBH buffer containing 2.8 mmol/l glucose and 100 nmol/l BafA1 (Sigma-Aldrich) for 30 min before perfusion with 16.7 mmol/l glucose. The insulin levels in the perfusate were measured using a mouse insulin ELISA kit (Morinaga, Tokyo, Japan).

Characterisation of impaired insulin secretion in Wfs1 deficiency. (a) Time course profiles of insulin secretion during perfusion, and (b) comparison of the amount of insulin secretion in the first phase (6–10 min) and second phase (11–20 min) on stimulation with 16.7 mmol/l glucose in 10-week-old male WT (grey circles) and Wfs1−/− (black squares) mice. (c) Time course profiles of insulin secretion during perfusion, and (d) amount of insulin secretion over 6–15 min on 30 mmol/l KCl stimulation in WT (grey circles) and Wfs1−/− (black squares). Data were obtained from six independent experiments and expressed as means ± SD. (e–h) Insulin granule dynamics in individual beta cells of 10-week-old male mice was assessed by TIRFM. Histogram of fusion events per cell surface area of 200 mm2 at 30 s intervals in primary cultured pancreatic beta cells from (e) WT and (f) Wfs1−/− mice stimulated with 16.7 mmol/l glucose. Old face (black bars): granules predocked to the plasma membrane and fused to the membrane by stimulation. Restless newcomer (dark grey bars): granules newly recruited and immediately fused to the plasma membrane by stimulation. Resting newcomer (light grey bars): granules newly recruited, docked and fused to the plasma membrane by stimulation. Times are presented in the format min:s. (g) Mean number of fusion events during the first phase (0:00–0:30 to 05:01–05:30 min) and second phase (05:30–06:00 to 15:01–15:30 min). (h) Mean number of fusion events in each mode of insulin granule exocytosis. Grey circles, WT beta cells (n = 3); black squares, Wfs1−/− beta cells (n = 4). Data are expressed as means ± SD. *p < 0.05, ***p < 0.001 (unpaired Student’s t test)

Effects of Ex-4 on GSIS in perfused pancreas in mice lacking Wfs1 and on pharmacological inhibition of V-ATPase. (a, b) Pancreases of 10-week-old male Wfs1−/− mice were perfused with KRBH buffer containing either 10 nmol/l Ex-4 or volume-matched saline in the presence of glucose at the concentrations indicated. (a) Changes in insulin levels in the perfusate, and (b) first-phase (6–10 min), second-phase (11–20 min) and total insulin secretion after initiation of perfusion with 16.7 mmol/l glucose in WT mice + saline (light grey circles), Wfs1−/− mice + saline (black squares) and Wfs1−/− mice +10 nmol/l Ex-4 (dark grey triangles). Data were obtained from six independent experiments (n = 6 each group) and expressed as means ± SD. Statistical analyses were performed with ANOVA followed by Bonferroni’s post hoc test. *p < 0.05, ***p < 0.001 compared with WT mice + saline or Wfs1−/− mice + saline. (c, d) The pancreases of WT mice were perfused with KRBH buffer containing 2.8 mmol/l glucose and 100 nmol/l BafA1 for 30 min, followed by 16.7 mmol/l glucose. (c) Changes in insulin levels in the perfusate, and (d) comparison of insulin secretion (6–15 min) in the presence of either vehicle (light grey circles, n = 5) or BafA1 (black squares, n = 4). (e, f) A 30 min perfusion with BafA1 was followed by perfusion with either Ex-4 or volume-matched saline. At 5 min after the initiation of Ex-4, insulin secretion was stimulated by perfusion with 16.7 mmol/l glucose. (e) Changes in insulin levels in the perfusate, and (f) comparison of insulin secretion in the first phase (6–10 min) and second phase (11–20 min) after initiation of perfusion with 16.7 mmol/l glucose in the presence of either saline (black squares, n = 5) or Ex-4 (dark grey triangles, n = 5). Data were obtained from five independent experiments and were expressed as means ± SD. **p < 0.01 (unpaired Student’s t test)
Total internal reflection fluorescence microscopy analysis
Primary cultured beta cells isolated from mouse pancreatic islets of 10-week-old male mice were infected with adenovirus carrying insulin-Venus generated as described previously [21] and were subjected to analysis by total internal reflection fluorescence microscopy (TIRFM) (Olympus, Tokyo, Japan) as previously described [21]. Briefly, cells were preincubated in KRBH containing 4.4 mmol/l glucose for 30 min, and glucose was then added to the chamber to a final concentration of 16.7 mmol/l. Images were acquired every 250 ms after glucose stimulation. The data analysis was performed using MetaMorph software version 6.1 (Universal Imaging, New York, NY, USA).
Ex-4 treatment in mice
For single treatments, we gave an intraperitoneal injection of Ex-4 at a dose of 24 nmol/kg body weight to 10-week-old Wfs1−/− mice. For chronic administration, 5-week-old Wfs1−/− mice were given an intraperitoneal injection of 24 nmol/kg Ex-4 twice daily for 4 weeks.
IPGTT
Ten-week-old male Wfs1−/− and littermate WT mice were subjected to a 6 h fast followed by an intraperitoneal glucose injection (2.0 g/kg). To assess the acute effects of Ex-4, Wfs1−/− mice were given a single injection of Ex-4 60 min before glucose loading. To assess the chronic effects of Ex-4, mice were given a 48 h drug washout period after the last Ex-4 injection and then underwent an IPGTT after a 12 h fast. Blood glucose levels were measured at 0, 2, 5, 15, 30 and 60 min after injection using an Antsense III (Horiba, Kyoto, Japan), and blood samples were collected from tail vein at the same time intervals. Plasma insulin levels were measured as described above.
Immunohistochemical analysis
Mouse pancreas was fixed in 4% paraformaldehyde, embedded in paraffin and sectioned in thicknesses of 4 μm. Serial sections were stained with antibodies (ESM Table 2). Immunodetection of phosphorylated AMP-activated kinase (AMPK) required amplification of the primary signal using the TSA kit (Perkin Elmer, Waltham, MA, USA). Images were captured using a Keyence Biozero microscope with the BZ-II software (Keyence, Osaka, Japan). Quantitation of beta cell mass was performed as previously described [22].
Isolation of islets from mice
Islets were isolated from 10- to 12-week-old male mice by ductal collagenase digestion of the pancreas as previously described [10]. All experiments on isolated islets were carried out after 24 h culture following isolation. Isolated islets were incubated with or without 10 nmol/l Ex-4 for 24 h, and gene expression and protein analyses were then carried out.
Protein analysis
Proteins extracted from isolated islets were resolved on 4–20% gradient polyacrylamide gels, blotted on to a nitrocellulose membrane. Protein-band densitometry was determined using the same membrane by pixel intensity using NIH Image J software version 1.51s (freely available at http://rsb.info.nih.gov/ij/index.html) [23]. Antibodies used for immunoblotting are included in ESM Table 3. The antibodies against WFS1 were generated as described previously [9].
Gene expression analysis
We isolated RNA using the PicoPure RNA isolation kit (Thermo Fisher Scientific, Waltham, MA, USA) and DNase I digestion, and synthesised cDNA using the High-Capacity cDNA Reverse Transcription kit (Applied Biosystems, Waltham, MA, USA). We then performed quantitative real-time PCR with PowerUp SYBR Green Master Mix (Applied Biosystems) in an ABI StepOnePlus Real-Time PCR system. We calculated relative gene expression levels by the ΔΔCt method using Cypa (encoding cyclophilin A) as an internal control. Primer sequences are listed in ESM Table 4.
Statistical analysis
Quantitative data are presented as means ± SD. Results were evaluated with a two-tailed Student’s t test or one-way ANOVA, as appropriate, with the use of GraphPad Prism software version 7 (GraphPad Software, La Jolla, CA, USA). Significant differences revealed by ANOVA were assessed with Bonferroni’s test. A p value <0.05 was considered statistically significant.
Result
Liraglutide modulated beta cell function and improved glycaemic control in an insulin-dependent woman with Wolfram syndrome
The woman presented typical disease-related clinical features other than neuropsychiatric symptoms (ESM Table 1). She carried the WFS1 mutation of the 5-amino-acid in-frame deletion located between a predicted transmembrane domain and a luminal domain of WFS1 (ESM Fig. 1a). This mutation has deleterious effects on protein expression, accounting for loss of function of WFS1 (ESM Fig. 1b). The woman weighed 52.6 kg and was not obese. Her daily insulin dose was 40 U, given as a bolus of injection of 18 U, with 22 U as basal insulin, and her glycaemic status was indicated by an HbA1c of 60 mmol/mol (7.6%) (Table 1). Fasting CPR was very low but detectable (0.077 nmol/l), yielding CPI and SUIT index values of 0.65 nmol/mmol and 2.13, respectively (Table 2). In addition, an evaluation of residual insulin reserve yielded ΔCPR and urinary CPR excretion of 0.08 nmol/l and 2.16 nmol/24 h, respectively (Table 2).
After initiation of liraglutide treatment, her fasting plasma glucose levels were maintained in the range 7.3–9 mmol/l, and HbA1c gradually decreased from the baseline of 60 mmol/mol (7.6%) to a minimum of 52 mmol/mol (6.9%) (Table 1); there was also a lower incidence of hypoglycaemia. Although her body weight initially showed a 1.2 kg decrease from baseline, it later returned to baseline values. As her glycaemic control was improving, the daily insulin dose was titrated to achieve a 20% reduction from the baseline dose. Secretory function was assessed by measuring CPR after a 48 h drug washout period. Despite decreased fasting plasma glucose levels, fasting CPR values were maintained above the baseline level (Table 2.) At 4 weeks after the start of the intervention, the SUIT index and CPI had increased 3.6- and 2.6-fold, respectively, compared with baseline (Table 2). Increases in the SUIT index and CPI were also evident at 16 weeks. This suggested that clinical use of a GLP-1 receptor agonist might facilitate glycaemic control and even modulate beta cell function in individuals with Wolfram syndrome who are on long-standing insulin therapy.
Glucose and potassium-stimulated insulin secretion are impaired in Wfs1 −/− mice
The glucose-lowering effects of the GLP-1 receptor agonist observed in this woman with Wolfram syndrome suggested that its use at an earlier stage of the disease might possibly prevent a deterioration of beta cell function, and thereby modulate progression of diabetes. To better understand the role of WFS1 in regulating beta cell function, we analysed insulin secretion in the perfused pancreases of young Wfs1−/− mice at 10 weeks of age, in which both fasting and fed blood glucose levels are indistinguishable from those in WT mice. Perfusion with 16.7 mmol/l glucose elicited a marked insulin release in wild-type (WT) mice, whereas the insulin secretion rapidly triggered by glucose was apparently blunted in Wfs1−/− mice (Fig. 1a). Insulin secretion in the first phase (6–10 min) was decreased by 53% compared with WT mice (Fig. 1b). In contrast, insulin secretion in the second phase (11–20 min), was not affected, suggesting that WFS1 is essential for insulin secretion immediately triggered by glucose mediated-depolarisation. In support of this notion, perfusion with 30 mmol/l KCl also failed to stimulate insulin secretion in Wfs1−/− mice (Fig. 1c). Insulin secretion during the 10 min period after the initiation of perfusion with KCl was decreased by 59% compared with that in WT mice (Fig. 1d). To exclude in vivo factors affecting insulin secretion, we assessed insulin secretion from isolated islets. Although basal insulin secretion was not affected in Wfs1−/− islets, insulin secretion stimulated by glucose and by KCl was significantly decreased, by 36% and 40%, respectively (ESM Fig. 2).
We next investigated the dynamics of insulin granules in living pancreatic beta cells using TIRFM. We classified the modes of insulin granule exocytosis into three classes depending on insulin granule dynamics [21]: (1) old face; (2) restless newcomer; and (3) resting newcomer. Glucose-induced insulin granule exocytosis comprised mainly restless newcomer. The number of fusion events immediately triggered by glucose stimulation was specifically decreased in Wfs1−/− beta cells (Fig. 1e,f). The number of fusion events in the first phase (00:00–00:30 to 05:01–05:30 min; where times are presented as min:s) was decreased by 41% compared with that in WT beta cells (Fig. 1g), whereas the number in the second phase (05:30–06:00 to 15:01–15:30 min) was not affected. Among the three granule classes, only the restless newcomer was found to be involved in the reduced exocytosis (46% reduction; Fig. 1h).
Activation of GLP-1 receptor stimulates glucose-stimulated insulin secretion in Wfs1 −/− mice
We investigated the secretagogue effects of Ex-4 in the perfused pancreas. Ex-4 restored maximum insulin release and persistently potentiated subsequent insulin release (Fig. 2a). Insulin secretion in the first and second phases was significantly increased, by 3.9-fold and 5.6-fold, respectively, compared with perfusion with saline (154 mmol/l NaCl) as a control; it was 1.6-fold and 3.2-fold higher than that in WT mice, resulting in a significant increase in total insulin secretion (Fig. 2b). In addition, Ex-4 potentiated glucose-stimulated insulin secretion (GSIS) in isolated islets from Wfs1−/− mice, which was similar to its effects in WT islets (ESM Fig. 2).
WFS1 reportedly contributes to the protein stability of vacuolar-type H+-ATPase V1A (ATP6V1a) subunit in human clonal cells [24]. Wfs1−/− islets exhibited a 44% reduction in expression of this protein compared with WT islets (ESM Fig. 3). Because secretory granules are acidified via a proton gradient established and maintained by V-ATPase, inadequate granular acidification in Wfs1−/− beta cells is probably caused by a failure of V-ATPase function. We therefore examined whether disturbed granular acidification affected GSIS and whether Ex-4 could potentiate GSIS under such conditions. Pretreatment with BafA1, a chemical inhibitor of V-ATPase, severely attenuated GSIS (Fig. 2c) and resulted in a 67% reduction in insulin secretion (Fig. 2d). Importantly, Ex-4 apparently potentiated GSIS in the presence of BafA1 (Fig. 2e). Insulin secretion in the first and second phases increased by 4.8-fold and 22.4-fold, respectively (Fig. 2f).
We next assessed the acute effects of activating GLP-1 receptors in mice under physiological conditions. The 10-week-old Wfs1−/− mice showed normal baseline blood glucose levels, but an IPGTT revealed that they had glucose intolerance (Fig. 3a). Ex-4 corrected the glucose excursion in Wfs1−/− mice, and they then showed better glucose tolerance than WT mice (Fig. 3a,b). In terms of insulin secretion, Wfs1−/− mice showed a blunted rapid insulin secretory response (Fig. 3c). Although the maximal secretory response appeared with a slight delay, Ex-4 potentiated GSIS (Fig. 3c), resulting in a significantly increased AUC for insulin release during the 15 min period after glucose loading (Fig. 3d).

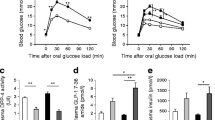
Acute effects of Ex-4 on glucose tolerance and GSIS in Wfs1−/− mice. Ten-week-old male Wfs1−/− mice were intraperitoneally injected with either 24 nmol/kg body weight Ex-4 or control saline at 60 min before glucose loading for an IPGTT. (a) Changes in blood glucose levels and (b) comparison of the AUC for glucose (0–60 min). (c) Change in plasma insulin levels and (d) comparison of the AUC for insulin secretion (0–15 min) in WT mice + saline (light grey circles), Wfs1−/− mice + saline (black squares) and Wfs1−/− mice + Ex-4 (dark grey triangles). Data are means ± SD (n = 6 for each group). Statistical analyses were performed with ANOVA followed by Bonferroni’s post hoc test. *p < 0.05, **p < 0.01, ***p < 0.001 compared with WT mice + saline or Wfs1−/− mice + saline
Beta cell function in Wfs1 −/− mice is essentially restored by prolonged activation of GLP-1 receptors
To assess the effects of prolonged activation of GLP-1 receptors, Wfs1−/− mice at 5 weeks of age were given Ex-4 for 4 weeks. Following a 48 h drug washout period, we performed IPGTTs after the mice had undergone a 12 h fast (Fig. 4a). After this intervention, there was an amelioration of the glucose excursion in Wfs1−/− mice (Fig. 4b,c), indicating an off-drug effect of Ex-4. Importantly, the insulin secretory dynamics were indistinguishable from those in WT mice, and, notably, maximal insulin responses appeared at 2 and 15 min, essentially as in WT mice (Fig. 4d). The AUC for insulin increased by 1.39-fold relative to the control treatment (Fig. 4e). Treatment with Ex-4 did not alter beta cell mass (Fig. 4f, g), body weight (ESM Fig. 4a) or insulin sensitivity (ESM Fig. 4b). Therefore, improved glucose tolerance with prolonged activation of GLP-1 receptors is likely to be attributable to restored beta cell function.

Chronic effects of Ex-4 on glucose tolerance, GSIS and beta cell mass in Wfs1−/− mice. (a) The experimental procedure presented schematically. Five-week-old male Wfs1−/− mice received either a 4 week Ex-4 treatment regimen or saline (n = 8 for each group). Following a 48 h drug washout period, an IPGTT was performed after 12 h of fasting. (b) Changes in blood glucose levels, and (c) comparison of the AUC (0–60 min) for blood glucose among the study groups. (d) Changes in plasma insulin levels, and (e) comparison of the AUC for plasma insulin (0–30 min) among the study groups. (f) Immunohistochemical analysis using anti-insulin and anti-glucagon antibodies was performed on pancreatic sections. Representative images under low magnification (upper; scale bar, 200 μm) and high magnification (lower; scale bar, 20 μm) are shown. (g) Total pancreas area and islet area were determined using ImageJ software. Beta cell mass was measured using the calculation: ([islet area/total pancreas area] × pancreatic weight). Light grey circles, WT mice + saline; black squares, Wfs1−/− mice + saline; dark grey triangles, Wfs1−/− mice + Ex-4 (n = 5 for each group). Data are means ± SD. Statistical analyses were performed with ANOVA followed by Bonferroni’s post hoc test. *p < 0.05, **p < 0.01, compared with WT mice + saline or Wfs1−/− mice + saline
Ex-4 alleviates beta cell stress resulting from Wfs1 deficiency
The restoration of beta cell function suggested that, in addition to its acute amplification of insulin secretion, Ex-4 might also alleviate fundamental defects leading to beta cell dysfunction. To elucidate the effects of Ex-4 on beta cell stress, we assessed phosphorylation on Ser51 of eukaryotic initiation factor 2α (p-EIF2α), which mediates the attenuation of global protein translation in response to various cellular stresses [25]. As shown in Fig. 5a, the overall intensity of immunoreactivity for p-EIF2α appeared to be increased in Wfs1−/− islet cells compared with WT cells. This was particularly apparent when the relative fluorescence intensities of p-EIF2α were compared between islet cells and acinar cells in the WT and Wfs1−/− mice (Fig. 5a). Ex-4 treatment specifically reduced p-EIF2α in islet cells without affecting neighbouring exocrine acinar cells. Concomitantly, the apparently reduced immunoreactivity of insulin in Wfs1−/− beta cells was partly restored.

Ex-4 alleviates beta cell stress and produces a reversal of reduced p-AMPK and increased TXNIP expression. (a) Immunohistochemical analysis using anti-p-EIF2α and anti-insulin antibodies was performed on pancreatic sections. The experiments were conducted three times for each group, and representative data are shown. The white arrows indicate exocrine acinar cells, and white lines denote islet borders. (b–d) Isolated islets from 10-week-old male WT and Wfs1−/− mice were treated with saline or 10 nmol/l Ex-4 for 24 h. Quantitative real-time PCR of islets for genes involved in oxidative stress (b), ER stress (c), and beta cell function (d). (e, f) Immunohistochemical analysis of pancreatic sections using anti-insulin antibodies with anti-p-AMPK (e) and anti-TXNIP (f) antibodies. (g) Isolated islets of 10- to 12-week-old male WT and Wfs1−/− mice were treated with either vehicle (saline) or 10 nmol/l Ex-4 for 24 h. Protein extracts were analysed by immunoblotting using the indicated antibodies. Representative images of three independent experiments are shown. Densitometry was performed, and p-AMPK and TXNIP were measured and normalised to total-AMPK and α-tubulin, respectively. Relative p-AMPK/total-AMPK and TXNIP/α-tubulin are shown graphically. (h) Relative mRNA levels for Txnip, Atf5 and Chrebp in islets were determined by quantitative real-time PCR. (a, e, f) Scale bars, 20 μm. (b–d, g, h) Quantitative data were obtained from at least three independent experiments. Bar charts show means ± SD, and individual data points are shown in the graphs. White bars, WT islets + saline; dark grey bars, Wfs1−/− islets + saline; light grey bars, Wfs1−/− islets + Ex-4. Statistical analyses were performed with ANOVA followed by Bonferroni’s post hoc test. *p < 0.05, **p < 0.01, ***p < 0.001 compared with WT mice + saline or Wfs1−/− mice + saline. INS, insulin
We next tested whether Ex-4 was directly modulating stress responses in isolated islets. Oxidative stress-responsive genes including Nrf2 (also known as Nfe2l2), Sod1, Hmox1 and Txnrd1 were significantly elevated in Wfs1−/− islets (Fig. 5b). Although increases in Cat (encoding catalase), Sod1 and Txnrd1 were not affected, Ex-4 treatment lowered the overall magnitude of oxidative stress in Wfs1−/− islets. Moreover, ER stress-induced transcripts appeared to increase in the Wfs1−/− islets. Ex-4 significantly lowered Chop (also known as Ddit3) and spliced Xbp1 (sXbp1), but did not affect Bip (also known as Hspa5) and Atf4 (Fig. 5c). In relation to genes involved in beta cell function, Ex-4 reversed the reductions in Ins2 and Pdx1, and the increase in Ucp2, although the change in Ucp2 was not significant (Fig. 5d). Collectively, activation of GLP-1 receptors was shown to modulate stress responses against the oxidative and ER stresses imposed by Wfs1 deficiency, and was associated with improved beta cell function.
Ex-4 modulates p-AMPK and thioredoxin interacting protein in Wfs1 −/− beta cells
Cell functions under various stressed conditions can be restored by alleviating ER stress, via activation of AMPK [26,27,28]. The phosphorylation at Thr172 of AMPK (p-AMPK), which is involved in increasing its own kinase activity, was apparently decreased in Wfs1−/− beta cells, and was partly restored by Ex-4 (Fig. 5e). We tested the direct effects of Ex-4 on p-AMPK in isolated islets. Wfs1−/− islets showed a 30% decrease in p-AMPK/total-AMPK compared with WT islets. Ex-4 significantly increased p-AMPK in both WT and Wfs1−/− islets, by 46% and 37%, respectively, compared with control, without affecting AMPK expression (Fig. 5g). Thioredoxin interacting protein (TXNIP), one of the downstream targets of AMPK [29, 30], is a key mediator of beta cell dysfunction in diabetes and is induced by oxidative and ER stress [31]. Whereas Wfs1−/− beta cells showed nuclear accumulation of TXNIP, indicating its activation [32], this was inhibited by Ex-4 (Fig. 5f). Production of TXNIP increased by 3.3-fold in Wfs1−/− islets relative to that in WT islets. Ex-4 significantly reduced TXNIP production in Wfs1−/− islets, along with increasing p-AMPK (Fig. 5g). Activation of AMPK reportedly decreases Txnip transcription in clonal beta cells [29]. Although gene expression of key transcriptional mediators of Txnip, including Atf5 and Chrebp (also known as Mlxipl), was not affected, increased Txnip transcription in Wfs1−/− islets was reversed by Ex-4 (Fig. 5h). These results suggest a correlation between decreased p-AMPK and increased TXNIP in Wfs1−/− beta cells, and raise the possibility of therapeutic applications of activating GLP-1 receptors through their modulation in association with the alleviation of cellular stress (Fig. 6).

Hypothetical actions of Ex-4 in Wfs1-deficient pancreatic beta cells. Lack of WFS1 in the ER and insulin granules may cause beta cell stress and disturbed granular acidification, resulting in impaired GSIS and eventual beta cell loss. Ex-4 may ameliorate aberrant beta cell function through modulation of insulin secretion pathways, which require proper granular acidification, and alleviation of cellular stress. GLP-1R, GLP-1 receptor
Discussion
In Wolfram syndrome, diabetes mellitus progresses together with declining functional beta cell mass, perhaps through processes that initially cause individual cellular dysfunctions. The young Wfs1−/− mice in this experiment demonstrated impaired GSIS despite beta cell mass being maintained. Although chronic elevations of oxidative and ER stress in vivo lead to beta cell loss as well as dysfunction, we investigated the impact of Wfs1 deficiency on beta cell function and the effects of the GLP-1 receptor agonist as well as its direct actions on beta cells, irrespective of anti-apoptotic actions, in young Wfs1−/− mice in which glycaemic homeostasis has not yet noticeably deteriorated.
Our data clearly show WFS1 to be essential for the rapid insulin secretion triggered by membrane depolarisation, and that acute administration of Ex-4 potentiates GSIS in Wfs1−/− mice, thereby lowering glucose levels. WFS1 deficiency may affect the docking and fusion steps of exocytosis through perturbations of Ca2+ homeostasis and impaired intragranular acidification [15, 33, 34]. GLP-1 receptor agonists are known to modulate Ca2+ signalling through an elevation of cytosolic Ca2+. In addition, we demonstrated that Ex-4 reversed severely impaired GSIS caused by the pharmacological inhibition of V-ATPase, which mimics the disturbed intragranular acidification resulting from WFS1 deficiency [10]. This observation may provide insights into the pathophysiological implications of beta cell dysfunction and the acute actions of GLP-1 receptor agonists in Wfs1−/− mice. However, the precise function of WFS1 and its roles in secretagogue pathways, including GLP-1 receptor signalling, have yet to be fully elucidated. Future investigations are needed to address these fundamental issues.
Chronic administration of Ex-4 restored GSIS in Wfs1−/− mice via improved functioning of existing beta cells rather than having an effect through increments in beta cell mass. We demonstrated that treatment with Ex-4 decreased stress-related markers in Wfs1-deficient beta cells and isolated islets, while exerting opposing effects on insulin production. These findings may, at least partly, support previous literature reporting that Ex-4 prevents ER stress-mediated apoptosis by modulating the unfolded protein response in clonal beta cells [35]. However, whereas Wfs1 deficiency increased cellular stress, the rate of beta cell apoptosis did not increase significantly in Wfs1−/− mice in which normoglycaemia was still maintained (K. Amo-Shiinoki, K. Tanabe, M. Hatanaka and Y. Tanizawa, unpublished data). We therefore speculated that Wfs1−/− beta cells may adapt to persistent cellular stress through as yet unknown processes, which may be involved in the regulation of cell viability and, perhaps, identity [36]. Anti-stress effects of Ex-4 correlate with reduced TXNIP and partial restoration of p-AMPK, which at least partially elucidates the mechanisms of this reversal [26, 31]. As consistent effects of Ex-4 on TXNIP and p-AMPK were observed in isolated islets, future investigations are needed to elucidate whether these actions of the GLP-1 receptor agonist are direct or a consequence of the mitigation of cellular stress.
In our insulin-dependent patient with Wolfram syndrome, liraglutide modulated beta cell function and improved glycaemic control, with a slight reduction in the insulin dose required. These effects persisted, at least for the 6 month observation period, suggesting a possible clinical role of GLP-1-based therapy in Wolfram syndrome. Residual beta cell function was, however, very limited, despite being improved by liraglutide. Moreover, beta cell function peaked at 4 weeks of intervention, and then declined at 16 weeks. This suggests that activation of GLP-1 receptors might only temporarily affect beta cells. Although glucagon levels were not examined in the present study, it is more likely that the effects of liraglutide on gastric emptying rate and alpha cells rather than beta cells may have contributed to the improvement in glycaemic control. On the other hand, whether the endogenous GLP-1 response is affected by WFS1 deficiency has not been investigated. This issue requires further examination to elucidate the significance and efficacy of GLP-1-based therapy in Wolfram syndrome.
The finding that Ex-4 mitigated oxidative and ER stress may have implications for understanding the mechanisms of its action in diabetes mellitus and, more importantly, for treating the neurodegeneration that is a major determinant of the deleterious consequences of this syndrome. In support of this hypothesis, the neuroprotective effects of GLP-1 receptor agonist have been demonstrated in various murine models, and clinical trials have recently been initiated to investigate them in Parkinson’s disease and Alzheimer’s disease, in which oxidative and ER stress may have pathogenic roles [37]. Based on our finding that Ex-4 mitigated cellular stress while improving function in Wfs1-deficient beta cells, we suggest that GLP-1 receptor agonist therapy may provide a means of attenuating or even slowing the progression of diabetes and neuronal degeneration in individuals with Wolfram syndrome. In addition, a recent study has demonstrated that dantrolene, which targets ryanodine receptors localised to the ER, could be a potential treatment for Wolfram syndrome [18, 38]. Future investigations are needed to elucidate whether a combination of a GLP-1 receptor agonist and dantrolene would have greater therapeutic efficacy.
In conclusion, our experimental data and clinical observations may have important clinical implications because there is no preventive treatment for Wolfram syndrome. The information obtained here may therefore aid in designing future clinical studies to assess the effects of GLP-1 receptor agonists on the natural history and prognosis of the disease. The use of GLP-1 receptor agonists in paediatric patients also merits careful consideration given the myriad potential impacts of this agent.
Data availability
All data generated and analysed during this study are included in this published article and its supplementary information files.
Abbreviations
- AMPK:
-
AMP-activated kinase
- ATP6V1a:
-
Vacuolar-type H+-ATPase V1A
- BafA1:
-
Bafilomycin A1
- CPI:
-
CPR index
- CPR:
-
C-peptide immunoreactivity
- EIF2α:
-
Eukaryotic initiation factor 2α
- ER:
-
Endoplasmic reticulum
- EX-4:
-
Exendin-4
- GLP-1:
-
Glucagon-like peptide-1
- GSIS:
-
Glucose-stimulated insulin secretion
- KRBH:
-
KRB HEPES
- SUIT:
-
Secretory units of islets in transplantation
- TIRFM:
-
Total internal reflection fluorescence microscopy
- TXNIP:
-
Thioredoxin interacting protein
- WT:
-
Wild-type
- WFS1:
-
Wolfram syndrome 1 protein
References
Barrett TG, Bundey SE, Macleod AF (1995) Neurodegeneration and diabetes: UK nationwide study of Wolfram (DIDMOAD) syndrome. Lancet 346:1458–1463
Minton JA, Rainbow LA, Ricketts C, Barrett TG (2003) Wolfram syndrome. Rev Endocr Metab Disord 4:53–59
Marshall BA, Permutt MA, Paciorkowski AR et al (2013) Phenotypic characteristics of early Wolfram syndrome. Orphanet J Rare Dis 8:64
Matsunaga K, Tanabe K, Inoue H et al (2014) Wolfram syndrome in the Japanese population; molecular analysis of WFS1 gene and characterization of clinical features. PLoS One 9:e106906
Karasik A, O'Hara C, Srikanta S et al (1989) Genetically programmed selective islet beta-cell loss in diabetic subjects with Wolfram’s syndrome. Diabetes Care 12:135–138
Hardy C, Khanim F, Torres R et al (1999) Clinical and molecular genetic analysis of 19 Wolfram syndrome kindreds demonstrating a wide spectrum of mutations in WFS1. Am J Hum Genet 65:1279–1290
Hofmann S, Philbrook C, Gerbitz KD, Bauer MF (2003) Wolfram syndrome: structural and functional analyses of mutant and wild-type wolframin, the WFS1 gene product. Hum Mol Genet 12:2003–2012
Inoue H, Tanizawa Y, Wasson J et al (1998) A gene encoding a transmembrane protein is mutated in patients with diabetes mellitus and optic atrophy (Wolfram syndrome). Nat Genet 20:143–148
Takeda K, Inoue H, Tanizawa Y et al (2001) WFS1 (Wolfram syndrome 1) gene product: predominant subcellular localization to endoplasmic reticulum in cultured cells and neuronal expression in rat brain. Hum Mol Genet 10:477–484
Hatanaka M, Tanabe K, Yanai A et al (2011) Wolfram syndrome 1 gene (WFS1) product localizes to secretory granules and determines granule acidification in pancreatic beta-cells. Hum Mol Genet 20:1274–1284
Cagalinec M, Liiv M, Hodurova Z et al (2016) Role of mitochondrial dynamics in neuronal development: mechanism for Wolfram syndrome. PLoS Biol 14:e1002511
Ishihara H, Takeda S, Tamura A et al (2004) Disruption of the WFS1 gene in mice causes progressive beta-cell loss and impaired stimulus-secretion coupling in insulin secretion. Hum Mol Genet 13:1159–1170
Takei D, Ishihara H, Yamaguchi S, Yamada et al (2006) WFS1 protein modulates the free Ca(2+) concentration in the endoplasmic reticulum. FEBS Lett 580:5635–5640
Fonseca SG, Ishigaki S, Oslowski CM et al (2010) Wolfram syndrome 1 gene negatively regulates ER stress signaling in rodent and human cells. J Clin Invest 120:744–755
Deriy LV, Gomez EA, Jacobson DA et al (2009) The granular chloride channel ClC-3 is permissive for insulin secretion. Cell Metab 10:316–323
Orci L, Halban P, Perrelet A, Amherdt M, Ravazzola M, Anderson RG (1994) pH-independent and -dependent cleavage of proinsulin in the same secretory vesicle. J Cell Biol 126:1149–1156
Fonseca SG, Fukuma M, Lipson KL et al (2005) WFS1 is a novel component of the unfolded protein response and maintains homeostasis of the endoplasmic reticulum in pancreatic beta-cells. J Biol Chem 280:39609–39615
Lu S, Kanekura K, Hara T et al (2014) A calcium-dependent protease as a potential therapeutic target for Wolfram syndrome. Proc Natl Acad Sci U S A 111:E5292–E5301
Yamada Y, Fukuda K, Fujimoto S et al (2006) SUIT, secretory units of islets in transplantation: an index for therapeutic management of islet transplanted patients and its application to type 2 diabetes. Diabetes Res Clin Pract 74:222–226
Miki T, Minami K, Shinozaki H et al (2005) Distinct effects of glucose-dependent insulinotropic polypeptide and glucagon-like peptide-1 on insulin secretion and gut motility. Diabetes 54:1056–1063
Shibasaki T, Takahashi H, Miki T et al (2007) Essential role of Epac2/Rap1 signaling in regulation of insulin granule dynamics by cAMP. Proc Natl Acad Sci U S A 104:19333–19338
Tanabe K, Liu Z, Patel S et al (2008) Genetic deficiency of glycogen synthase kinase-3beta corrects diabetes in mouse models of insulin resistance. PLoS Biol 6:e37
Girish V, Vijayalakshmi A (2004) Affordable image analysis using NIH Image/ImageJ. Indian J Cancer 41:47
Gharanei S, Zatyka M, Astuti D et al (2013) Vacuolar-type H+-ATPase V1A subunit is a molecular partner of Wolfram syndrome 1 (WFS1) protein, which regulates its expression and stability. Hum Mol Genet 22:203–217
Volchuk A, Ron D (2010) The endoplasmic reticulum stress response in the pancreatic beta-cell. Diabetes Obes Metab 12(Suppl 2):48–57.z
Matsuda T, Takahashi H, Mieda Y et al (2015) Regulation of pancreatic beta cell mass by cross-interaction between CCAAT enhancer binding protein beta induced by endoplasmic reticulum stress and AMP-activated protein kinase activity. PLoS One 10:e0130757
Nyblom HK, Sargsyan E, Bergsten P (2008) AMP-activated protein kinase agonist dose dependently improves function and reduces apoptosis in glucotoxic beta-cells without changing triglyceride levels. J Mol Endocrinol 41:187–194
Steinberg GR, Kemp BE (2009) AMPK in health and disease. Physiol Rev 89:1025–1078
Shaked M, Ketzinel-Gilad M, Cerasi E, Kaiser N, Leibowitz G (2011) AMP-activated protein kinase (AMPK) mediates nutrient regulation of thioredoxin-interacting protein (TXNIP) in pancreatic beta-cells. PLoS One 6:e28804
Wu N, Zheng B, Shaywitz A et al (2013) AMPK-dependent degradation of TXNIP upon energy stress leads to enhanced glucose uptake via GLUT1. Mol Cell 49:1167–1175
Shalev A (2014) Minireview: Thioredoxin-interacting protein: regulation and function in the pancreatic beta-cell. Mol Endocrinol 28:1211–1220
Saxena G, Chen J, Shalev A (2010) Intracellular shuttling and mitochondrial function of thioredoxin-interacting protein. J Biol Chem 285:3997–4005
Li DQ, Jing X, Salehi A et al (2009) Suppression of sulfonylurea- and glucose-induced insulin secretion in vitro and in vivo in mice lacking the chloride transport protein ClC-3. Cell Metab 10:309–315
Verhage M, Sorensen JB (2008) Vesicle docking in regulated exocytosis. Traffic 9:1414–1424
Yusta B, Baggio LL, Estall JL et al (2006) GLP-1 receptor activation improves beta cell function and survival following induction of endoplasmic reticulum stress. Cell Metab 4:391–406
Dor Y, Glaser B (2013) beta-cell dedifferentiation and type 2 diabetes. N Engl J Med 368:572–573
Muscogiuri G, DeFronzo RA, Gastaldelli A, Holst JJ (2017) Glucagon-like peptide-1 and the central/peripheral nervous system: crosstalk in diabetes. Trends Endocrinol Metab 28:88–103
Urano F (2016) Wolfram Syndrome: diagnosis, management, and treatment. Curr Diab Rep 16:6
Acknowledgements
The authors would like to thank members of the division of Yamaguchi University Graduate School of Medicine for their helpful discussion with preparing the manuscript.
Funding
KT is supported by a Grant-in-Aid for Scientific Research (grant number 16K09752). YT is supported by a Grant-in-Aid for Scientific Research (grant numbers 23390080 and 15H04849) and by Takeda Science Foundation. KA-S is supported by a Grant-in-Aid for Scientific Research (grant number 15K21198), a Japan Diabetes Society Junior Scientist Development Grant supported by Novo Nordisk Pharma Ltd., a Grant for Front Runner of Future Diabetes Research, and Grants for young researchers from the Japan Association for Diabetes Education and Care and from Banyu Life Science Foundation International. MH is supported by a Grant-in-Aid for Scientific Research (grant number 15K09390).
Author information
Authors and Affiliations
Contributions
All authors participated substantially in the investigations reported here as indicated. KT and YT conceived and designed experimental studies, collected, analysed and interpreted data, and drafted and critically revised the manuscript. MK designed the study, conducted experiments, collected, analysed and interpreted data and wrote and revised the manuscript. KA-S, MH, TM, and HT designed experimental studies and collected, analysed and interpreted data; they also critically reviewed the manuscript and gave valuable suggestions for its revision. SS and YY assisted in the design of the study and contributed significantly to data interpretation, critical reading and revision of the manuscript. All authors carefully read and approved the final version to be published. YT is the guarantor of this work.
Corresponding authors
Ethics declarations
The authors declare that there is no duality of interest associated with this manuscript.
Additional information
Manabu Kondo and Katsuya Tanabe are joint first authors.
Electronic supplementary material
ESM
(PDF 1.67 mb)
Rights and permissions
About this article

Cite this article
Kondo, M., Tanabe, K., Amo-Shiinoki, K. et al. Activation of GLP-1 receptor signalling alleviates cellular stresses and improves beta cell function in a mouse model of Wolfram syndrome. Diabetologia 61, 2189–2201 (2018). https://doi.org/10.1007/s00125-018-4679-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-018-4679-y