
Abstract
In recent years, CRISPR–Cas9 technology is widely acknowledged for having major applications in the field of biotechnology for editing genome of any organism to treat a variety of complex diseases and for other purposes. The acronym ‘CRISPR–Cas’ stands for clustered regularly interspaced short palindromic repeats–CRISPR-associated genes. This genetic organization exists in prokaryotic organisms and aids in the development of adaptive immunity since a protein called Cas9 nuclease cleaves specific target nucleic acid sequences from foreign invaders and destroys them. This mode of action has gained interest of the researchers to understand the insights of CRISPR–Cas9 technology. Here, we review that CRISPR–Cas organization is restricted to two classes and possesses different protein effectors. We also review the architecture of CRISPR loci, mechanism involved in genome editing by CRISPR–Cas9 technology and pathways of repairing double-strand breaks (DSBs) generated during the process of genome editing. This review also presents the strategies to increase the Cas9 specificity and reduce off-target activity to achieve accurate genome editing. Further, this review provides information on CRISPR tools used for genome editing, databases that are required for storing data on loci, strategies for delivering CRISPR–Cas9 to cells under study and applications of CRISPR–Cas9 to various fields. Safety measures are implemented on this technology to avoid misuse or ethical issues. We also discuss about the future aspects and potential applications of CRISPR–Cas9 technology required mainly for the treatment of dreadful diseases, crop improvement as well as genetic improvement in human.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
‘CRISPR–Cas9’ stands for ‘Clustered Regularly Interspaced Short Palindromic Repeats-Cas CRISPR-associated protein 9’. CRISPR–Cas9 technology has immense potential in genome editing as well as gene therapy for treating diseases related to cancer, infections and genetic disorders (Doudna and Charpentier 2014; Hsu et al. 2014). This technology serves as a framework to carry out genome editing for the purpose of investigating and studying various diseases (Cox et al. 2015; Jinek et al. 2013; Mali et al. 2013). CRISPR–Cas9 organization helps in determining the activity of a gene during any illness state, rectifying a gene that causes harmful mutation and switching off/on cancer causing genes or switching on tumor suppressors (Doudna and Charpentier 2014; Hsu et al. 2014; Charpentier and Marraffini 2014; Wang et al. 2016). This technology possesses enormous capability to cure patients suffering from cancer by increasing the efficacy of immunotherapy and minimizing the price of T cell treatment (Eyquem et al. 2017; Ren et al. 2017; Legut et al. 2018). CRISPR–Cas9 technology can also cure many diseases related to nerve cells such as Duchenne muscular dystrophy, heart related diseases and diseases arising from the breakdown of immune system such as autoimmune disorders (Barrangou and Doudna 2016; Heidenreich and Zhang 2016; Strong and Musunuru 2017; Xiong et al. 2016). As this technology has a wide range of applications in various fields, it becomes essential to have a detailed understanding of this technology and its future aspects.
A wide range of strategies exist in prokaryotes such as archaea and bacteria that provide resistance against invading foreign agents mainly viruses and plasmids. Prokaryotic organisms have the two strategies of immunity, viz. innate immunity and adaptive (acquired) immunity. In innate immunity, prokaryotes recognize the foreign invaders on encounter and contribute to first line of defense mechanism whereas adaptive immunity serves as the second line of defense mechanism by providing immunological responses and storing immunogenic memory for their defense during second encounter. Adaptive immunity in archaeal and bacterial genomes is due to the existence of CRISPR–Cas structure because CRISPR along with Cas proteins target foreign mobile genetic elements (MGEs) and hence, they are eradicated eventually (Barrangou et al. 2007; Van Der Oost et al. 2014). CRISPR–Cas organization provides immunogenic memory to bacteria in order to protect themselves from foreign invaders during second exposure (Marraffini and Sontheimer 2010). CRISPR–Cas system exists in prokaryotic genomes, and thereby characterizes about 83% of archaea and 45% of bacteria (Barrangou and Marraffini 2014).
CRISPR loci were reported to be nearly palindromic and were first observed in iap gene (gene causing alkaline phosphatase isozyme conversion) of Escherichia coli in an intergenic region upstream to the gene (Ishino et al. 1987). These DNA repeats were also studied in many bacterial species like Mycobacterium tuberculosis (Groenen et al. 1993), Streptococcus pyogenes (Hoe et al. 1999), Methanocaldococcus jannaschii (Bult et al. 1996), in archaeal species namely Haloferax mediterranei, Haloferax volcanii (Mojica et al. 1995), Thermotoga maritima (Nelson et al. 1999) and in filamentous cyanobacterium Anabaena sp. (Masepohl et al. 1996) as well as in other archaeal and bacterial species. The acronym ‘CRISPR’ was coined in 2002 (Jansen et al. 2002). In 2005, three autonomous groups reported that CRISPR–Cas organization occurs in prokaryotes (Bolotin et al. 2005; Mojica et al. 2005; Pourcel et al. 2005) and its contribution to adaptive immunity was proved in 2007 (Barrangou et al. 2007). Several studies on the mechanism of CRISPR–Cas system were experimentally carried out in vitro by infecting different hosts with different kinds of bacteriophages and plasmids (Barrangou et al. 2007; Díez-Villaseñor et al. 2010; Pougach et al. 2010; Westra et al. 2010). It was first reported in Streptococcus thermophilus that the bacterium integrated nucleotide sequences termed ‘protospacer’ of foreign MGEs into its CRISPR locus and developed protection against invaders and these sequences are termed as ‘spacers’ (Barrangou et al. 2007). As viruses evolve rapidly, the bacteria also need to develop effective defense mechanism against these viruses. Therefore, the cas genes of the CRISPR–Cas system evolve with magnificent variations in their gene repertoires and loci structure (Makarova et al. 2011, 2015).
In recent years, Cpf1 nuclease, also known as Cas12a, was identified in Lachnospiraceae bacterium ND2006 (LbCpf1) and Acidaminococcus sp. BV3L6 (AsCpf1) (Zetsche et al. 2015) and has also paved the way for their potential applicability in the field of genome editing (Zetsche et al. 2015, 2017; Kim et al. 2016a, b; Hur et al. 2016; Kleinstiver et al. 2016; Tang et al. 2017). Cas12a can effectively modify genomes of microorganisms with reduced destructive outcome compared to Cas9 and as a result, the enzyme can be largely implemented in the area of biotechnology (Swarts and Jinek 2018). CRISPR–Cas12a has also major applicability for manipulating genomes of plants (Kim et al. 2017), non-mammalian vertebrates (Moreno-Mateos et al. 2017), mammals (Kim et al. 2016; Hur et al. 2016), yeasts (Świat et al. 2017) and insects (Port and Bullock 2016). Another enzyme called Cas13a can only cleave target sites of RNA sequences. CRISPR–Cas13a can be useful for editing RNA sequences and developing RNA interference in higher class organisms to provide protection from viruses (Aman et al. 2018). This enzyme also helps in understanding the insights of RNA in eukaryotic organisms and treatment of diseases (Abudayyeh et al. 2017). CRISPR–Cas13a is also utilized for diagnostic test for identifying genetic materials within an organism that possess pathogenicity (Knott and Doudna 2018).
Genome engineering was first performed in bacteria (Jiang et al. 2013) and mammalian cells (Mali et al. 2013; Cong et al. 2013) by using CRISPR–Cas9 system. Gene editing technologies that are accomplished based on restriction enzymes, e.g., transcription activator-like effector nucleases (TALENs) and zinc-finger nuclease (ZFNs) depend on binding of protein–DNA (Gaj et al. 2013). The disadvantage of these technologies is that proteins are needed to be designed for each experiment (Barrangou and Doudna 2016). The Cas9 endonuclease from the bacterium, Streptococcus pyogenes, termed SpCas9 is widely employed for genome engineering where the gRNA guides the SpCas9 for cleaving the target sites based on DNA–RNA hybridization (Sander and Joung 2014). In addition to genome editing and disease treatment, CRISPR–Cas toolbox has recently emerged to provide various strategies for functional genomics screening, point-of-care diagnosis as well as live-cell imaging (Knott and Doudna 2018). In this review, we explain the mechanism involved in CRISPR–Cas9, its classification, its tools and databases developed for acquiring precise gene editing, increase in Cas9 specificity, off-target activity reduction, its delivery strategies to cultured cells, its applications and future perspectives.
Classification of CRISPR–Cas system
CRISPR–Cas organization occurs in two forms of classes, and is further categorized into six types (I–VI) and 27 subtypes (Makarova et al. 2015; Shmakov et al. 2017; Koonin et al. 2017). Large number of archaea (such as in entire hyperthermophiles) and bacteria are known to consist of Class 1 CRISPR–Cas system in their genomes, whereas the Class 2 system is known to exist in bacteria, but not in hyperthermophiles (Makarova et al. 2015; Chylinski et al. 2014). Based on the character of nuclease effector, Class 1 system comprises types I, III and IV which have multi-subunit Cas protein effector complexes whereas Class 2 system includes types II, V and VI with single protein effector modules. The nuclease effector proteins are necessary at interference stage (Makarova et al. 2011, 2013; Shmakov et al. 2015). CRISPR–Cas systems targeting the DNA viruses are type I, II and V, whereas type VI targets the RNA viruses. However, type III is both DNA and RNA targeting CRISPR–Cas system, though target for type IV system has not yet been identified (Koonin et al. 2017). Table 1 represents the classification of CRISPR–Cas organization and their few subtypes and effectors.
Structure of CRISPR loci
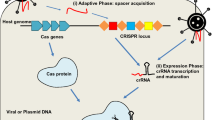
In CRISPR loci, a series of repeats are present that flank the ‘spacer’ sequence and this spacer sequence matches with the sequences in virus, plasmid or other pathogen genomic elements (Bolotin et al. 2005; Van der Oost et al. 2009; Horvath and Barrangou 2010; Terns and Terns 2011; Deveau et al. 2010). Generally, an AT-rich leader sequence is located in the upstream position of the CRISPR array (Jansen et al. 2002). On one end of the array, a set of conserved genes coding for varieties of Cas proteins, called CRISPR-associated (cas) genes are present (Marraffini and Sontheimer 2010). The structure of CRISPR loci is shown in Fig. 1.

Structure of CRISPR locus. In CRISPR locus, each ‘repeat’ sequence is flanked by ‘spacer’ sequence and these spacers match with the genomic sequences found in virus, plasmid or pathogen. Upstream to CRISPR array, leader sequence and CRISPR-associated (cas) genes are located
Mechanism involved in CRISPR–Cas system
The mechanism behind CRISPR–Cas organization operates in three noticeable phases: (1) adaptation, (2) expression and maturation, and (3) interference (Amitai and Sorek 2016; Puschnik et al. 2017).
Adaptation of CRISPR–Cas spacer sequences
The adaptation phase occurs in two steps; firstly, Cas proteins of the bacterium identify the invader and acquire specific sequences from foreign nucleic acids and these sequences are termed as ‘protospacer’ and secondly, the protospacer is incorporated in the extremity of the leader sequence in the CRISPR array as ‘spacer’ and this causes the first repeat of the CRISPR array to be extended (Pourcel et al. 2005; Yosef et al. 2012; Mojica et al. 2009). These spacers are responsible for creating immunological memory to archaea and bacteria for defense, in case, they encounter the MGEs for the second time (Bolotin et al. 2005; Mojica et al. 2005; Pourcel et al. 2005). Cas1 and Cas2 are essentially involved in this phase (Yosef et al. 2012).
Expression and maturation of CRISPR–Cas system
During the expression and maturation phase, the leader sequence situated upstream to the CRISPR loci, acts as a promoter and initiates transcription of the loci, giving rise to long precursor CRISPR RNA or pre-crRNA and subsequently, processing of this pre-crRNA into small and mature units, known as crRNA takes place (Pougach et al. 2010; Yosef et al. 2012; Wei et al. 2015). Representation of crRNA is exhibited by joining of a spacer region (sequence showing complementarity to the foreign nucleic acid) at the 5′ end to repeat sequence at the 3′ end (Garneau et al. 2010; Barrangou 2015).
Interference of CRISPR–Cas system
During the interference phase, Cas–crRNA complex formed as a result of recruitment of Cas proteins to crRNA, detects the foreign MGEs via Watson–Crick base pairing of sequences that is complementary to the crRNA and hence, the targeted element is subjected to cleavage (Amitai and Sorek 2016). Existence of a small conserved sequence (2–5 bp) called protospacer adjacent motif (PAM) juxtaposed to target site in the invading nucleic acid is essential for identification between self and non-self nucleic acids by the Cas–crRNA complex (Mojica et al. 2009; Deveau et al. 2008; Westra et al. 2013).
Structure of Cas9 enzyme
Cas9 enzyme has enormous possibilities in genome engineering (Wilkinson et al. 2019). Cas9 is a DNA endonuclease and it possesses two RNA molecules, i.e., crRNA and transactivating crRNA (tracrRNA). It can detect and degrade any foreign nucleic acids and so, Cas9 is extensively used in the area of biotechnology for genome editing (Mali et al. 2013; Cong et al. 2013). Structurally, Cas9 has two lobes, i.e., nuclease (NUC) lobe and recognition (REC) lobe (Jinek et al. 2014; Nishimasu et al. 2014). NUC lobe comprises two nuclease domains, i.e., HNH and RuvC and a PAM interacting domain (PI) (Nishimasu et al. 2014). REC lobe consists of a Bridge Helix (BH) which is rich in arginine and is divided into three α-helical sub-domains, i.e., REC1, REC2 and REC3 (Wilkinson et al. 2019).
Biology of CRISPR–Cas9 belonging to type II
Type II organization represents the following vital elements: genes, i.e., cas1, cas2 and cas9, CRISPR array, as well as a tracrRNA that shows complementarity to the sequence of CRISPR repeat (Chylinski et al. 2014; Deltcheva et al. 2011). During the acquisition of spacers, all the Cas signature proteins are associated (Heler et al. 2015; Wei et al. 2015), whereas in interference stage, only the role of Cas9 is significantly involved (Jinek et al. 2012; Sapranauskas et al. 2011). Class 2 type II organization codes for endonucleases such as Cas9 signature protein and a non-coding RNA called tracrRNA in addition to crRNA (Barrangou and Doudna 2016). Base pairing of crRNA and tracrRNA results into crRNA: tracrRNA hybrid, following which RNase III cleaves the hybrid, thereby, forming a mature dual-RNA hybrid (Deltcheva et al. 2011) and subsequently, recruitment of Cas9 proteins occurs (Jinek et al. 2012). Chimeric single guide RNA (sgRNA) is constructed by hybridizing 5′ end of the tracrRNA with 3′ end of the crRNA, thereby, resulting into single guide RNA, which has potential use in genome engineering as it can degrade any target DNA sequence (Gasiunas et al. 2012). The type II includes two parts, Cas9 and sgRNA (Jinek et al. 2012).
The sgRNA guides the Cas9 endonuclease and recognizes G-rich PAM (i.e., 5ʹ-NGG) and then identifies the target DNA sequence that lies in the upstream position of the PAM sequence and causes melting of the target DNA (Sternberg et al. 2014). As a result, upstream to the PAM, the strands undergo directional separation, i.e., an R-loop is formed and subsequently, sgRNA strand is incorporated and thereby, forms RNA–DNA heteroduplex (Sternberg et al. 2014; Szczelkun et al. 2014; Anders et al. 2014). The duplex is formed by base pairing of the ~ 20 nt spacer sequence of the sgRNA with the protospacer of the target DNA as they are complementary to each other (Gasiunas et al. 2012; Jinek et al. 2012). One of the domains of Cas9 enzyme, HNH cleaves the DNA sequence that shares complementarity to the sequence (target sequence) in guide RNA and the other domain of the enzyme, RuvC cleaves the sequence that shares non-complementarity to the sequence (non-target sequence) in guide RNA (Jinek et al. 2012; Sapranauskas et al. 2011; Gasiunas et al. 2012). The two domains cleave the RNA–DNA hybrid at a site 3 bp upstream to the PAM and the outcome of cleavage is the creation of a double-strand break (DSB) with blunt ends (Gasiunas et al. 2012; Jinek et al. 2012). The three phases of mechanism of CRISPR–Cas9 organization and the process of genome editing by this organization are depicted in Fig. 2.

Mechanism of natural CRISPR–Cas9 system existing in prokaryotes and modified CRISPR–Cas9 technology used for genome editing. a–c There are three phases of mechanism of naturally existing CRISPR–Cas9 system: a adaptation: bacteria acquire specific genomic sequences termed ‘protospacer’ from phages and incorporate them in CRISPR array as ‘spacer’. b Expression and maturation: leader sequence situated in CRISPR loci initiates transcription of the loci, leading to the production of tracrRNA, Cas9 enzyme and crRNA. c Interference: base pairing of crRNA and tracrRNA takes place which results into crRNA: tracrRNA hybrid and subsequently, recruitment of Cas9 proteins occurs. The hybrid leads the Cas9 to cleave the protospacer and degrade it. The protospacer of the bacteriophage is identified as it is complementary to the spacer of crRNA and as a result, base pairing takes place between them. d In genome editing, chimeric single guide RNA (sgRNA) is constructed by hybridization of tracrRNA and crRNA. sgRNA identifies target DNA sequences that lie upstream to protospacer adjacent motif (PAM). Strands upstream to PAM undergo directional separation and subsequently, sgRNA strand is incorporated and form RNA–DNA heteroduplex as a result of base paring between sgRNA and protospacer of the target DNA as they are complementary to each other. The domains of Cas9 cleave the target and non-target DNA sequences and generate double-strand breaks (DSBs)
Pathways of DNA double-strand break (DSB) repair
Genome editing by CRISPR–Cas9 system primarily involves DNA DSB to be generated at the target gene locus (Carroll 2011). The target DNA strands cleaved by two nuclease domains of Cas9 results in the creation of DSBs at the sequences contained in crRNA (Nishimasu et al. 2014; Jinek et al. 2012). Repairing of DSBs is facilitated by either of the two pathways such as homology-directed repair (HDR) or non-homologous end-joining (NHEJ) (Sander and Joung 2014; Ghezraoui et al. 2014). NHEJ involves short insertions and/or deletions (termed as indels) to be incorporated and these cause disruption of the target locus due to shifting of the translational reading frame and consequently, NHEJ becomes a fallible pathway (Lieber et al. 2003). However, DSB repairing by HDR pathway occurs by external delivery of donor template DNA that possesses homology to the target locus, which then hybridizes and results in accurate mutations or incorporation of sequences of interest (Sander and Joung 2014). Figure 3 represents the pathways involved in DSB repair.

Pathways of DSB (double-strand break) repair. DSBs generated during genome editing can be repaired by either of the two pathways, i.e., non-homologous end-joining (NHEJ) and homology-directed repair (HDR). At DSB sites, NHEJ includes short insertions and/or deletions (termed as indels) while HDR includes accurate insertions or mutation using donor template
On-target activity and off-target activity
The comprehensive Cas9 specificity is subjected to the bases within the ~ 20 nucleotide sequence of the sgRNA, but at the time of hybridization of sgRNA and target DNA, multiple mismatches occur and Cas9 has the capability to tolerate about five such mismatches (Jiang et al. 2013; Cong et al. 2013; Fu et al. 2013; Hsu et al. 2013). These mismatches are the result of off-target sites (sites different from target sites in terms of few bases) contained within the target DNA that remains temporarily bounded to the sgRNA sequence (Wu et al. 2014). Studies reported that Cas9 also facilitates the binding of sgRNA with off-target sites and consequently, Cas9 cleaves these sites to form DSBs (Wu et al. 2014; Ran et al. 2013). In order to carry out genome editing, a Cas9 should cleave the target DNA sequence precisely (Hsu et al. 2013), and the off-target effect has to be minimized essentially.
Improving Cas9 specificity and reducing off-target activity
Upon inactivation of either HNH or RuvC nuclease domains of Cas9, Cas9 is converted into DNA nickase called Cas9 nickase (Cas9n) which cleaves the target DNA into single-strand break (SSB) instead of DSB (Sapranauskas et al. 2011; Gasiunas et al. 2012; Jinek et al. 2012). Cas9n has increased specificity for target sites and the repairing of SSB occurs by high-fidelity base excision repair (BER) (Dianov and Hübscher 2013). Using of ‘paired nickase’ was found to increase Cas9 specificity where two gRNA paired with Cas9n cleaved the target sites and generated DSB (Ran et al. 2013; Mali et al. 2013; Cho et al. 2014). Hybridization between inactive or dead Cas9 (dCas9) and FokI nuclease has also enhanced target sites cleavage (Guilinger et al. 2014; Tsai et al. 2014). Further, dCas9 can be used for silencing an undesirable or diseased gene as well as for activating a favorable gene (Qi et al. 2013; Gilbert et al. 2013; Perez-Pinera et al. 2013).
Inactivation of Cas9 immediately after target site cleavage was found to reduce off-target sites effect. A strategy termed as self-limiting circuit for enhanced safety and specificity (SLiCES) was developed for elimination of Cas9 action from cells (Petris et al. 2017). Anti-CRISPR proteins found in phages to escape from CRISPR–Cas immunity were also used for destroying Cas9 enzyme (Pawluk et al. 2016). Moreover, genetically engineered SpCas9 nucleases such as high-fidelity Cas9 (Cas9-HF1), hyper-accurate Cas9 (HypaCas9) as well as enhanced specificity Cas9 (eSpCas9) were created to facilitate genome editing (Kleinstiver et al. 2016; Slaymaker et al. 2016; Chen et al. 2017).
The sgRNA libraries and genome screening
With the help of genome-wide sgRNA libraries, genome screening can be achieved efficiently for determining and analyzing functional genes involved in a phenotype of interest (Gilbert et al. 2014; Shalem et al. 2015; Chen et al. 2015; Kampmann et al. 2014, 2015; Gilles and Averof 2014; Malina et al. 2014; Li et al. 2014; Wang et al. 2014; Koike-Yusa et al. 2014). Lentiviral genome-wide sgRNA library has found an extensive use for genome screening or mutagenesis screening because screening by CRISPR–Cas system is comparatively better than that by RNA interference (RNAi) (Koike-Yusa et al. 2014).
A study reported that the artificially created CRISPR–Cas9 paired gRNA (pgRNA) library induced the deletion of large genomic fragments and facilitated the detection of a long non-coding RNA (lncRNA) in cancer cells (Zhu et al. 2016). CRISPR–Cas9 mutant library created in rice is extensively useful in genome screening since functional genes as well as phenotype mutants can be determined, and hence paves the way to crop improvement (Meng et al. 2017).
CRISPR tools
Genome scan is carried out using a variety of bioinformatic tools as these tools function to detect specific target sites as well as off-target sites and design sgRNA for achieving high specificity for cleaving the target site. Bioinformatic tools are listed in Table 2.
Database for CRISPR–Cas system
Databases have been developed to provide information on cas genes and CRISPR loci. Table 3 provides the list of useful databases.
Strategies for delivering CRISPR
Researchers have experimentally proved that the delivery of Cas9 and gRNA can be carried out by nucleofection (Mali et al. 2013; Fu et al. 2013), lipofectamine-mediated transfection (Mali et al. 2013; Cong et al. 2013; Fu et al. 2013; Li et al. 2013), polyethylenimine-mediated transfection (Zuckermann et al. 2015) as well as electroporation (Ding et al. 2013; Straub et al. 2014). At present, the most reliable approach for CRISPR–Cas component delivery to cultured cells is by the use of viral vectors.
Lentivirus
Delivery of CRISPR system had been successfully achieved in model organisms by lentivirus vectors for inducing cancers such as brain (Zuckermann et al. 2015), colon (Roper et al. 2017; O’Rourke et al. 2017), lung (Sánchez-Rivera et al. 2014; Rogers et al. 2017, 2018; Walter et al. 2017), pancreatic cancer (Chiou et al. 2015) and breast (Annunziato et al. 2016) cancer. These findings have provided significant insights for understanding cancer in general.
Adenovirus-associated virus (AAV)
Till date, the most effective virus vector is AAV as it provides a long-term expression of CRISPR system and is safe for use because of its non-pathogenic nature (Burger et al. 2005; Taymans et al. 2007). Genome editing by these viral vectors is possible because AAV has many serotypes for inducing cancer in model organisms and this is useful for genome editing (Platt et al. 2014; Chow et al. 2017; Yin et al. 2017; Winters et al. 2017).
Adenovirus
Adenovirus has also been successful in delivery CRISPR–Cas9 in vivo (Wang et al. 2015). However, the use of this virus has now been restricted because the virus possesses immunogenic and adjuvant property (Nelson et al. 2017).
Applications of CRISPR–Cas technology
Gene therapy
Genome editing experiments based on CRISPR system have demonstrated that this technology has enormous potentiality in the field of gene therapy in order to modify or eliminate disease genes (Firth et al. 2015; Wu et al. 2013; Long et al. 2014, 2016; Osborn et al. 2014; Nelson et al. 2016; Tabebordbar et al. 2016). A study demonstrated that by the use of organoids of cultured intestinal stem cells, cystic fibrosis was treated in vitro by homologous recombination of CFTR (cystic fibrosis transmembrane conductance regulator) locus, a gene loci that cause the disease (Schwank et al. 2013). CRISPR–Cas based genome editing has successfully been carried out in diseases such as Fanconi anemia (Osborn et al. 2014), and crystalline gamma c (Crygc) associated cataract (Wu et al. 2013).
Neuroscience
Studies demonstrated that CRISP-Cas9 system has potential for treating the neurodegenerative disorders like Duchenne muscular dystrophy and for this purpose, AAV was used for delivering CRISPR–Cas9 to a model organism (Mendell and Rodino-Klapac 2016). Huntington disease, a neurodegenerative disease, is caused by the presence of mutant allele HTT and by applying CRISPR–Cas9 system, the allele was silenced or inactivated (Shin et al. 2016; Xu et al. 2017; Monteys et al. 2017). Disease causing gene in neurons can be identified from the cultures of induced pluripotent stem cells (iPSCs) by using CRISPR–Cas9 system (Polstein and Gersbach 2015; Zetsche et al. 2015).
Agriculture
In plant genomes the most extensively used CRISPR systems are CRISPR–Cas9 and CRISPR-Cpf1 (Zetsche et al. 2015; Jinek et al. 2012). This technology has been applied for editing the genomes in plants such as wheat (Shan et al. 2013), tobacco (Li et al. 2013), sweet orange (Jia and Wang 2014), rice (Shan et al. 2013; Miao et al. 2013; Xie and Yang 2013) and Arabidopsis (Li et al. 2013; Feng et al. 2014; Yin et al. 2017). The latest adapted two-step CRISPR–Cas9 technology permits genome editing to occur scarlessly. Scientists can rely on this stepwise method for performing successful genome editing with the purpose of analyzing results of phenotypic changes and correlation between genotype and phenotype. The approach can be useful for crop improvement (Elison and Acar 2018).
Microbiology
CRISPR–Cas9 system has also been successful in mutating or deleting genes in yeasts (Enkler et al. 2016; Vyas et al. 2015; Min et al. 2016; Grahl et al. 2017), molds (Fuller et al. 2015) and filamentous fungi (Liu et al. 2015, 2017) which may have industrial importance as well as pathogenicity. The genomes of industrially important bacteria such as Clostridium spp. (Huang et al. 2016; Wang et al. 2016; Nagaraju et al. 2016) and Streptomyces spp. (Cobb et al. 2014) were edited by this technology for efficient production of biofuels, anticancer agents and antibiotics. Yeast such as Saccharomyces cerevisiae possesses HDR repair pathway, which permits it to undergo genome editing accurately. CRISPR–Cas9 technology has been applied to these eukaryotic organisms for manipulating their strains so that they could be used in the field of synthetic biology and metabolic engineering. Based on this technology, genetic interaction screens could be accomplished for creating diversified mutant yeast strains (Adames et al. 2019). In S. cerevisiae, genetic manipulations can be achieved by associating the function of selectable marker integration and genome engineering potential of CRISPR–Cas9 organization. Using this application, researchers can investigate yeast cells for understanding the activity of any gene or promoter as well as for developing strains that possess replacements of gene or promoter (Soreanu et al. 2018).
Antiviral therapy
Earlier studies reported that with the help of CRISPR–Cas technology, diseases caused by the viruses such as hepatitis B (Dong et al. 2015; Kennedy et al. 2015; Kennedy and Cullen 2015; Liu et al. 2015; Ramanan et al. 2015; Wang et al. 2015; Lin et al. 2014; Zhen et al. 2015), papillomavirus (Kennedy et al. 2014; Hu et al. 2014a), herpes (Wang and Quake 2014), Porcine endogenous retroviruses (PERVs) (Yang et al. 2015) and Human Immunodeficiency Virus-1 (HIV-1) (Hu et al. 2014b; Li et al. 2015; Wang et al. 2014; Ye et al. 2014; Zhang and Sodroski 2015; Hou et al. 2015) were treated effectively.
Drug discovery and targets
CRISPR–Cas9 technology has great potential in the field of drug discovery and for generation of therapeutic drugs for treating heritable diseases (Fellmann et al. 2017). Genomic screens can be performed by CRISPR–Cas9 system to detect mutated genes that have become drug resistant (Wang et al. 2014; Koike-Yusa et al. 2014; Shalem et al. 2014; Zhou et al. 2014). Genomic screening by CRISPR–Cas9 has also been employed for analyzing the activity of drugs on infectious agents, cancer cells as well as proteins or genes involved (Deans et al. 2016; Marceau et al. 2016).
Antimicrobials
Sequence-specific antimicrobials act specifically to target any pathogenic microbe as well as sgRNA can be designed to target microbes on broad-scale (Barrangou and Doudna 2016). Studies reported that antimicrobial treatment-mediated CRISPR–Cas9 is exceptional when compared with traditional antimicrobials as well as antibiotics (Beisel et al. 2014). The main challenge of CRISPR-based antimicrobial lies in the advancement of delivery strategies of CRISPR–Cas9 system (Barrangou and Doudna 2016).
Cancer
Somatic genome editing mediated by CRISPR–Cas9 system has paved a way in cancer modeling and in the development of model organisms suffering from hematopoietic malignant tumors (Heckl et al. 2014; Chen et al. 2014). CRISPR system can also be used for cancer detection during initial stages of the disease in an individual because Cas13a has the ability to distinguish mutation that can lead to cancer (Gootenberg et al. 2017). Multiplexed CRISPR–Cas9 genome editing has great potential for analyzing susceptibility in cancer causing cells (Sánchez-Rivera and Jacks 2015). CRISPR system provides immense opportunity in cancer immunotherapy by compressing negative factors and embellishing effectives (Yin et al. 2019).
Other applications
In addition to genome editing, technology based on CRISPR–Cas9 system has recently been applied for live-cell chromatin imaging, chromatin topology manipulations, genome regulation, RNA targeting and epigenome editing (Adli 2018). Transposable elements (TEs) have contributed greatly during genome evolution. A gene of interest can be incorporated within the genome of any organism by altering the activity of these elements. CRISPR–Cas9 technology can be employed for making the best use of TEs in order to regulate transcription process in the organism (Vaschetto 2018).
Discussion and future perspectives
Here, we present deep understanding of the biology of CRISPR–Cas9 system in genome editing. The increase in cleavage specificity of Cas9 and the reduction of off-target activity of this enzyme enable to recognize particular target DNA sequences and then alter or manipulate genome correctly. CRISPR databases and tools provide information and proper facility for altering, manipulating or visualizing genomes to perform correct genome editing experiments. We show that CRISPR–Cas9 technology has been applied to various fields including disease treatment related to genetic disorders or pathogens, agriculture, genetic engineering, clinical applications. However, many challenges are still in the way and need to be overcome. Further studies on CRISPR–Cas9 organization and potential applications of this technology will aid in overcoming these challenges, thereby, leading to better and healthy lives of humans in the society by curing complex diseases and improving crop field.
Improvement of CRISPR tools is essential so that off-target cleavage activity by Cas9 can be reduced effectively. Delivery strategies of CRISPR–Cas9 system into cells of higher class organisms such as mammals and plants precisely are crucially important for improvement. Identification of drug targets by the use of CRISPR–Cas9 technology will aid in the development of new drugs for the emerging dreadful diseases. Insertion of altered TEs by this technology can lead to crop improvement and creation of new ornamental plants. As CRISPR–Cas system is found in bacteria, it undergoes evolution rapidly and thereby, may give rise to new cas genes which will encode new proteins and thus, these proteins may have potential for genome editing or other applications in near future. Recently, Cas12a has been known to have great application in genome editing. CRISPR technology has also been applied to treat cancer and moreover, Cas13a can detect mutations that can lead to cancer. This technology is on the brink of treating various cancer and related diseases. Experiments for this CRISPR–Cas9 technology are mostly performed in vitro in model organisms and stem cells such as human pluripotent stem cells (hPSCs). Gene editing by this technology may be performed in human embryonic stem cells (ESCs) in vitro for correcting mutations, but research in ESCs raises many ethical issues. However, genome editing of ESCs may have the potential to give rise to organisms possessing excellent desirable qualities. Safety measures are to be taken while using this technology to prevent its misuse or reduce the risk of negative impact of genome editing.
References
Abudayyeh OO et al (2016) C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science 353(6299):5573
Abudayyeh OO et al (2017) RNA targeting with CRISPR–Cas13. Nature 550(7675):280
Adames NR, Gallegos JE, Peccoud J (2019) Yeast genetic interaction screens in the age of CRISPR/Cas. Curr Genet 65(2):307–327
Adli M (2018) The CRISPR tool kit for genome editing and beyond. Nat Commun 9(1):1911
Aman R et al (2018) RNA virus interference via CRISPR/Cas13a system in plants. Genome Biol 19(1):1
Amitai G, Sorek R (2016) CRISPR–Cas adaptation: insights into the mechanism of action. Nat Rev Microbiol 14(2):67
Anders C et al (2014) Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature 513(7519):569
Annunziato S et al (2016) Modeling invasive lobular breast carcinoma by CRISPR/Cas9-mediated somatic genome editing of the mammary gland. Genes Dev 30(12):1470–1480
Bae S et al (2014) Microhomology-based choice of Cas9 nuclease target sites. Nat Methods 11(7):705
Barrangou R (2015) Diversity of CRISPR–Cas immune systems and molecular machines. Genome Biol 16(1):247
Barrangou R, Doudna JA (2016) Applications of CRISPR technologies in research and beyond. Nat Biotechnol 34(9):933
Barrangou R, Marraffini LA (2014) CRISPR–Cas systems: prokaryotes upgrade to adaptive immunity. Mol Cell 54(2):234–244
Barrangou R et al (2007) CRISPR provides acquired resistance against viruses in prokaryotes. Science 315(5819):1709–1712
Beisel CL, Gomaa AA, Barrangou R (2014) A CRISPR design for next-generation antimicrobials. Genome Biol 15(11):516
Blin K et al (2016) CRISPy-web: an online resource to design sgRNAs for CRISPR applications. Synth Syst Biotechnol 1(2):118–121
Bolotin A et al (2005) Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology 151(8):2551–2561
Bult CJ et al (1996) Complete genome sequence of the Methanogenic archaeon, Methanococcus jannaschii. Science 273(5278):1058–1073
Burger C, Nash K, Mandel RJ (2005) Recombinant adeno-associated viral vectors in the nervous system. Hum Gene Ther 16(7):781–791
Carroll D (2011) Genome engineering with zinc-finger nucleases. Genetics 188(4):773–782
Charpentier E, Marraffini LA (2014) Harnessing CRISPR–Cas9 immunity for genetic engineering. Curr Opin Microbiol 19:114–119
Chen B et al (2013) Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell 155(7):1479–1491
Chen C et al (2014) MLL3 is a haploinsufficient 7q tumor suppressor in acute myeloid leukemia. Cancer Cell 25(5):652–665
Chen S et al (2015) Genome-wide CRISPR screen in a mouse model of tumor growth and metastasis. Cell 160(6):1246–1260
Chen JS et al (2017) Enhanced proofreading governs CRISPR–Cas9 targeting accuracy. Nature 550(7676):407
Chiou S-H et al (2015) Pancreatic cancer modeling using retrograde viral vector delivery and in vivo CRISPR/Cas9-mediated somatic genome editing. Genes Dev 29(14):1576–1585
Cho SW et al (2014) Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res 24(1):132–141
Chow RD et al (2017) AAV-mediated direct in vivo CRISPR screen identifies functional suppressors in glioblastoma. Nat Neurosci 20(10):1329
Chylinski K et al (2014) Classification and evolution of type II CRISPR–Cas systems. Nucleic Acids Res 42(10):6091–6105
Clement K et al (2019) CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat Biotechnol 37(3):224
Cobb RE, Wang Y, Zhao H (2014) High-efficiency multiplex genome editing of Streptomyces species using an engineered CRISPR/Cas system. ACS Synth Biol 4(6):723–728
Cong L et al (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339(6121):819–823
Cox DBT, Platt RJ, Zhang F (2015) Therapeutic genome editing: prospects and challenges. Nat Med 21(2):121
Cradick TJ et al (2014) COSMID: a web-based tool for identifying and validating CRISPR/Cas off-target sites. Mol Ther Nucleic Acids 3:e214
Deans RM et al (2016) Parallel shRNA and CRISPR–Cas9 screens enable antiviral drug target identification. Nat Chem Biol 12(5):361
Deltcheva E et al (2011) CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471(7340):602
Deveau H et al (2008) Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J Bacteriol 190(4):1390–1400
Deveau H, Garneau JE, Moineau S (2010) CRISPR/Cas system and its role in phage–bacteria interactions. Annu Rev Microbiol 64:475–493
Dianov GL, Hübscher U (2013) Mammalian base excision repair: the forgotten archangel. Nucleic Acids Res 41(6):3483–3490
Díez-Villaseñor C et al (2010) Diversity of CRISPR loci in Escherichia coli. Microbiology 156(5):1351–1361
Ding Q et al (2013) Enhanced efficiency of human pluripotent stem cell genome editing through replacing TALENs with CRISPRs. Cell Stem Cell 12(4):393–394
Dong C et al (2015) Targeting hepatitis B virus cccDNA by CRISPR/Cas9 nuclease efficiently inhibits viral replication. Antiviral Res 118:110–117
Doudna JA, Charpentier E (2014) The new frontier of genome engineering with CRISPR–Cas9. Science 346(6213):1258096
Elison GL, Acar M (2018) Scarless genome editing: progress towards understanding genotype–phenotype relationships. Curr Genet 64(6):1229–1238
Enkler L et al (2016) Genome engineering in the yeast pathogen Candida glabrata using the CRISPR–Cas9 system. Sci Rep 6:35766
Eyquem J et al (2017) Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 543(7643):113
Fellmann C et al (2017) Cornerstones of CRISPR–Cas in drug discovery and therapy. Nat Rev Drug Discov 16(2):89
Feng Z et al (2014) Multigeneration analysis reveals the inheritance, specificity, and patterns of CRISPR/Cas-induced gene modifications in Arabidopsis. Proc Natl Acad Sci 111(12):4632–4637
Firth AL et al (2015) Functional gene correction for cystic fibrosis in lung epithelial cells generated from patient iPSCs. Cell Rep 12(9):1385–1390
Fu Y et al (2013) High-frequency off-target mutagenesis induced by CRISPR–Cas nucleases in human cells. Nat Biotechnol 31(9):822
Fuller KK et al (2015) Development of the CRISPR/Cas9 system for targeted gene disruption in Aspergillus fumigatus. Eukaryot Cell 14(11):1073–1080
Gaj T, Gersbach CA, Barbas CF III (2013) ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol 31(7):397–405
Garneau JE et al (2010) The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 468(7320):67
Gasiunas G et al (2012) Cas9–crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci 109(39):E2579–E2586
Ghezraoui H et al (2014) Chromosomal translocations in human cells are generated by canonical nonhomologous end-joining. Mol Cell 55(6):829–842
Gilbert LA et al (2013) CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154(2):442–451
Gilbert LA et al (2014) Genome-scale CRISPR-mediated control of gene repression and activation. Cell 159(3):647–661
Gilles AF, Averof M (2014) Functional genetics for all: engineered nucleases, CRISPR and the gene editing revolution. EvoDevo 5(1):43
Gootenberg JS et al (2017) Nucleic acid detection with CRISPR–Cas13a/C2c2. Science 356(6336):438–442
Grahl N et al (2017) Use of RNA–protein complexes for genome editing in non-albicans Candida species. mSphere 2(3):e00218–e00317
Grissa I, Vergnaud G, Pourcel C (2007) The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinform 8(1):172
Groenen PM et al (1993) Nature of DNA polymorphism in the direct repeat cluster of Mycobacterium tuberculosis; application for strain differentiation by a novel typing method. Mol Microbiol 10(5):1057–1065
Guilinger JP, Thompson DB, Liu DR (2014) Fusion of catalytically inactive Cas9 to FokI nuclease improves the specificity of genome modification. Nat Biotechnol 32(6):577
Heckl D et al (2014) Generation of mouse models of myeloid malignancy with combinatorial genetic lesions using CRISPR–Cas9 genome editing. Nat Biotechnol 32(9):941
Heidenreich M, Zhang F (2016) Applications of CRISPR–Cas systems in neuroscience. Nat Rev Neurosci 17(1):36
Heigwer F, Kerr G, Boutros M (2014) E-CRISP: fast CRISPR target site identification. Nat Methods 11(2):122
Heler R et al (2015) Cas9 specifies functional viral targets during CRISPR–Cas adaptation. Nature 519(7542):199
Hodgkins A et al (2015) WGE: a CRISPR database for genome engineering. Bioinformatics 31(18):3078–3080
Hoe N et al (1999) Rapid molecular genetic subtyping of serotype M1 group A Streptococcus strains. Emerg Infect Dis 5(2):254
Horvath P, Barrangou R (2010) CRISPR/Cas, the immune system of bacteria and archaea. Science 327(5962):167–170
Hou P et al (2015) Genome editing of CXCR189 by CRISPR/cas9 confers cells resistant to HIV-1 infection. Sci Rep 5:15577
Hsu PD et al (2013) DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol 31(9):827
Hsu PD, Lander ES, Zhang F (2014) Development and applications of CRISPR–Cas9 for genome engineering. Cell 157(6):1262–1278
Hu Z, Yu L, Zhu D, Ding W, Wang X, Zhang C, Wang L, Jiang X, Shen H, He D, Li K, Xi L, Ma D, Wang H (2014a) Disruption of HPV16-E7 by CRISPR/Cas system induces apoptosis and growth inhibition in HPV16 positive human cervical cancer cells. BioMed Res Int. https://doi.org/10.1155/2014/612823
Hu W et al (2014) RNA-directed gene editing specifically eradicates latent and prevents new HIV-1 infection. Proc Natl Acad Sci 111(31):11461–11466
Huang H et al (2016) CRISPR/Cas9-based efficient genome editing in Clostridium ljungdahlii, an autotrophic gas-fermenting bacterium. ACS Synth Biol 5(12):1355–1361
Hur JK et al (2016) Targeted mutagenesis in mice by electroporation of Cpf1 ribonucleoproteins. Nat Biotechnol 34(8):807
Ishino Y et al (1987) Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J Bacteriol 169(12):5429–5433
Jansen R et al (2002) Identification of genes that are associated with DNA repeats in prokaryotes. Mol Microbiol 43(6):1565–1575
Jia H, Wang N (2014) Targeted genome editing of sweet orange using Cas9/sgRNA. PLoS One 9(4):e93806
Jiang W et al (2013) RNA-guided editing of bacterial genomes using CRISPR–Cas systems. Nat Biotechnol 31(3):233
Jinek M et al (2012a) A programmable dual-RNA—guided DNA endonuclease in adaptive bacterial immunity. Science 337:816
Jinek M et al (2012b) A programmable dual-RNA—guided DNA endonuclease in adaptive bacterial immunity. Science 337(6096):816–821
Jinek M et al (2013) RNA-programmed genome editing in human cells. Life 2:00471
Jinek M et al (2014) Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science 343(6176):1247997
Kampmann M, Bassik MC, Weissman JS (2014) Functional genomics platform for pooled screening and generation of mammalian genetic interaction maps. Nat Protoc 9(8):1825
Kampmann M et al (2015) Next-generation libraries for robust RNA interference-based genome-wide screens. Proc Natl Acad Sci 112(26):E3384–E3391
Kennedy EM, Cullen BR (2015) Bacterial CRISPR/Cas DNA endonucleases: a revolutionary technology that could dramatically impact viral research and treatment. Virology 479:213–220
Kennedy EM et al (2014) Inactivation of the human papillomavirus E6 or E7 gene in cervical carcinoma cells by using a bacterial CRISPR/Cas RNA-guided endonuclease. J Virol 88(20):11965–11972
Kennedy EM et al (2015) Suppression of hepatitis B virus DNA accumulation in chronically infected cells using a bacterial CRISPR/Cas RNA-guided DNA endonuclease. Virology 476:196–205
Kim Y et al (2016a) Generation of knockout mice by Cpf1-mediated gene targeting. Nat Biotechnol 34(8):808
Kim D et al (2016b) Genome-wide analysis reveals specificities of Cpf1 endonucleases in human cells. Nat Biotechnol 34(8):863
Kim H et al (2017) CRISPR/Cpf1-mediated DNA-free plant genome editing. Nat Commun 8:14406
Kleinstiver BP et al (2016a) Genome-wide specificities of CRISPR–Cas Cpf1 nucleases in human cells. Nat Biotechnol 34(8):869
Kleinstiver BP et al (2016b) High-fidelity CRISPR–Cas9 nucleases with no detectable genome-wide off-target effects. Nature 529(7587):490
Knott GJ, Doudna JA (2018) CRISPR–Cas guides the future of genetic engineering. Science 361(6405):866–869
Koike-Yusa H et al (2014) Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nat Biotechnol 32(3):267
Koonin EV, Makarova KS, Zhang F (2017) Diversity, classification and evolution of CRISPR–Cas systems. Curr Opin Microbiol 37:67–78
Labun K et al (2016) CHOPCHOP v2: a web tool for the next generation of CRISPR genome engineering. Nucleic Acids Res 44(W1):W272–W276
Legut M et al (2018) CRISPR-mediated TCR replacement generates superior anticancer transgenic T cells. Blood 131(3):311–322
Lei Y et al (2014) CRISPR-P: a web tool for synthetic single-guide RNA design of CRISPR-system in plants. Mol Plant 7(9):1494–1496
Lenoir WF, Lim TL, Hart T (2017) PICKLES: the database of pooled in vitro CRISPR knockout library essentiality screens. Nucleic Acids Res 46(D1):D776–D780
Li J-F et al (2013) Multiplex and homologous recombination–mediated genome editing in Arabidopsis and Nicotiana benthamiana using guide RNA and Cas9. Nat Biotechnol 31(8):688
Li W et al (2014) MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol 15(12):554
Li C et al (2015) Inhibition of HIV-1 infection of primary CD4 + T-cells by gene editing of CCR185 using adenovirus-delivered CRISPR/Cas9. J Gen Virol 96(8):2381–2393
Liang G et al (2016) Selection of highly efficient sgRNAs for CRISPR/Cas9-based plant genome editing. Sci Rep 6:21451
Lieber MR et al (2003) Mechanism and regulation of human non-homologous DNA end-joining. Nat Rev Mol Cell Biol 4(9):712
Lin S-R et al (2014) The CRISPR/Cas9 system facilitates clearance of the intrahepatic HBV templates in vivo. Mol Ther Nucleic Acids 3:e186
Liu R et al (2015a) Efficient genome editing in filamentous fungus Trichoderma reesei using the CRISPR/Cas9 system. Cell Discov 1:15007
Liu X et al (2015b) Inhibition of hepatitis B virus by the CRISPR/Cas9 system via targeting the conserved regions of the viral genome. J Gen Virol 96(8):2252–2261
Liu Q et al (2017a) Development of a genome-editing CRISPR/Cas9 system in thermophilic fungal Myceliophthora species and its application to hyper-cellulase production strain engineering. Biotechnol Biofuels 10(1):1
Liu H et al (2017b) CRISPR-P 2.0: an improved CRISPR–Cas9 tool for genome editing in plants. Mol Plant 10(3):530–532
Long C et al (2014) Prevention of muscular dystrophy in mice by CRISPR/Cas9–mediated editing of germline DNA. Science 345(6201):1184–1188
Long C et al (2016) Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science 351(6271):400–403
Makarova KS et al (2011a) Evolution and classification of the CRISPR–Cas systems. Nat Rev Microbiol 9(6):467
Makarova KS et al (2011b) Unification of Cas protein families and a simple scenario for the origin and evolution of CRISPR–Cas systems. Biol Direct 6(1):38
Makarova KS, Wolf YI, Koonin EV (2013) The basic building blocks and evolution of CRISPR–Cas systems. Portland Press Limited, London
Makarova KS et al (2015) An updated evolutionary classification of CRISPR–Cas systems. Nat Rev Microbiol 13(11):722
Mali P et al (2013a) RNA-guided human genome engineering via Cas9. Science 339(6121):823–826
Mali P et al (2013b) CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat Biotechnol 31(9):833
Malina A et al (2014) Adapting CRISPR/Cas9 for functional genomics screens. Methods in enzymology. Elsevier, Amsterdam, pp 193–213
Marceau CD et al (2016) Genetic dissection of Flaviviridae host factors through genome-scale CRISPR screens. Nature 535(7610):159
Marraffini LA, Sontheimer EJ (2010) CRISPR interference: RNA-directed adaptive immunity in bacteria and archaea. Nat Rev Genet 11(3):181
Masepohl B, Görlitz K, Böhme H (1996) Long tandemly repeated repetitive (LTRR) sequences in the filamentous cyanobacterium Anabaena sp. PCC 7120. Biochim Biophys Acta (BBA) Gene Struct Expr 1307(1):26–30
Mendell JR, Rodino-Klapac LR (2016) Duchenne muscular dystrophy: CRISPR/Cas9 treatment. Cell Res 26(5):513
Meng X et al (2017) Construction of a genome-wide mutant library in rice using CRISPR/Cas9. Mol Plant 10(9):1238–1241
Miao J et al (2013) Targeted mutagenesis in rice using CRISPR–Cas system. Cell Res 23(10):1233
Min K et al (2016) Candida albicans gene deletion with a transient CRISPR–Cas9 system. mSphere 1(3):e00130–e00216
Mojica F et al (1995) Long stretches of short tandem repeats are present in the largest replicons of the Archaea Haloferax mediterranei and Haloferax volcanii and could be involved in replicon partitioning. Mol Microbiol 17(1):85–93
Mojica FJ, García-Martínez J, Soria E (2005) Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J Mol Evol 60(2):174–182
Mojica FJ et al (2009) Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology 155(3):733–740
Monteys AM et al (2017) CRISPR/Cas9 editing of the mutant huntingtin allele in vitro and in vivo. Mol Ther 25(1):12–23
Moreno-Mateos MA et al (2017) CRISPR-Cpf1 mediates efficient homology-directed repair and temperature-controlled genome editing. Nat Commun 8(1):2024
Nagaraju S et al (2016) Genome editing of Clostridium autoethanogenum using CRISPR/Cas9. Biotechnol Biofuels 9(1):219
Nelson KE et al (1999) Evidence for lateral gene transfer between Archaea and bacteria from genome sequence of Thermotoga maritima. Nature 399(6734):323
Nelson CE et al (2016) In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science 351(6271):403–407
Nelson CE, Robinson-Hamm JN, Gersbach CA (2017) Genome engineering: a new approach to gene therapy for neuromuscular disorders. Nat Rev Neurol 13(11):647
Nishimasu H et al (2014) Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 156(5):935–949
O’Rourke KP et al (2017) Transplantation of engineered organoids enables rapid generation of metastatic mouse models of colorectal cancer. Nat Biotechnol 35(6):577
Osborn MJ et al (2014) Fanconi anemia gene editing by the CRISPR/Cas9 system. Hum Gene Ther 26(2):114–126
Park J, Bae S, Kim J-S (2015) Cas-Designer: a web-based tool for choice of CRISPR–Cas9 target sites. Bioinformatics 31(24):4014–4016
Pawluk A et al (2016) Naturally occurring off-switches for CRISPR–Cas9. Cell 167(7):1829–1838
Peng D, Tarleton R (2015) EuPaGDT: a web tool tailored to design CRISPR guide RNAs for eukaryotic pathogens. Microb Genom 1(4):e000033. https://doi.org/10.1099/mgen.0.000033
Perez-Pinera P et al (2013) RNA-guided gene activation by CRISPR–Cas9—based transcription factors. Nat Methods 10(10):973
Petris G et al (2017) Hit and go CAS9 delivered through a lentiviral based self-limiting circuit. Nat Commun 8:15334
Platt RJ et al (2014) CRISPR–Cas9 knockin mice for genome editing and cancer modeling. Cell 159(2):440–455
Polstein LR, Gersbach CA (2015) A light-inducible CRISPR–Cas9 system for control of endogenous gene activation. Nat Chem Biol 11(3):198
Port F, Bullock SL (2016) Augmenting CRISPR applications in Drosophila with tRNA-flanked sgRNAs. Nat Methods 13(10):852
Pougach K et al (2010) Transcription, processing and function of CRISPR cassettes in Escherichia coli. Mol Microbiol 77(6):1367–1379
Pourcel C, Salvignol G, Vergnaud G (2005) CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology 151(3):653–663
Prykhozhij SV et al (2015) CRISPR multitargeter: a web tool to find common and unique CRISPR single guide RNA targets in a set of similar sequences. PLoS One 10(3):e0119372
Puschnik AS et al (2017) A CRISPR toolbox to study virus–host interactions. Nat Rev Microbiol 15(6):351
Qi LS et al (2013) Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152(5):1173–1183
Ramanan V et al (2015) CRISPR/Cas9 cleavage of viral DNA efficiently suppresses hepatitis B virus. Sci Rep 5:10833
Ran FA et al (2013) Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154(6):1380–1389
Rauscher B, Heigwer F, Breinig M, Winter J, Boutros M (2016) GenomeCRISPR-a database for high-throughput CRISPR/Cas9 screens. Nucleic Acids Res. https://doi.org/10.1093/nar/gkw997
Ren X et al (2014) Enhanced specificity and efficiency of the CRISPR/Cas9 system with optimized sgRNA parameters in Drosophila. Cell Rep 9(3):1151–1162
Ren J et al (2017) Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clin Cancer Res 23(9):2255–2266
Rogers ZN et al (2017) A quantitative and multiplexed approach to uncover the fitness landscape of tumor suppression in vivo. Nat Methods 14(7):737
Rogers ZN et al (2018) Mapping the in vivo fitness landscape of lung adenocarcinoma tumor suppression in mice. Nat Genet 50(4):483
Ronda C et al (2014) Accelerating genome editing in CHO cells using CRISPR Cas9 and CRISPy, a web-based target finding tool. Biotechnol Bioeng 111(8):1604–1616
Roper J et al (2017) In vivo genome editing and organoid transplantation models of colorectal cancer and metastasis. Nat Biotechnol 35(6):569
Rousseau C et al (2009) CRISPI: a CRISPR interactive database. Bioinformatics 25(24):3317–3318
Sánchez-Rivera FJ, Jacks T (2015) Applications of the CRISPR–Cas9 system in cancer biology. Nat Rev Cancer 15(7):387
Sánchez-Rivera FJ et al (2014) Rapid modelling of cooperating genetic events in cancer through somatic genome editing. Nature 516(7531):428
Sander JD, Joung JK (2014) CRISPR–Cas systems for editing, regulating and targeting genomes. Nat Biotechnol 32(4):347
Sapranauskas R et al (2011) The Streptococcus thermophilus CRISPR/Cas system provides immunity in Escherichia coli. Nucleic Acids Res 39(21):9275–9282
Schwank G et al (2013) Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell 13(6):653–658
Shalem O et al (2014) Genome-scale CRISPR–Cas9 knockout screening in human cells. Science 343(6166):84–87
Shalem O, Sanjana NE, Zhang F (2015) High-throughput functional genomics using CRISPR–Cas9. Nat Rev Genet 16(5):299
Shan Q et al (2013) Targeted genome modification of crop plants using a CRISPR–Cas system. Nat Biotechnol 31(8):686
Shin JW et al (2016) Permanent inactivation of Huntington’s disease mutation by personalized allele-specific CRISPR/Cas9. Hum Mol Genet 25(20):4566–4576
Shmakov S et al (2015) Discovery and functional characterization of diverse class 2 CRISPR–Cas systems. Mol Cell 60(3):385–397
Shmakov S et al (2017) Diversity and evolution of class 2 CRISPR–Cas systems. Nat Rev Microbiol 15(3):169
Slaymaker IM et al (2016) Rationally engineered Cas9 nucleases with improved specificity. Science 351(6268):84–88
Smargon AA et al (2017) Cas13b is a type VI-B CRISPR-associated RNA-guided RNase differentially regulated by accessory proteins Csx27 and Csx28. Mol Cell 65(4):618–630
Soreanu I et al (2018) Marker-free genetic manipulations in yeast using CRISPR/CAS9 system. Curr Genet 64(5):1129–1139
Stemmer M et al (2015) CCTop: an intuitive, flexible and reliable CRISPR/Cas9 target prediction tool. PLoS One 10(4):e0124633
Sternberg SH et al (2014) DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 507(7490):62
Straub C et al (2014) CRISPR/Cas9-mediated gene knock-down in post-mitotic neurons. PLoS One 9(8):e105584
Strong A, Musunuru K (2017) Genome editing in cardiovascular diseases. Nat Rev Cardiol 14(1):11
Swarts DC, Jinek M (2018) Cas9 versus Cas12a/Cpf1: structure–function comparisons and implications for genome editing. Wiley Interdiscip Rev RNA 9(5):e1481
Świat MA et al (2017) FnCpf1: a novel and efficient genome editing tool for Saccharomyces cerevisiae. Nucleic Acids Res 45(21):12585–12598
Szczelkun MD et al (2014) Direct observation of R-loop formation by single RNA-guided Cas9 and Cascade effector complexes. Proc Natl Acad Sci 111(27):9798–9803
Tabebordbar M et al (2016) In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science 351(6271):407–411
Tang X et al (2017) A CRISPR–Cpf1 system for efficient genome editing and transcriptional repression in plants. Nat Plants 3(3):17018
Taymans J-M et al (2007) Comparative analysis of adeno-associated viral vector serotypes 1, 2, 5, 7, and 8 in mouse brain. Hum Gene Ther 18(3):195–206
Terns MP, Terns RM (2011) CRISPR-based adaptive immune systems. Curr Opin Microbiol 14(3):321–327
Tsai SQ et al (2014) Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat Biotechnol 32(6):569
Van der Oost J et al (2009) CRISPR-based adaptive and heritable immunity in prokaryotes. Trends Biochem Sci 34(8):401–407
Van Der Oost J et al (2014) Unravelling the structural and mechanistic basis of CRISPR–Cas systems. Nat Rev Microbiol 12(7):479
Varshney GK et al (2015) CRISPRz: a database of zebrafish validated sgRNAs. Nucleic Acids Res 44(D1):D822–D826
Vaschetto LM (2018) Modulating signaling networks by CRISPR/Cas9-mediated transposable element insertion. Curr Genet 64(2):405–412
Vyas VK, Barrasa MI, Fink GR (2015) A Candida albicans CRISPR system permits genetic engineering of essential genes and gene families. Sci Adv 1(3):e1500248
Walter DM et al (2017) Systematic in vivo inactivation of chromatin-regulating enzymes identifies Setd2 as a potent tumor suppressor in lung adenocarcinoma. Cancer Res 77(7):1719–1729
Wang J, Quake SR (2014) RNA-guided endonuclease provides a therapeutic strategy to cure latent herpesviridae infection. Proc Natl Acad Sci 111(36):13157–13162
Wang T et al (2014a) Genetic screens in human cells using the CRISPR–Cas9 system. Science 343(6166):80–84
Wang W et al (2014b) CCR186 gene disruption via lentiviral vectors expressing Cas9 and single guided RNA renders cells resistant to HIV-1 infection. PLoS One 9(12):e115987
Wang D et al (2015a) Adenovirus-mediated somatic genome editing of Pten by CRISPR/Cas9 in mouse liver in spite of Cas9-specific immune responses. Hum Gene Ther 26(7):432–442
Wang J et al (2015b) Dual gRNAs guided CRISPR/Cas9 system inhibits hepatitis B virus replication. World J Gastroenterol 21(32):9554
Wang H, La Russa M, Qi LS (2016a) CRISPR/Cas9 in genome editing and beyond. Annu Rev Biochem 85:227–264
Wang Y et al (2016b) Bacterial genome editing with CRISPR–Cas9: deletion, integration, single nucleotide modification, and desirable “clean” mutant selection in Clostridium beijerinckii as an example. ACS Synth Biol 5(7):721–732
Wei Y et al (2015a) Sequences spanning the leader-repeat junction mediate CRISPR adaptation to phage in Streptococcus thermophilus. Nucleic Acids Res 43(3):1749–1758
Wei Y, Terns RM, Terns MP (2015b) Cas9 function and host genome sampling in type II-A CRISPR–Cas adaptation. Genes Dev 29(4):356–361
Westra ER et al (2010) H-NS-mediated repression of CRISPR-based immunity in Escherichia coli K12 can be relieved by the transcription activator LeuO. Mol Microbiol 77(6):1380–1393
Westra ER et al (2013) Type IE CRISPR–Cas systems discriminate target from non-target DNA through base pairing-independent PAM recognition. PLoS Genet 9(9):e1003742
Wilkinson RA, Martin C, Nemudryi AA, Wiedenheft B (2019) CRISPR RNA-guided autonomous delivery of Cas9. Nat Struct Mol Biol 26(1):14–24
Winters IP et al (2017) Multiplexed in vivo homology-directed repair and tumor barcoding enables parallel quantification of Kras variant oncogenicity. Nat Commun 8(1):2053
Wong N, Liu W, Wang X (2015) WU-CRISPR: characteristics of functional guide RNAs for the CRISPR/Cas9 system. Genome Biol 16(1):218
Wu Y et al (2013) Correction of a genetic disease in mouse via use of CRISPR–Cas9. Cell Stem Cell 13(6):659–662
Wu X et al (2014) Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nat Biotechnol 32(7):670
Xie K, Yang Y (2013) RNA-guided genome editing in plants using a CRISPR–Cas system. Mol Plant 6(6):1975–1983
Xiong X et al (2016) CRISPR/Cas9 for human genome engineering and disease research. Annu Rev Genom Hum Genet 17:131–154
Xu X et al (2017) Reversal of phenotypic abnormalities by CRISPR/Cas9-mediated gene correction in Huntington disease patient-derived induced pluripotent stem cells. Stem Cell Rep 8(3):619–633
Yang L et al (2015) Genome-wide inactivation of porcine endogenous retroviruses (PERVs). Science 350(6264):1101–1104
Ye L et al (2014) Seamless modification of wild-type induced pluripotent stem cells to the natural CCR187Δ32 mutation confers resistance to HIV infection. Proc Natl Acad Sci 111(26):9591–9596
Yin H, Kauffman KJ, Anderson DG (2017a) Delivery technologies for genome editing. Nat Rev Drug Discov 16(6):387
Yin K, Gao C, Qiu J-L (2017b) Progress and prospects in plant genome editing. Nat Plants 3(8):17107
Yin H, Xue W, Anderson DG (2019) CRISPR–Cas: a tool for cancer research and therapeutics. Nat Rev Clin Oncol 16:281
Yosef I, Goren MG, Qimron U (2012) Proteins and DNA elements essential for the CRISPR adaptation process in Escherichia coli. Nucleic Acids Res 40(12):5569–5576
Zetsche B et al (2015a) Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR–Cas system. Cell 163(3):759–771
Zetsche B, Volz SE, Zhang F (2015b) A split-Cas9 architecture for inducible genome editing and transcription modulation. Nat Biotechnol 33(2):139
Zetsche B et al (2017) Multiplex gene editing by CRISPR–Cpf1 using a single crRNA array. Nat Biotechnol 35(1):31
Zhang S, Sodroski J (2015) Efficient human immunodeficiency virus (HIV-1) infection of cells lacking PDZD8. Virology 481:73–78
Zhang F et al (2018) CRISPRminer is a knowledge base for exploring CRISPR–Cas systems in microbe and phage interactions. Commun Biol 1(1):180
Zhen S et al (2015) Harnessing the clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated Cas9 system to disrupt the hepatitis B virus. Gene Ther 22(5):404
Zhou Y et al (2014) High-throughput screening of a CRISPR/Cas9 library for functional genomics in human cells. Nature 509(7501):487
Zhu LJ et al (2014) CRISPRseek: a bioconductor package to identify target-specific guide RNAs for CRISPR–Cas9 genome-editing systems. PLoS One 9(9):e108424
Zhu S et al (2016) Genome-scale deletion screening of human long non-coding RNAs using a paired-guide RNA CRISPR–Cas9 library. Nat Biotechnol 34(12):1279
Zuckermann M et al (2015) Somatic CRISPR/Cas9-mediated tumour suppressor disruption enables versatile brain tumour modelling. Nat Commun 6:7391
Acknowledgements
We are grateful to Assam University, Silchar, Assam, India, for providing necessary facilities for this review work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Authors declare no conflict of interest in this work.
Additional information
Communicated by M. Kupiec.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Barman, A., Deb, B. & Chakraborty, S. A glance at genome editing with CRISPR–Cas9 technology. Curr Genet 66, 447–462 (2020). https://doi.org/10.1007/s00294-019-01040-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00294-019-01040-3