
Abstract
Genome editing is a recent technological advancement in life sciences that is being used to create novel genetic changes in the genome across different species, including plants, bacteria, and animals. Site-directed nucleases were earlier used for genome editing, and nowadays, CRISPR/Cas (clustered regularly interspaced short palindromic repeats)-based genome editing technology is popular among scientists due to its simplicity, flexibility, and ease of access. In this review, mechanisms such as repairing double-stranded breaks through non-homologous end joining and homologous recombination as well as history of genome editing and different genome-editing approaches, including ZFNs, TALENs, meganucleases, base editing, prime editing, and CRISPR/Cas, are discussed. CRISPR/Cas have been successfully used for treating human diseases and crop improvement. But despite numerous advantages of using CRISR/Cas as a tool of gene modification, it is also facing major hurdles. The review highlights complex designing, inefficient delivery systems, selection of target sites, design of guide RNA (gRNA), occurrence of off-targets, weak efficiency of repair of eukaryotes, endonuclease activity, and cytotoxicity of Cas9 as the major challenges. The information provided in this review will facilitate in understanding genome editing and approaches that it includes along with their technological advancements and challenges.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
1 Introduction
Plant breeders have tried to improve crop varieties in order to satisfy the hunger of exponentially growing human population. Furthermore, steady changes in climatic conditions and reduction in natural resources gave birth to new problems which limited the scientists to achieve desirable outcomes within time. Traditional breeding methods utilize already available genetic variation in natural population to produce a new variety which takes around 8–10 years, lacking behind in the race to feed the ever-growing population. In addition, it leads to degradation of genomic diversity, ultimately resulting in the generation of vulnerable genetic stock (Haroon et al. 2020). Genome-editing (GE) technology creates fundamental insight into biology of crop plants ultimately revolutionizing the agriculture sector at commercial scale (Chen et al. 2019). The GE techniques including the custom-based site-specific nucleases (SSNs), such as meganucleases, zinc finger nucleases (ZFNs), and transcription activator like effector nucleases (TALENs), come under the traditional techniques. However, GE came into the limelight after CRISPR/Cas9 was developed and was considered as a modern technique (Chen et al. 2020). CRISPR/Cas9 technique is mostly utilized in plant breeding programs for the sake of its high efficiency, easy to perform, and high flexibility in comparison to conventional GE techniques. Plants generated through these techniques are almost similar to their wild types except the corresponding trait allowing them to be separate from the genetically modified organisms (GMOs) legislation (Ran et al. 2017). The GE techniques are now known as new breeding techniques (NBT) which influenced the academic institutions, legislation authorities, and government bodies to rewrite the regulation document. Together with conventional plant breeding methods, NBT have shown immense potential in future of trait improvement of elite cultivars. SSNs directed generation of double-stranded breaks (DSBs) are simultaneously repaired by two natural mechanisms either non-homologous end joining (NHEJ) or homologous recombination (HR), resulting into a loss of function or replacement of gene, respectively (Yin et al. 2017). Using the idea of natural mechanism, plant breeders are exploiting NHEJ pathway for the production of knock-out mutants and synthetically deriving homology-directed repair (HDR) pathway for development of knock-in mutants. However, GE offers great opportunities to basic and applied research areas, but on the other hand it is also arousing many dynamic challenges. Independent responses of a particular plant species or cultivar against in vitro processes, transformations, and survival rate are its major drawbacks. Furthermore, off-targets and unintended modification due to integration of cassettes in plant genomic background affect the productivity of GE techniques (Ellison et al. 2020). Since the beginning of CRISPR in plant GE, there has been tremendous improvement in this technique with the introduction of novel tools. These include DNA free editing, base editing, prime editing, epigenome editing, CRISPRa (gene activation by CRISPR), CRISPRi (gene induction by CRISPR), etc. (Zhang et al. 2019, 2020). The future of plant science is looking promising, although substitution of every novel technique needs to be simultaneously addressed to avoid any delay for betterment of agricultural sciences. This chapter highlights the current scenario of GE techniques along with their challenges and new approaches with future perspective.
2 Mechanisms of Repairing Double-Stranded Breaks
2.1 Non-Homologous End Joining (NHEJ)
NHEJ is a type of DSB repair which is not a result of immense homology. There are two types of NHEJ – classical NHEJ and alternative NHEJ. The former one needs a lot of factors such as Ligase 4, KU70/80, XRCC4, etc. (Burma et al. 2006), whereas the latter one will lead to the least of the DSB repair that is required, and it doesn’t need any of the mentioned factors that are needed in classical NHEJ. Alternative NHEJ usually results in minimum homology and a deletion at the repair junction. It is not clear up to what extent is alternative-NHEJ different from homology directed repair (Guirouilh-Barbat et al. 2004). NHEJ can lead to mutations and the error rates can be as high as 50% (Paris et al. 2015).
The DNA repair mechanisms either NHEJ or HDR play a role in genome editing. In bacteria, the DSB can be repaired by either HDR or NHEJ. In eukaryotes, breaks by CRISPR/Cas can be most effectively repaired by NHEJ, which leads to indel mutations (Bernheim et al. 2017). For example, different pathways can affect how CRISPR/Cas will perform. These pathways lead to the regulation of the DSB, if it will be available or if it will compete with the CRISPR/Cas machinery for DNA substrate. Also, as the DNA substrate becomes available for the CRISPR/Cas mechanism, it might inhibit the DNA repair mechanism pathways to work. It has been observed that after a DSB, spacers from CRISPR/Cas have been obtained from a DNA repair mechanism called RecBCD pathway (Levy et al. 2015).
2.2 Genome editing and Homologous Recombination
Homologous recombination refers to the exchange of identical DNA sequences. This mechanism makes sure that the precise replacement and joining of DNA molecules happen; however, the exchange might not be possible if there is less homology. This process is very helpful when certain mutations are required to be brought into the organism’s system or when certain mutations are needed out of the same system. Different kinds of mutations can be introduced into the DNA with the help of certain nucleases called SSNs (sequence-specific nucleases). As the name suggests, SSNs are very specific in cutting the double-stranded DNA at a particular targeted sequence. The natural DSB repair mechanisms of the host come into play afterwards and have been studied in yeast and bacteria (Doudna and Charpentier 2014). This indispensable mechanism has a lot of applications in the biological systems. This is an efficient and simple method for gene deletion as a minimal level of gene homology would also lead to targeting a specific gene. Gene targeting is very successful in mouse model system. Mammalian cells were not targeted that often but as such techniques improved, it leads to manipulating the non-selectable genes more frequently and with much higher efficiency (Müller et al. 1999; Sedivy and Dutriaux 1999). Once damage is done to the DNA, the DSB can be repaired by SDSA (synthesis dependent strand annealing) pathway, or by the formation of a DSBR (double-stranded break repair) which follows either a non-crossover or a crossover approach, or SDSA, which follows only the non-crossover approach (San Filippo et al. 2008).
2.2.1 History of Genome Editing
Gene targeting was first done in animal cells, which were earlier considered hard to work with. But shortly after the creation of first knockouts in animal cells, it was seen that the same can be done in plant cells as well. Nicotiana spp. was used as a transformation system using PEG (polyethylene glycol)-mediated transformation, which had led to low transformation efficiency and is also time consuming (Paszkowski et al. 1988); however, Agrobacterium-mediated transformation has been later proved to be more efficient (Offringa et al. 1990). Agrobacterium-mediated transformation method is more efficient in terms of transformation efficiencies but less efficient in terms of targeting efficiencies, and it is also considered to be less labor intensive. With Agrobacterium-mediated transformation, transformation efficiency increased and the targeting efficiency decreased (Offringa et al. 1990). False positives can be a problem. Some gene targeting products that were thought to be positive proved to be random integrations. It may have happened because of the cell’s repair mechanism that resulted in the integration of a random sequence in place of the target sequence. Agrobacterium-mediated transformation has also shown low transformation efficiencies as PEG (Hrouda and Paszkowski 1994). Lower organisms such as Chlamydomonas also show low transformation efficiencies as higher organisms such as tobacco or Arabidopsis (Smart and Selman 1991; Sodeinde and Kindle 1993; Gumpel et al. 1994).
Most of the time, the tissues used were from the mesophyll protoplasts of leaf from Arabidopsis or tobacco, and sometimes the root tissue from Arabidopsis was also used (Miao and Lam 1995). Vacuum infiltration is another method that made use of inflorescence of Arabidopsis (Bechtold 1993). It was hypothesized that the positive-negative selection as well as the endogenous genes might also have caused the efficiency to be low. By using a negative selection system, a very high efficiency has been seen in rice (Terada et al. 2002).
2.2.2 Homologous Recombination in E. coli
It is common knowledge that prokaryotic systems are easier to understand as compared to eukaryotes (Roca and Cox 1997). In eukaryotes, as well as prokaryotes, there are different kinds of enzymes involved in HR. DSBs are identified and repaired by the RecBCD pathway (Kowalczykowski et al. 1994). The heterotrimer of RecB, RecC, and RecD proteins recognizes the break and uses its exonuclease and helicase activity, which also requires Mg2+ ions. Recombination hot spots are created by Chi(χ)-site sequences, with which the said heterotrimer complex interacts and the enzyme degrades the 3′ terminal strand, followed by 5′-terminal degradation. Single-stranded DNA on the 3′ terminal is produced by RecBCD complex, followed by RecQ helicase providing a substrate for RecA to produce a nucleoprotein filament by coating the tail of 3′ssDNA (Bianco and Kowalczykowski 1997; Arnold and Kowalczykowski 2000). RecBCD displaces single-stranded binding proteins from the ssDNA, which is stabilized with the binding of RecBCD (Meyer and Laine 1990).
Branch migration is promoted by proteins such as RuvA, RuvB, and RecG. RuvA identifies the holliday junction, RuvB is important for the migration of branch, and RecG is also important for branch migration, but at a smaller scale (West 1996; Whitby and Lloyd 1998). Mutants of RecA proteins show no recombination events (Cox 1999).
2.2.3 HR in Saccharomyces cerevisiae
HR is more important when it comes to S. cerevisiae as compared to NHEJ. The cells that were competent for HR didn’t show any DSB. NHEJ activity could only be seen when the HR system is disabled, acting as a backup system (Siede et al. 1996). RAD genes such as RAD54, RAD55, RAD52, RAD50, RAD50, RAD 57, RAD55, and MRE11, XRS2 are involved in HR. The function of these genes is written in Table 1. The mutants of these genes are not affected by infrared but by ultraviolet light (Petes 1991; Pâques and Haber 1999; Pastink et al. 2001; Symington 2002; van den Bosch et al. 2002). RAD52 is the most important of all the said genes for HR. Mutant of rad52 shows IR (Infrared) sensitivity. But when a double mutant is made with rad52 and one of rad52, rad54, rad55, or rad57 genes, the phenotype is consistent with each other. Other than RAD52, RAD51 is also needed for some HR events. The mutants of other genes involved in HR in S. cerevisiae such as xrs2, mre11, and rad50 also show similar phenotypes as described before (Game and Mortimer 1974).
Different genes in S. cerevisiae that are involved in HR are connected by networks.
2.2.4 HR in Higher Organisms
Genes involved in homologous recombination in higher organisms are a little different than the ones found in lower organisms. Gene targeting in mice helped scientists generate thousands of mutations, which lead to loss of function of a protein (Smithies 1987). One integration event in a hundred could be seen in embryonic stem cells (Jasin et al. 1996). The same has not been seen in plants, where the transformation frequencies are much lower (Paszkowski et al. 1988). Generally, transformation methods such as using polyethylene glycol, electroporation, and Agrobacterium-mediated transformation are employed; however, the gene transformation efficiencies are always as low as 1 in 10,000 or 1 in 100,000 (Zupan et al. 2000; Potrykus and Spangenberg 2013). Up to 22 kb DNA has been transferred (Thykjær et al. 1997). Recombination can be increased in plants by induction of DSBs (Pâques and Haber 1999).
Transformation mediated by Agrobacterium in Arabidopsis to target TGA (TGA1A-related gene 3) locus has been used. The number of calluses that were used was 2580, whereas only one of them showed the targeted TGA locus (Miao and Lam 1995). In Arabidopsis, MADS-box gene AGL5 (Agamous-like 5) was knocked out. Out of 750 events, one showed to have the said gene actually targeted, when vacuum infiltration was used (Kempin et al. 1997). Out of the two models of recombination double-stranded break repair (DSBR) and SDSA, it is observed that chromosomal rearrangements, specifically translocations can occur, according to the DSBR model, whereas translocations are completely avoided according to the SDSA model (Gorbunova and Levy 1999; Puchta 1999).
Gene targeting has also been done in moss, Physcomitrella patens, where DNA was very efficiently integrated into the organism using homologous recombination (Schaefer 2001). The reason for more efficient gene transfer in moss compared to plants can be given to the fact that gene transfer occurs at a particular stage in the life cycle of a moss, which is the G2/M phase (Reski 1999).
3 Different Genome Editing Techniques
3.1 Meganucleases (MNs)
Meganucleases or homing endonucleases are the type of endonucleases that cleaves DNA at a larger recognition site of around 14–40 bp (Iqbal et al. 2020). They are naturally occurring restriction enzymes that are found in prokaryotic and unicellular eukaryotic organisms (Carroll 2017). The recognition site of MNs is bigger than normal type II restriction enzymes and can alter the target sequence in a highly efficient manner. MNs are encoded by mobile genetic elements and are composed of both DNA binding and DNA cleavage domains. The double-stranded breaks formed by MNs are repaired by NHEJ or HDR process (Silva et al. 2011).
Based on their structural and sequence motifs, MNs have been characterized into five families: HNH, His-Cys box, GIG-YIG, PD-(D/E) XK, and LAGLIDADG (Zhao et al. 2007). Among all these families, the LAGLIDADG family is well characterized and is highly used for genome modification purposes. I-SceI (Saccharomyces cerevisiae), I-CreI (Chlamydomonas reinhardtii), and I-DmoI (Desulfurococcus mobilis) are widely used meganucleases of the LAGLIDADG family (Khandagale and Nadaf 2016). They can withstand site-specific polymorphism without loss of binding and cleavage activity. This technology has been successful with I-SceI-mediated transformation in prokaryotes and eukaryotes, but the structure of I-SceI is quite complex which makes it difficult for re-engineering to target genes of interest (Zaman et al. 2019). The structure of I-CreI is less complex and has been widely used to knockout genes in several organisms (Arnould et al. 2007). For example, The Cre-I-based meganuclease was used to target two maize loci, namely, liguless 1 and ms26, which upon treatment induced the insertion and deletion mutations at target loci (Gao et al. 2010; Djukanovic et al. 2013). Furthermore, it has been shown that the DSB repair by NHEJ led to gene knockout in Arabidopsis and tobacco (Kirik et al. 2000). Thus, overall meganucleases are easy to use and can be used to edit the genomes of plants and animals. They also possess a small size (40 kD) which makes them compatible with viral vectors with shorter coding sequences (Iqbal et al. 2020). Despite these advantages, they have not been commonly used in genome engineering as other genome-editing tools due to certain limitations. The first limitation is that the DNA binding domain and catalytic domain are overlapping. To edit the target gene, one has to engineer the DNA recognition sites of MNs but as both domains overlap each other, it is really hard to re-engineer the MNs compared to other genome-editing tools (Khandagale and Nadaf 2016; Iqbal et al. 2020). Second, meganucleases are prone to sequence degeneracy which can highly result in off-target binding and cleavage (Argast et al. 1998). So, in order to overcome the limitations of MNs, researchers were focusing on other simple and efficient methods of gene editing which gave rise to ZFN, TALENs, and CRISPR.
3.2 Zinc Finger Nucleases (ZFNs)
ZFNs are the proteins, designed to cut the DNA at specific sites known as DSBs, which subsequently leads to induction of HR or NHEJ. These repair mechanisms result in deletions, insertions, and base mutations at the site of cleavage (Carroll 2011). Hence, this technology has been employed for the editing of plant and mammalian genomes. ZFNs have different DNA-binding and DNA-cleavage domains (Li et al. 1992).
Fok1 is a type IIS restriction enzyme (Kim and Chandrasegaran 1994). It consists of N-terminal DNA-binding domain and non-specific DNA cleavage domain at the C-terminal end. The cleavage domain has no sequence specificity and hence, can be redirected by substituting with the alternative recognition domains and the most useful for these were Cys2His2 zinc fingers (Kim and Chandrasegaran 1994). Various sequences can be attacked by using novel assemblies of ZFNs. When both sets of ZFNs bind to their recognition sequences on the DNA, dimerization and cleavage is achieved. Short linkers of 5–6 bp (base pair) are generally used between the domains of the protein and binding sites (Bibikova et al. 2001; Händel et al. 2009; Shimizu et al. 2009).
Kim et al. (1996) created first ZFNs as chimeric restriction endonucleases, and the first success was achieved using a ZFN pair that targeted the genome of Drosophila. However, frequency of target modification varies, and ZFN pairs have been successfully used in a wide range of organisms and cell types (Carroll 2011). The success of this technique of genome editing depends on the delivery method used to deliver ZFNs into the host cell. Earlier experiments were dependent on the genomic integration of ZFN-coding sequences and donor DNA via P-element-mediated transformation, (Bibikova et al. 2002, 2003; Beumer et al. 2006) which used to require elaborate and complex construction of delivery system of the ZFNs. A major breakthrough in this technology occurred when it was demonstrated that both DSB repair mechanisms could be obtained through injecting ZFN mRNAs and donor DNA into the host embryo (Beumer et al. 2006). This method is well-established in zebrafish, rat (Geurts et al. 2009; Mashimo et al. 2010), frog (Young et al. 2011) as well as sea urchin (Ochiai et al. 2010), and ZFN-induced mutagenesis had been achieved in a number of genes (Doyon et al. 2008; Meng et al. 2008; Foley et al. 2009). In the higher organisms such as plants, including Arabidopsis thaliana and several crop species, Agrobacterium-mediated transformation had been used through the delivery of coding sequences which are under the control of viral promoter (Lloyd et al. 2005; Cai et al. 2009; De Pater et al. 2009; Osakabe et al. 2010; Zhang et al. 2010). In addition to this, direct transfer of DNA (Wright et al. 2005; Cai et al. 2009; Shukla et al. 2009; Townsend et al. 2009) and viral delivery has been a success in the plants as well (Ira et al. 2010).
The design of ZFNs to target genetic modifications is smooth; however, substantial proportion of ZFN pairs fail (Ramirez et al. 2008; Joung et al. 2010; Carroll 2011). This is the reason that scientists at Sigma-Aldrich and Sangamo Biosciences always practice making multiple pairs for sequences within a single target gene to do extensive testing. There are various methods to select three sets of ZFNs from partially randomized libraries, which can be quite time consuming (Meng et al. 2007). ZFNs for some DNA triplets derived by ToolGen explain the individual finger in their collection that behaves best in modular assembly (Kim et al. 2011).
The modular structure of ZF motifs and recognition by ZF domains allows designing of the artificial DNA binding domains to facilitate genetic modification at the specific target site in the genome (Pabo et al. 2001; Beerli and Barbas 2002). The ZF motifs binds the DNA through insertion of its α-helix into the major groove of helical structure of DNA (Pavletich and Pabo 1991). Key amino acids are present at −1, +1, +2, +3, +4, +5, and +6 positions (relative to the start of α -helix) of ZF motif (Pavletich and Pabo 1991; Shi and Berg 1995; Elrod-Erickson and Pabo 1999). In order to bind long sequences of DNA, several ZF motifs are linked in a tandem fashion which forms zinc finger proteins (ZFPs) (Kim et al. 1996; Liu et al. 1997; Beerli et al. 1998). Zinc finger activators (ZFAs), zinc finger transcription repressors (ZFRs), and zinc finger methylases (ZFMs) make the whole ZFP platform (Xu and Bestor 1997; Bartsevich and Juliano 2000; Zhang et al. 2000; Liu et al. 2001; McNamara et al. 2002; Rebar et al. 2002; Ren et al. 2002; Bartsevich et al. 2003; Snowden et al. 2003; Dai et al. 2004; Rebar 2004).
ZF recognition depends upon the match to the target DNA sequence as well as mechanisms being employed for DSB repair. This ability of ZFNs to modify specific sequences of genes in order to create variants with loss-in-function is a powerful tool for investigating the function of genes as well as development of new products (Osakabe et al. 2010). Gene knockouts have been prepared by the scientists in zebrafish. Similarly, mutagenesis and gene replacement had been achieved in mice (Carbery et al. 2010). Alterations in the genomic loci had been also achieved in the crop plants. Tobacco (Townsend et al. 2009) and maize (Shukla et al. 2009) can be regrown again from the callus, modified in culture by ZFNs. ZFN knockout of CCR5 gene is one of the therapeutic application in humans, which provide resistance to HIV with an improved immune system (Urnov et al. 2005). Similarly, efforts are being made to knock out the targeted genes in order to treat the neurodegenerative diseases in humans such as Huntington’s, Parkinson’s, Schizophrenia, and amyotrophic lateral necrosis (Swarthout et al. 2011).
This technology is broadly applicable and versatile in the genetic engineering of plants. Before the emergence of this technology, targeted gene modification was difficult in plants, engineering of plant traits had always been laborious, time consuming, and unpredictable (Puchta 2002). Zinc finger consortium (ZFC) had been established to ensure the development of ZFNs technology through creation of software, resources, and other required tools for engineering zinc fingers for performing genome editing (Wright et al. 2005; Maeder et al. 2008). For instance, using this publicly available consortium, ZFNs were engineered to recognize SuR loci to achieve high-frequency modification of plant genes (Townsend et al. 2009). Jeffrey A. Townsend et al. (2009) used ZFN to target acetolactate synthase genes (ALS, SuRA, SuRB) in tobacco, which resulted in herbicide-resistance mutations. ZFC had developed a method, called oligomerized pool engineering (OPEN), that uses genetic selections in bacteria for identifying variants of zinc finger arrays (ZFAs) that recognize specific target sequences in the genome (Maeder et al. 2008). These ZFAs function as ZFNs. Similarly, other researchers also used ZFNs to modify endogenous loci in plants, for instance, Shukla et al. (2009), described the use of this technology for genome editing in the crop species of Zea mays. Furthermore, ZFNs is a powerful tool for genome modification of animals as well (Rémy et al. 2010). Initially, genetic manipulations of embryonic cells were done by cloning through nuclear transfer which was limited to only some species and modification at specific loci started with emergence of ZFNs. It has been used to modify Drosophila, zebra fish, and rats (Beumer et al. 2008; Ekker 2008; Geurts et al. 2009). Mammalian cells, including the human genome had been modified permanently via HR of targeted DSB (Durai et al. 2005).
Scientists have also faced several challenges with delivery of the targeting materials. High level of somatic mutagenesis in the targets of genomes as well as extra chromosomal arrays was achieved using heat shock promoter for driving ZFN expression from a DNA template in Caenorhabditis elegans. Due to RNA interference, parallel expression in the germline was undetectable (Morton et al. 2006). Apart from the use of ZFN for mutagenesis, there have been other studies reporting gene replacement using ZFNs. Some genomic regions and sequences within a single gene are sometimes inaccessible due to compact chromatin structure or modifications in the DNA. For instance, chromatin structure prevents cleavage of intact recognition sites during mating-type switching in Saccharomyces cerevisiae (Rusche et al. 2003). DSB repair mechanisms differ with cell types and their developmental stages; hence, understanding of the biological system of every organism is essential to overcome these limitations. Another challenge is specificity of ZF binding, as some bind equally well to triplets other than their supposed preference. The addition of fingers can improve both, specificity and affinity; however, it might lead to binding at off-target sites. The separation of two-finger modules with a short linker was shown to improve specificity (Moore et al. 2001). Death of the host cells is the ultimate result due to off-target cleavage, as the number of breaks outstrips the DSB repair capacity of the DNA (Bibikova et al. 2002; Porteus and Baltimore 2003; Alwin et al. 2005).
3.3 Transcription Activator-Like Effector Nucleases (TALENs)
TALENs are the restriction enzymes which can be engineered to cut the DNA at specific sequences; hence, they are being used as site specific nucleases for targeted genome editing. TALENs are the fusions between non-specific DNA cleavage domain and a custom-designed DNA binding domain (Miller et al. 2011; Wood et al. 2011). DSBs are induced at the desired site on the DNA, which can be repaired by HDR or NHEJ to create small insertions or deletions at the cleavage sites. This technology emerged as an alternative to one of the similar genome-editing technologies, i.e., ZFNs. It contains DNA-binding domains which contain highly conserved repeats, derived from TALENs. These are proteins secreted by Xanthomonas spp. (Boch and Bonas 2010). Both ZFNs and TALENs can cleave the DNA at similar efficiency (Hockemeyer et al. 2011; Tesson et al. 2011; Reyon et al. 2012). The difference between them is that, TALENs more site specific with lesser off-target effects as compared to ZFNs (Chandrasegaran and Carroll 2016).
TALENs can be easily and rapidly designed using “protein-DNA code,” relating DNA-binding TALE repeat domains to the target-binding site. One TAL effector repeatedly binds to one base pair of DNA (Boch et al. 2009; Moscou and Bogdanove 2009). These TAL effector repeats can also be joined together to develop extended arrays that can recognize new targets in DNA (Boch et al. 2009; Morbitzer et al. 2010; Miller et al. 2011; Weber et al. 2011). Most of the methods that are used to construct TALENs, use golden gate cloning method with some variations (Morbitzer et al. 2010; Cermak et al. 2011; Huang et al. 2011; Li et al. 2011; Sander et al. 2011; Weber et al. 2011). However, none of them are adaptable for automated high-throughput production (Maeder et al. 2008; Morbitzer et al. 2010; Cermak et al. 2011; Li et al. 2011; Sander et al. 2011; Weber et al. 2011). DNA-binding domains with highly conserved repeats of 33–35 amino acids are transferred into the host cells via Type III secretion system of the bacteria, Xanthomonas spp., hence, facilitating bacterial colonization to alter the transcription of genomic DNA of the host cells. The two hypervariable residues identify the site at the DNA where the TALE repeats bind. Hypervariable residues, viz., NN, NI, HD, and NG, in nearly all engineered TALE repeats recognize guanine, adenine, cytosine, and thymine respectively (Joung and Sander 2013).
Fast ligation-based automatable solid-phase high throughput (FLASH) is a rapid and cost-effective technology for assembly of TALENs at large scale (Reyon et al. 2012). This technology had been used previously to construct 48 TALEN pairs which were targeted to diverse range of gene sequences in human beings and 100% of nucleases were active in human cells, similarly FLASH TALEN pairs had been targeted to 96 genes involved in epigenetic regulation in humans, out of which targeted alterations arose in 84 genes (Reyon et al. 2012).
Genes in a wide range of cell types or organisms can be engineered using this technology; thus, this technology has significant effects on biological research and has a potential to treat genetic diseases as well as has applications in crop improvement (Joung and Sander 2013). For reflecting its wide importance, it was named the 2011 “Method of the Year” by the journal “Nature Methods” (Baker 2011). This technique had been employed in a variety of organisms, including yeast (Li et al. 2011), zebrafish (Sander et al. 2011), frog (Lei et al. 2012), roundworm (Wood et al. 2011), rat (Tesson et al. 2011), cow, pig (Carlson et al. 2012), rice (Li et al. 2012), thale cress (Cermak et al. 2011), silkworm (Ma et al. 2012), cricket (Watanabe et al. 2012), fruit fly (Liu et al. 2012), and somatic and pluripotent stem cells of human beings (Cermak et al. 2011; Hockemeyer et al. 2011; Miller et al. 2011; Reyon et al. 2012).
Construction of TALE repeat arrays can be challenging due to the need for assembling multiple, identical repeat sequences. Different platforms have been designed, including “Golden Gate” cloning; solid phase assemble; standard restriction enzyme; and ligation-based cloning to facilitate the assembly of plasmids that encode TALE repeat arrays (Joung and Sander 2013). Usually, TALENs are built to bind 18-bp sequences or even longer than that; however, recent studies have suggested that use of larger TALENs may result in less specificity (Guilinger et al. 2014). Off-target effects are also one of the major concerns regarding TALENs, as, in one of the studies where this technology was used in human pluripotent stem cells, mutagenesis at 19 possible off-target sites was reported (Hockemeyer et al. 2011). The size of cDNA encoding TALEN is approximately 3 kb. This large size is also one of the disadvantages of TALENs, which makes it harder to deliver and express TALENs into the host cells. The ability of the TALENs to get delivered by some of the viral vectors is often impaired due to their highly repetitive nature (Holkers et al. 2013); however, this limitation can be overcome through diversification of the coding sequences of the TALE repeats (Yang et al. 2013).
3.4 Clustered Regularly Interspaced Short Palindromic Repeats/CRISPR Associated Protein
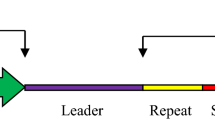
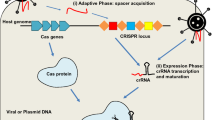
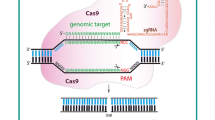
TALENs came as an alternative approach to the less accurate and error prone ZFNs. CRISPR is even easier to be prosecuted and is more efficient compared to TALENs and ZFNs. CRISPR and different types of Cas have been widely adapted as a gene-editing technology, which shows a lot of applications and many promising results. Emmanuelle Charpentier and Jennifer Doudna won the 2020 Noble prize in chemistry for CRISPR discovery (NoblePrize.org). Different types of Cas proteins are: Cas1, Cas2, Cas3, Cas5, Cas6, Cas7, Cas9, Cas10, etc. (Makarova et al. 2011). CRISPR/Cas is part of the immune system of bacteria to provide resistance against viruses. Once the bacteria encounter the same virus again, it attacks it with the memory from before (Horvath and Barrangou 2010). It consists of Cas endonuclease, a variety of which, i.e., Cas9 is derived from Streptococcus pyogenes and sgRNA (Heler et al. 2015). sgRNA consists of a tracrRNA and a crRNA (Deltcheva et al. 2011), also known as trans-activating crRNA and CRISPR RNA respectively. The sgRNA has a part that binds to the target sequence of the host, and another part that loops on itself (Cui et al. 2018), the former can be made specific for any target sequence (Cong and Zhang 2015). The target sequence consists of the region to be targeted by sgRNA, and a PAM (protospacer adjacent motif) sequence, which usually is NGG, where N pertains to any nucleotide, and G is guanosine. The target sequence is usually 20 nucleotides. If the PAM site is absent, the Cas9 endonuclease will not be able to recognize and hence cleave the target sequence. If the PAM site is present right before the sgRNA, the Cas9 endonuclease can bind to it and leads to the creation of a DSB exactly the size of the target sequence. Once that sequence is cut, the natural mechanisms of the host cells come into play to repair the DSB, by the mechanisms such as NHEJ and HDR (Wyman and Kanaar 2006). The latter will not, but the former one might lead to the creation of insertions and deletions, which leads to the loss of gene function.
Figure 1 shows different parts of CRISPR. CRISPR cannot only lead to loss of gene function, i.e., gene knockouts (CRISPRko) (Mali et al. 2013) but is also helpful in inducing the gain of function of a gene. CRISPRi (Qi et al. 2013) includes a non- functional Cas9 endonuclease, which doesn’t cleave the target sequence. CRISPRa (Gilbert et al. 2014) can be used for the activation of the gene when non-functional Cas9 is attached to a transcriptional activation domain. The sgRNA can be designed in various ways. Along with binding to the target sequence, sgRNA also works in recognizing the target sequence in the whole genome. A good sgRNA will have specificity as well as efficacy.

Mechanism of CRISPR/Cas. (Modified from https://www.labiotech.eu/in-depth/crispr-cas9-drug-discovery/)
The mutations that are created by NHEJ can be identified by Sanger sequencing, AFLP (amplified fragment length polymorphism), and restriction enzyme assays (Belhaj et al. 2013; Belhaj et al. 2015). Instead of targeting only one gene, multiple genes can be targeted together, which is called target gene multiplexing (Cong et al. 2013; Čermák et al. 2017). This can be used either to target different genes or different sequences in the same gene to increase efficiency, by having multiple sgRNAs either under different promoters or under the same promoter (Čermák et al. 2017). The other versions include Cys4-gRNA or tRNA-gRNA (Xie et al. 2015). The former is an endoribonuclease from Pseudomonas aeruginosa. It has been applied in plants such as wheat, Arabidopsis, tomato, potato, rice, banana, and tobacco (Upadhyay et al. 2013; Andersson et al. 2018; Kaur et al. 2018; Castel et al. 2019).
4 Challenges in the CRISPR/cas9 System
Despite having several advantages, CRISPR/Cas9 system in gene modification faces major hurdles, lowering the efficiency or complete failure of genome editing.
4.1 Complex Designing
Despite of being highly specific, the conventional techniques are restricted to only research laboratories due to the inefficient understanding of their complex nature. The customized protein engineering varies from species to species and is more difficult in polyploidy genomes. Use of meganucleases is limited because of a rare homing site present in particular genome which requires extensive research to design DNA-binding domains ultimately narrowing their application in plant science (Wright et al. 2014). Additionally, it is also time consuming and costly in comparison to other GE techniques (Aglawe et al. 2018). ZFNs overcome few of the problems of meganucleases and broaden the scope of plant genome editing in various agriculturally important crops. However, the selection-based fabrication of large sized libraries for different traits make it inadequate to be used in diverse laboratories, since it requires high technical expertise (Maeder et al. 2008; Nelson and Gersbach 2016). The most specific technique, i.e., TALENs, requires monomer DNA-binding domains; perhaps, comprehensive knowledge is needed to achieve significant target efficiency. Various methods of TALEs construction have been developed using 20–30 monomers involving ligation, cloning, to generate dimers library, and subsequently, golden gate assembly is used which is very tedious and time-consuming process (Abdallah et al. 2015). Currently, CRISPR/Cas9 technique is a preferred approach, bypassing the meganuclease, ZFNs, and TALENS due to its simple RNA-dependent DNA binding followed by cleavage of target site via single protein (Zhang et al. 2014; Sharma et al. 2017).
4.2 Inefficient Delivery
To modify plant genome, the construct of GE machinery needs to be effectively introduced in plant cell, making it a very crucial step to achieve beneficial outcome. The methods available for delivery of cassettes into plants are based on Agrobacterium, viral vector, gene gun mediated, lipofection, etc. (Nelson and Gersbach 2016, Yin et al. 2017, Liu et al. 2020). The target tissues for transformation being utilized are callus, immature embryos, protoplast, shoot apical meristem, etc. (Altpeter et al. 2016, Ran et al. 2017). Delivery of construct is very challenging because of the specific requirement of independent genotypes, explants, and type of in vitro culture conditions (Ran et al. 2017). Additionally, the requirement of transgene free altered plants at commercial scale implies restriction on the most frequently used methods of delivery of GE cassettes, creating a tough task for the researchers. This emerges as a principal obstacle toward the generation of novel traits in plants (Baltes et al. 2014). Although considerable achievements have been obtained for transformation or delivery of GE reagent in various crops, their low regeneration ability and unstable integration are hindering the scientific endeavors (Altpeter et al. 2016).
4.3 Selection of Target Site and gRNA Design
The major advantage of CRISPR machinery is its ability to target ~21–23 base pair (bp) DNA sequence containing a PAM sequence on forward or reverse strand. The PAM motif on average occurs every 8bps which provides higher flexibility in target site selection (Ramakrishna et al. 2014). In addition, Cas9 proteins from several other organisms have been studied and different PAM sites have been identified. This diversity in PAM sites have increased choices for choosing target sequences. However, recent reports have shown that the target site selection and designing of guide RNA is not as simple as it was previously assumed. Due to post-transcriptional modifications of mRNA transcribed by RNA polymerase II, it is not possible to use RNA polymerase II for sgRNA construction (Zhang et al. 2014). Currently, RNA polymerase III along with Ubiquitin3 (U3) and U6 small nuclear RNA (snRNA) promoters are used for guide RNA production. The Ubiquitin genes are housekeeping genes and cannot be used to generate tissue specific or cell specific gRNAs (Gao and Zhao 2014). As the RNA polymerase III is not commercially available, this also limits the production U3- and U6-based guide RNA. Therefore, various approaches should be developed to overcome the application of ubiquitin promoters-based gRNA construction.
4.4 Off-Target Effect
In additional to the rational design of sgRNA, off-target DNA cleavage by Cas9 endonuclease is a major challenge, reducing the efficiency of the machinery. During the process of CRISPR/Cas9 gene editing, the interaction of endonuclease and target strand initiated by the recognition of PAM motif, which denatures DNA upstream of the motif, therefore allowing the binding of target sequence and sgRNA to form R-loop (Ebrahimi and Hashemi 2020). The Cas9 enzyme then cleaves the target DNA having less number of mismatches, ultimately resulting in the off-target cleavage (Herai 2019; Newton et al. 2019). This off-target cleavage came from natural combat between bacteria and virus where the DNA sequence of virus mutates itself to escape from the chopping effect of Cas9 nuclease, but Cas9 in return is capable to bind and chop target virus with minimum number of mismatches (Li et al. 2019). Based on these accepted number of mismatches, various algorithms have been developed to design guide RNA and to check off-target sequences. In general, highly specific sequence with zero mismatch is required for cleavage of DNA at 7–9 PAM proximal bases, and 3–4 mismatches away from PAM site is admissible for attachment of Cas9 but not cleavage (Dagdas et al. 2017; Singh et al. 2017).
The off-target effect is solely not based on the Cas9 endonuclease, but also depends highly on sgRNA design. Therefore, modifications in Cas9 protein and sgRNA can limit these off-target effects respectively. One approach is the modification of sgRNA backbone chemically. The introduction of 2′O-methyl-3′-phosphonoacetate at particular sites of sgRNA or partial substitution of ribonucleotides with deoxyribonucleotides can reduce off-target effects respectively (Ryan et al. 2018). Another approach includes creating mutant Cas9 systems where ZFNs and TALENs serve as an inspiration for improved target precision. This mutant Cas9 system involves the fusion of dcas9 (deactivated) and a Fok1 nuclease dimer. Here, the 20–21bp guide RNAs binds the target strand. After binding of sgRNA, the fok1 nuclease dimers become functional Fok1 and cause double-strand breaks (Guilinger et al. 2014; Tsai et al. 2014). This system significantly reduces off-target cleavage but increases the size of genome-editing tool, thereby reducing the efficiency of transformation in vivo. Another mutant approach involves the fusion of Cas9 protein with ZFPs or TALEs, which can target the genomic loci with better precision (Bolukbasi et al. 2015).
4.5 Weak Repair Efficiency of HDR in Eukaryotes
In eukaryotic cells, the double-strand breaks are repaired by NHEJ mechanism with higher efficiency. This mechanism commonly repairs the DNA without the use of template DNA resulting in indel mutations, which causes the initiation of frameshift resulting in establishment of gene knockout or knockdown (Shalem et al. 2014). On the other hand, the efficiency of HDR-mediated repair from double-strand break is very low in mammalian cells. It has been reported that the HDR repair in mice after cas9-based genome editing is 0.5–20% whereas the repair efficiency through NHEJ is ~20–60% (Maruyama et al. 2015).
Various strategies have been developed to increase the HDR repair process and reduce NHEJ efficacy. The use of tiny molecules known as inhibitors (for NHEJ) or inducers (for HDR) is one of the approach for improving recombination frequency (Aird et al. 2018). The application of SCR-7 (NHEJ inhibitor) or RS-1(HDR enhancer) have been reported to increase the HDR efficiency by several folds (Yu et al. 2015; Vartak and Raghavan 2015; Song et al. 2016). In addition, the other approaches include the gene silencing, use of cell lines deficit in NHEJ machinery (Weinstock and Jasin 2006), cell cycle synchronization, or controlled cas9 expression (Weber et al. 2015). The controlled delivery of CRISPR/Cas9 machinery along with cell cycle has increased the CRISPR/Cas9 mediated DNA repair. The synchronization of Cas9 protein with cell cycle progression can be made by fusing Cas9 protein with human DNA replication inhibitor (geminin), which modifies Cas9 endonuclease post-translationally (Gutschner et al. 2016). While the use of NHEJ inhibitors or HDR inducers have resulted in increased HDR mediated genome editing, they are really toxic to host cells and cause numerous problems for sufficient cell growth. To overcome these problems, another approach has been developed which rely on covalent tethering of repair template to ribonucleoprotein complex. The utilization of this approach led to the increase in HDR repair by ~30 folds, and it was proved that this strategy can be applied in various other organisms and target loci (Aird et al. 2018).
4.6 Cas9 Endonuclease Activity and Cytotoxicity
Several Cas9 proteins from different species have been found and used in genome editing, including Staphylococcus aureus (SaCas9) (Ran et al. 2013a, b), S. thermophiles (StCas9) (Kleinstiver et al. 2016), and Streptococcus pyogenes (SpCas9) (Vento et al. 2019). Among all, SpCas9 is an endonuclease enzyme commonly used for genome editing in prokaryotic bacteria. SpCas9 is a well-characterized endonuclease with simple PAM site and has high expression rate in various prokaryotes. On the other hand, different Cas9 proteins have been used in eukaryotes and the selection of specific Cas9 ortholog for each organism showed improved efficiency of editing for a specific sequence. In addition to the selection, many factors have shown to affect the activity of Cas9 protein. For gene editing to occur in eukaryotes, the Cas9 protein must translocate in nucleus, and the nuclear location signal (NLS) should be associated with Cas9 protein. It has been reported that by reducing the proximity of NLS and Cas9 using 32 amino acid spacer, the DNA cleavage activity increased to a higher extent (Shen et al. 2013). In addition, by increasing the guide RNA:Cas9 ratio was shown to increase on target chopping activity, by ensuring all Cas9 protein form active R loop with the sgRNA and DNA complex (Kim et al. 2014). Unlike other endonucleases, the activity of Cas9 protein is significantly less with a single turnover rate of ~0.4–1.0 per min (Jinek et al. 2012). Also, when bind to the target DNA, the displacement rate of Cas9 protein is quite challenging. It has been reported that even after DSB formation, the 1 nm Cas9 enzyme cleaved ~2 nm plasmid DNA after 2 h (Jinek et al. 2012). This shows Cas9 enzyme works more like actuator rather than a catalytic enzyme.
In addition to enzyme activity, the cytotoxic effect of Cas9 protein can be considered as a crucial obstacle in gene editing (Vento et al. 2019). Various attempts have been made to reduce this toxic effect of Cas9 protein for efficient genome editing using programmable DNA cleavage. The first approach includes the usage of inducible expression system for Cas9, where the activity of Cas9 protein is highly reduced when no inducer is present (Reisch and Prather 2015). Another approach includes the use of toxin-free endonucleases or variant nucleases. The variant of cas9 protein: cas9n has shown reduced toxicity because it targets only single strand of DNA (Standage-Beier et al. 2015).
5 Approaches
5.1 Base Editing
Base editing allows nucleotide substitutions in the genome by using modified Cas effectors without the requirement of producing DSBs (Komor et al. 2016). There are two types of base-editing tools, cytidine base editor (CBE) and adenine base-editor (ABE), that enable cytosine-guanine to thymine-adenine and adenine-thymine to guanine-cytosine transitions (Chen et al. 2019). These CBE and ABE tools make use of cytosine and adenosine deaminases for cytosine and adenine base editing (Komor et al. 2016; Nishida et al. 2016; Gaudelli et al. 2017; Ren et al. 2018; Wang et al. 2018). In the CBE system, nick/dead Cas protein fuses to cytidine deaminase, catalyzing conversion of cytidine (C) to uracil (U). In the ABE system, engineered Escherichia coli RNA adenosine deaminase (TadA) fuses to nick/dead Cas protein, leading to conversion of adenine (A) to inosine (I), which is recognized as guanine by DNA polymerase during replication of DNA. Plasmid transfection and viral delivery are the common methods of delivery of the base editors in the living cells (122,124). Hence, they introduce targeted substitutions in the genes, and it had been intensively employed in model plants and crops for the improvement of agricultural traits, including flowering, plant height, disease, and herbicide resistance (Chen et al. 2017; Shimatani et al. 2017; Kang et al. 2018; Tian et al. 2018; Chen et al. 2019; Wu et al. 2020). Other major applications are the study or treatment of disease-associated point mutations, use of base editors as the recorder of cellular events in biomedical research, and introduction of premature stop codons by CBA to disrupt genes in homogenous manner (Landrum et al. 2014, 2016; Farzadfard and Lu 2018). However, in mammalian cells, challenge is to circumvent DNA repair processes that oppose target base pair conversion (Rees and Liu 2018). Human cells undergo effective cellular repair of U.G intermediate through base excision repair of U.G in DNA, in which uracil N-glycosylate (UNG) recognizes U.G mismatch and cleaves glycosidic bond between uracil and backbone of deoxyribose (Kunz et al. 2009). Generation of indels, targeting limitations and off-target editing DNA base pairs, had been also reported (Rees and Liu 2018), and various scientists are doing efforts worldwide to overcome these limitations. Base editing in RNA is also possible that provide powerful capabilities to life sciences and medicine. To date, deamination of A to I has been only reported (Vogel and Stafforst 2019).
5.2 Prime Editing
Prime editors are being employed for precise editing, employs same mechanism as conventional CRISPR/Cas systems but does not require DSBs (Anzalone et al. 2019). It involves longer-than-usual guide-RNA, commonly known as pegRNA. Fusion of Moloney murine leukemia virus (M-MLV) reverse transcriptase (RT) and Cas9 nickase (nCas9) is the major component of prime editor. Complex of nCas/M-MLV/pegRNA mediates site-specific nicking by nCas9, which is then served as a template for RT and then at the end reverse transcriptions leads to production of stable edited DNA (Zhan et al. 2020). Base conversions and small insertions can be achieved through the use of prime editors (Anzalone et al. 2019). There are three prime editors: first is PE1 that is a combination of Cas9 H840A nickase and wild type (WT) M-MLV RT enzyme; second is PE2, improved thermostability, processivity, and DNA-RNA substrate affinity of the RT component; and third is PE3, in which second gRNA was introduced in addition to pegRNA (Anzalone et al. 2019). Prime-editing systems have the capability of performing precise genome editing in human cells, and scientists also tried to create mutations in the genome for treating rare genetic diseases, including SCD, Tay-Sach, and prion diseases in humans (Matsoukas 2020). Recently, Xu et al. (2020) has developed plant prime-editing system, plant prime editor 2 which was tested through targeted mutation on an HPT-ATG reporter in rice. Its development is an essential addition to the genome-editing technologies and also addresses CRISPR/Cas limitations. However, this technology also has some challenges, it may not be able to create large DNA insertions or deletions as compared to conventional CRISPR/Cas systems, possibility of addition of cDNAs due to presence of RT, and large protein constructs may affect the delivery of full-length therapeutic protein (Matsoukas 2020). Hence, further research is required for optimizing prime editors and maximizing their efficiency in different cell types.
6 Conclusion and Future Prospects
The steady and undesirable variations in climatic conditions, supplemented with depletion of the natural resources and biodiversity, are creating new challenges toward sustainable crop production. The rapid and constantly evolving nature of GE techniques assists plant scientists via numerous applications in crop improvement. These advances widen the scope of trait refinement in the diverse genetic background, irrespective of their natural mechanism. However, the efficient utilization of GE techniques seeks a deep understanding of the interaction between the genotype and their respective phenotype under the different environmental conditions as most of the agronomically important plant features are controlled through a complex genetic mechanism. Conventional GE techniques initially gained the attention of researchers due to their ability to induce directed DSBs followed by the natural repair mechanism. But, the natural weapon of the bacterial immune system transformed the era of GE and emerged as a principle genome modifying tool. The necessity of short sgRNA and single unit Cas9 nuclease protein makes it the first preference over the conventional methods. CRISPR opens up tremendous opportunities in the living world beyond the DSB mediated SDNs, including the study of regulatory elements, complex genetic mechanisms, cell signaling, chromatin modeling, etc. These developments broadened the way of assessment in various agriculturally important monocot and dicot species as well as the plants having multiple copies of genomes. The adequate information of functional genomics is a prerequisite to target any specific genotype. GE techniques, especially CRISPR, have shown the ability of precise mutation through knock-out, knock-in, and knock-down mutation resulting in loss of function, the gain of function, and specific transient modulation, respectively. The rapid advancement in CRISPR techniques accompanied by dynamic natural variations display enormous opportunities for the betterment of plant sciences. On the other hand, the technical difficulties and various shortcomings such as off-targets, genotype dependency, construct delivery, unintended effects, etc. take time to translate these techniques from basic research to applied studies. The techniques are already standardized in model plants such as Arabidopsis and Rice, but their exploitation in other important plant species needs to be simultaneously addressed. The exponential growth in recent years in the field of GE promises to plant scientists in providing customized and flexible solutions of their beneficial thoughts with regards to the nourishment of agricultural sciences. Conclusively, this handy approach has shown the potential to counter the upcoming challenges of agricultural, environmental, social, and geological issues ultimately hindering the fate of the food crisis in the scenario of climate alterations.
References
Abdallah NA et al (2015) Genome editing for crop improvement: Challenges and opportunities. GM Crops Food 6(4):183–205. https://doi.org/10.1080/21645698.2015.1129937
Aglawe SB et al (2018) New breeding technique “genome editing” for crop improvement: applications, potentials and challenges. 3. Biotech 8(8):1–20. https://doi.org/10.1007/s13205-018-1355-3
Aird EJ, Lovendahl KN, Martin AS, Harris RS, Gordon WR (2018) Increasing Cas9-mediated homology-directed repair efficiency through covalent tethering of DNA repair template. Commun Biol 1:1–6. https://doi.org/10.1038/s42003-018-0054-2
Altpeter F et al (2016) Advancing crop transformation in the era of genome editing. Plant Cell 28(7):1510–1520. https://doi.org/10.1105/tpc.16.00196
Alwin S, Gere MB, Guhl E, Effertz K, Barbas CF III, Segal DJ et al (2005) Custom zinc-finger nucleases for use in human cells. Mol Ther 12:610–617. https://doi.org/10.1016/j.ymthe.2005.06.094
Andersson M, Turesson H, Olsson N, Fält AS, Ohlsson P, Gonzalez MN et al (2018) Genome editing in potato via CRISPR-Cas9 ribonucleoprotein delivery. Physiol Plant 164:378–384. https://doi.org/10.1111/ppl.12731
Anzalone AV et al (2019) Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576(7785):149–157. https://doi.org/10.1038/s41586-019-1711-4
Argast GM et al (1998) I-PpoI and I-CreI homing site sequence degeneracy determined by random mutagenesis and sequential in vitro enrichment. J Mol Biol 280(3):345–353. https://doi.org/10.1093/nar/gkl645
Arnold DA, Kowalczykowski SC (2000) Facilitated loading of RecA protein is essential to recombination by RecBCD enzyme. J Biol Chem 275:12261–12265. https://doi.org/10.1074/jbc.275.16.12261
Arnould S et al (2007) Engineered I-CreI derivatives cleaving sequences from the human XPC gene can induce highly efficient gene correction in mammalian cells. J Mol Biol 371(1):49–65. https://doi.org/10.1016/j.jmb.2007.04.079
Baker M (2011) Gene-editing nucleases. In: Nature Publishing Group. V8 (12). https://doi.org/10.1101/cshperspect.a023754
Baltes NJ et al (2014) DNA replicons for plant genome engineering. Plant Cell 26(1):151–163. https://doi.org/10.1105/tpc.113.119792
Bartsevich VV, Juliano R (2000) Regulation of the MDR1 gene by transcriptional repressors selected using peptide combinatorial libraries. Mol Pharmacol 58:1–10. https://doi.org/10.1124/mol.58.1.1
Bartsevich VV, Miller JC, Case CC, Pabo CO (2003) Engineered zinc finger proteins for controlling stem cell fate. Stem Cells 21:632–637. https://doi.org/10.1634/stemcells.21-6-632
Bechtold N (1993) In planta Agrobacterium-mediated gene transfer by infiltration of adult Arabidopsis plants. CR Acad Sci Ser III Sci Vie 316:1194–1199
Beerli RR, Barbas CF (2002) Engineering polydactyl zinc-finger transcription factors. Nat Biotechnol 20:135–141. https://doi.org/10.1038/nbt0202-135
Beerli RR, Segal DJ, Dreier B, Barbas CF (1998) Toward controlling gene expression at will: specific regulation of the erbB-2/HER-2 promoter by using polydactyl zinc finger proteins constructed from modular building blocks. Proc Natl Acad Sci 95:14628–14633
Belhaj K, Chaparro-Garcia A, Kamoun S, Nekrasov V (2013) Plant genome editing made easy: targeted mutagenesis in model and crop plants using the CRISPR/Cas system. Plant Methods 9:1–10. https://doi.org/10.1186/1746-4811-9-39
Belhaj K, Chaparro-Garcia A, Kamoun S, Patron NJ, Nekrasov V (2015) Editing plant genomes with CRISPR/Cas9. Curr Opin Biotechnol 32:76–84. https://doi.org/10.1016/j.copbio.2014.11.007
Bernheim A et al (2017) Inhibition of NHEJ repair by type II-A CRISPR-Cas systems in bacteria. Nat Commun 8(1):1–9. https://doi.org/10.1038/s41467-017-02350-1
Beumer K, Bhattacharyya G, Bibikova M, Trautman JK, Carroll D (2006) Efficient gene targeting in Drosophila with zinc-finger nucleases. Genetics 172:2391–2403. https://doi.org/10.1016/j.copbio.2014.11.007
Beumer KJ, Trautman JK, Bozas A, Liu J-L, Rutter J, Gall JG, Carroll D (2008) Efficient gene targeting in Drosophila by direct embryo injection with zinc-finger nucleases. Proc Natl Acad Sci 105:19821–19826. https://doi.org/10.1073/pnas.0810475105
Bianco PR, Kowalczykowski SC (1997) The recombination hotspot Chi is recognized by the translocating RecBCD enzyme as the single strand of DNA containing the sequence 5′-GCTGGTGG-3′. Proc Natl Acad Sci 94:6706–6711. https://doi.org/10.1073/pnas.94.13.6706
Bibikova M, Carroll D, Segal DJ, Trautman JK, Smith J, Kim Y-G, Chandrasegaran S (2001) Stimulation of homologous recombination through targeted cleavage by chimeric nucleases. Mol Cell Biol 21:289–297. https://doi.org/10.1128/MCB.21.1.289-297.2001
Bibikova M, Golic M, Golic KG, Carroll D (2002) Targeted chromosomal cleavage and mutagenesis in Drosophila using zinc-finger nucleases. Genetics 161:1169–1175. https://doi.org/10.1093/genetics/161.3.1169
Bibikova M, Beumer K, Trautman JK, Carroll D (2003) Enhancing gene targeting with designed zinc finger nucleases. Science 300:764–764. https://doi.org/10.1126/science.1079512
Boch J, Bonas U (2010) Xanthomonas AvrBs3 family-type III effectors: discovery and function. Annu Rev Phytopathol 48:419–436. https://doi.org/10.1146/annurev-phyto-080508-081936
Boch J, Scholze H, Schornack S, Landgraf A, Hahn S, Kay S et al (2009) Breaking the code of DNA binding specificity of TAL-type III effectors. Science 326:1509–1512. https://doi.org/10.1126/science.1178811
Bolukbasi, M. F., Gupta, A., Oikemus, S., Derr, A. G., Garber, M., Brodsky, M. H., ... & Wolfe, S. A. (2015). DNA-binding-domain fusions enhance the targeting range and precision of Cas9. Nat Methods 12(12):1150–1156.
Burma S et al (2006) Role of non-homologous end joining (NHEJ) in maintaining genomic integrity. DNA Repair 5(9-10):1042–1048. https://doi.org/10.1016/j.dnarep.2006.05.026
Cai CQ, Doyon Y, Ainley WM, Miller JC, DeKelver RC, Moehle EA et al (2009) Targeted transgene integration in plant cells using designed zinc finger nucleases. Plant Mol Biol 69:699–709. https://doi.org/10.1007/s11103-008-9449-7
Carbery ID, Ji D, Harrington A, Brown V, Weinstein EJ, Liaw L, Cui X (2010) Targeted genome modification in mice using zinc-finger nucleases. Genetics 186:451–459. https://doi.org/10.1093/genetics/186.2.NP
Carlson DF, Tan W, Lillico SG, Stverakova D, Proudfoot C, Christian M et al (2012) Efficient TALEN-mediated gene knockout in livestock. Proc Natl Acad Sci 109:17382–17387. https://doi.org/10.1073/pnas.1211446109
Carroll D (2011) Genome engineering with zinc-finger nucleases. Genetics 188:773–782. https://doi.org/10.1534/genetics.111.131433
Carroll, D. (2017). Focus: Genome editing: genome editing: past, present, and future. Yale J Biol Med 90(4): 653.
Castel B, Tomlinson L, Locci F, Yang Y, Jones JD (2019) Optimization of T-DNA architecture for Cas9-mediated mutagenesis in Arabidopsis. PLoS One 14. https://doi.org/10.1371/journal.pone.0204778
Cermak T, Doyle EL, Christian M, Wang L, Zhang Y, Schmidt C et al (2011) Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res 39:e82–e82. https://doi.org/10.1093/nar/gkr218
Čermák T, Curtin SJ, Gil-Humanes J, Čegan R, Kono TJ, Konečná E et al (2017) A multipurpose toolkit to enable advanced genome engineering in plants. Plant Cell 29:1196–1217. https://doi.org/10.1105/tpc.16.00922
Chandrasegaran S, Carroll D (2016) Origins of programmable nucleases for genome engineering. J Mol Biol 428:963–989. https://doi.org/10.1016/j.jmb.2015.10.014
Chen Y et al (2017) CRISPR/Cas9-mediated base-editing system efficiently generates gain-of-function mutations in Arabidopsis. Sci China Life Sci 60(5):520–523. https://doi.org/10.1007/s11427-017-9021-5
Chen K et al (2019) CRISPR/Cas genome editing and precision plant breeding in agriculture. Annu Rev Plant Biol 70:667–697. https://doi.org/10.1146/annurev-arplant-050718-100049
Chen G et al (2020) Gene editing to facilitate hybrid crop production. Biotechnol Adv 107676. https://doi.org/10.1016/j.biotechadv.2020.107676
Cong L, Zhang F (2015) Genome engineering using CRISPR-Cas9 system. In: Chromosomal mutagenesis, vol 8. Humana Press, Totowa, pp 2281–2308. https://doi.org/10.1038/nprot.2013.143
Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N et al (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339:819–823. https://doi.org/10.1126/science.1231143
Cox MM (1999) Recombinational DNA repair in bacteria and the RecA protein. In: Progress in nucleic acid research and molecular biology, vol 63. Academic, New York, pp 311–366. https://doi.org/10.1016/S0079-6603(08)60726-6
Cui Y, Xu J, Cheng M, Liao X, Peng S (2018) Review of CRISPR/Cas9 sgRNA design tools. Interdiscip Sci Comput Life Sci 10:455–465. https://doi.org/10.1007/s12539-018-0298-z
Dagdas YS, Chen JS, Sternberg SH, Doudna JA, Yildiz A (2017) A conformational checkpoint between DNA binding and cleavage by CRISPR-Cas9. Sci Adv 3:eaao0027. https://doi.org/10.1126/sciadv.aao0027
Dai Q, Huang J, Klitzman B, Dong C, Goldschmidt-Clermont PJ, March KL et al (2004) Engineered zinc finger–activating vascular endothelial growth factor transcription factor plasmid DNA induces therapeutic angiogenesis in rabbits with hindlimb ischemia. Circulation 110:2467–2475. https://doi.org/10.1161/01.CIR.0000145139.53840.49
De Pater S, Neuteboom LW, Pinas JE, Hooykaas PJ, Van Der Zaal BJ (2009) ZFN-induced mutagenesis and gene-targeting in Arabidopsis through Agrobacterium-mediated floral dip transformation. Plant Biotechnol J 7:821–835. https://doi.org/10.1111/j.1467-7652.2009.00446.x
Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao Y, Pirzada ZA et al (2011) CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471:602–607. https://doi.org/10.1038/nature09886
Djukanovic V et al (2013) Male-sterile maize plants produced by targeted mutagenesis of the cytochrome P 450-like gene (MS 26) using a re-designed I–C reI homing endonuclease. Plant J 76(5):888–899. https://doi.org/10.1111/tpj.12335
Doudna JA, Charpentier E (2014) The new frontier of genome engineering with CRISPR-Cas9. Science 346:6213. https://doi.org/10.1126/science.1258096
Doyon Y, McCammon JM, Miller JC, Faraji F, Ngo C, Katibah GE et al (2008) Heritable targeted gene disruption in zebrafish using designed zinc-finger nucleases. Nat Biotechnol 26:702–708. https://doi.org/10.1038/nbt1409
Dudáš A, Chovanec M (2004) DNA double-strand break repair by homologous recombination. Mutat Res Rev Mutat Res 566:131–167. https://doi.org/10.1016/j.mrrev.2003.07.001
Durai S, Mani M, Kandavelou K, Wu J, Porteus MH, Chandrasegaran S (2005) Zinc finger nucleases: custom-designed molecular scissors for genome engineering of plant and mammalian cells. Nucleic Acids Res 33:5978–5990. https://doi.org/10.1093/nar/gki912
Ebrahimi V, Hashemi A (2020) Challenges of in vitro genome editing with CRISPR/Cas9 and possible solutions: A review. Gene 144813. https://doi.org/10.1016/j.gene.2020.144813
Ekker SC (2008) Zinc Finger–based knockout punches for Zebrafish genes. Zebrafish 5:121–123. https://doi.org/10.1089/zeb.2008.9988
Ellison EE et al (2020) Multiplexed heritable gene editing using RNA viruses and mobile single guide RNAs. Nat Plant 6(6):620–624. https://doi.org/10.1038/s41477-020-0670-y
Elrod-Erickson M, Pabo CO (1999) Binding studies with Mutants of Zif268: contribution of individual side chains to binding affinity and specificity in the Zif268 zinc finger-DNA complex. J Biol Chem 274:19281–19285. https://doi.org/10.1074/jbc.274.27.19281
Farzadfard F, Lu TK (2018) Emerging applications for DNA writers and molecular recorders. Science 361(6405):870–875. https://doi.org/10.1126/science.aat9249
Foley JE, Yeh J-RJ, Maeder ML, Reyon D, Sander JD, Peterson RT, Joung JK (2009) Rapid mutation of endogenous zebrafish genes using zinc finger nucleases made by Oligomerized Pool ENgineering (OPEN). PLoS One 4:e4348. https://doi.org/10.1371/journal.pone.0004348
Game J, Mortimer R (1974) A genetic study of X-ray sensitive mutants in yeast. Mutat Res Fundam Mol Mech Mutagen 24:281–292. https://doi.org/10.1016/0027-5107(74)90176-6
Gao Y, Zhao Y (2014) Self-processing of ribozyme-flanked RNAs into guide RNAs in vitro and in vivo for CRISPR-mediated genome editing. J Integr Plant Biol 56:343–349. https://doi.org/10.1111/jipb.12152
Gao H et al (2010) Heritable targeted mutagenesis in maize using a designed endonuclease. Plant J 61(1):176–187. https://doi.org/10.1111/j.1365-313X.2009.04041.x
Gaudelli NM et al (2017) Programmable base editing of A• T to G• C in genomic DNA without DNA cleavage. Nature 551(7681):464–471. https://doi.org/10.1038/nature24644
Geurts AM, Cost GJ, Freyvert Y, Zeitler B, Miller JC, Choi VM et al (2009) Knockout rats via embryo microinjection of zinc-finger nucleases. Science 325:433–433. https://doi.org/10.1126/science.1172447
Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH et al (2014) Genome-scale CRISPR-mediated control of gene repression and activation. Cell 159:647–661. https://doi.org/10.1016/j.cell.2014.09.029
Gorbunova V, Levy AA (1999) How plants make ends meet: DNA double-strand break repair. Trends Plant Sci 4:263–269. https://doi.org/10.1016/S1360-1385(99)01430-2
Guilinger JP, Thompson DB, Liu DR (2014) Fusion of catalytically inactive Cas9 to FokI nuclease improves the specificity of genome modification. Nat Biotechnol 32:577. https://doi.org/10.1038/nbt.2909
Guirouilh-Barbat J et al (2004) Impact of the KU80 pathway on NHEJ-induced genome rearrangements in mammalian cells. Mol Cell 14(5):611–623. https://doi.org/10.1016/j.molcel.2004.05.008
Gumpel NJ, Rochaix J-D, Purton S (1994) Studies on homologous recombination in the green alga Chlamydomonas reinhardtii. Curr Genet 26:438–442. https://doi.org/10.1007/BF00309931
Gutschner T, Haemmerle M, Genovese G, Draetta GF, Chin L (2016) Post-translational regulation of Cas9 during G1 enhances homology-directed repair. Cell Rep 14:1555–1566. https://doi.org/10.1016/j.celrep.2016.01.019
Händel E-M, Alwin S, Cathomen T (2009) Expanding or restricting the target site repertoire of zinc-finger nucleases: the inter-domain linker as a major determinant of target site selectivity. Mol Ther 17:104–111. https://doi.org/10.1038/mt.2008.233
Haroon M, et al (2020) Conventional breeding, molecular breeding and speed breeding; brave approaches to Revamp the production of cereal crops. https://doi.org/10.20944/preprints202011.0667.v1
Heler R, Samai P, Modell JW, Weiner C, Goldberg GW, Bikard D, Marraffini LA (2015) Cas9 specifies functional viral targets during CRISPR–Cas adaptation. Nature 519:199–202. https://doi.org/10.1038/nature14245
Herai RH (2019) Avoiding the off-target effects of CRISPR/cas9 system is still a challenging accomplishment for genetic transformation. Gene 700:176–178. https://doi.org/10.1016/j.gene.2019.03.019
Hockemeyer D, Wang H, Kiani S, Lai CS, Gao Q, Cassady JP et al (2011) Genetic engineering of human pluripotent cells using TALE nucleases. Nat Biotechnol 29:731–734. https://doi.org/10.1038/nbt.1927
Holkers M, Maggio I, Liu J, Janssen JM, Miselli F, Mussolino C et al (2013) Differential integrity of TALE nuclease genes following adenoviral and lentiviral vector gene transfer into human cells. Nucleic Acids Res 41:e63–e63. https://doi.org/10.1093/nar/gks1446
Horvath P, Barrangou R (2010) CRISPR/Cas, the immune system of bacteria and archaea. Science 327:167–170. https://doi.org/10.1126/science.1179555
Hrouda M, Paszkowski J (1994) High fidelity extrachromosomal recombination and gene targeting in plants. Mol Gen Genet MGG 243:106–111. https://doi.org/10.1007/BF00283882
Huang P, Xiao A, Zhou M, Zhu Z, Lin S, Zhang B (2011) Heritable gene targeting in zebrafish using customized TALENs. Nat Biotechnol 29:699–700. https://doi.org/10.1038/nbt.1939
Iqbal Z et al (2020) New prospects on the horizon: genome editing to engineer plants for desirable traits. Curr Plant Biol 100171. https://doi.org/10.1016/j.cpb.2020.100171
Ira M, Zuker A, Shklarman E, Zeevi V, Tovkach A, Roffe S et al (2010) Non-transgenic genome modification in plant cells. Plant Physiol. https://doi.org/10.1104/pp.110.164806
Jasin M, Moynahan ME, Richardson C (1996) Targeted transgenesis. Proc Natl Acad Sci U S A 93:8804. https://doi.org/10.1073/pnas.93.17.8804
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E (2012) A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science 337:816–821. https://doi.org/10.1126/science.1225829
Joung JK, Sander JD (2013) TALENs: a widely applicable technology for targeted genome editing. Nat Rev Mol Cell Biol 14:49–55. https://doi.org/10.1038/nrm3486
Joung JK, Voytas DF, Cathomen T (2010) Reply to “Genome editing with modularly assembled zinc-finger nucleases”. Nat Methods 7:91–92. https://doi.org/10.1038/nmeth0210-91b
Kang B-C et al (2018) Precision genome engineering through adenine base editing in plants. Nat Plant 4(7):427–431. https://doi.org/10.1038/s41477-018-0178-x
Kaur N, Alok A, Kaur N, Pandey P, Awasthi P, Tiwari S (2018) CRISPR/Cas9-mediated efficient editing in phytoene desaturase (PDS) demonstrates precise manipulation in banana cv. Rasthali genome. Funct Integr Genomic 18:89–99. https://doi.org/10.1007/s10142-017-0577-5
Kempin SA, Liljegren SJ, Block LM, Rounsley SD, Yanofsky MF, Lam E (1997) Targeted disruption in Arabidopsis. Nature 389:802–803. https://doi.org/10.1038/39770
Khandagale K, Nadaf A (2016) Genome editing for targeted improvement of plants. Plant Biotechnol Rep 10(6):327–343. https://doi.org/10.1007/s11816-016-0417-4
Kim Y-G, Chandrasegaran S (1994) Chimeric restriction endonuclease. Proc Natl Acad Sci 91:883–887. https://doi.org/10.1073/pnas.91.3.883
Kim H, Kim J-S (2014) A guide to genome engineering with programmable nucleases. Nat Rev Genet 15:321–334. https://doi.org/10.1038/nrg3686
Kim Y-G, Cha J, Chandrasegaran S (1996) Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc Natl Acad Sci 93:1156–1160. https://doi.org/10.1073/pnas.93.3.1156
Kim S, Lee MJ, Kim H, Kang M, Kim J-S (2011) Preassembled zinc-finger arrays for rapid construction of ZFNs. Nat Methods 8:7. https://doi.org/10.1038/nmeth0111-7a
Kim S, Kim D, Cho SW, Kim J, Kim J-S (2014) Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res 24:1012–1019. https://doi.org/10.1101/gr.171322.113
Kirik A et al (2000) Species-specific double-strand break repair and genome evolution in plants. EMBO J 19(20):5562–5566. https://doi.org/10.1093/emboj/19.20.5562
Kleinstiver BP, Pattanayak V, Prew MS, Tsai SQ, Nguyen NT, Zheng Z, Joung JK (2016) High-fidelity CRISPR–Cas9 nucleases with no detectable genome-wide off-target effects. Nature 529:490–495. https://doi.org/10.1038/nature16526
Komor AC et al (2016) Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533(7603):420–424. https://doi.org/10.1038/nature24644
Kowalczykowski SC, Dixon DA, Eggleston AK, Lauder SD, Rehrauer WM (1994) Biochemistry of homologous recombination in Escherichia coli. Microbiol Rev 58:401–465
Kunz C et al (2009) DNA repair in mammalian cells. Cell Mol Life Sci 66(6):1021–1038. https://doi.org/10.1007/s00018-009-8739-9
Landrum MJ et al (2014) ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res 42(D1):D980–D985. https://doi.org/10.1093/nar/gkt1113
Landrum MJ et al (2016) ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res 44(D1):D862–D868. https://doi.org/10.1093/nar/gkv1222
Lei Y, Guo X, Liu Y, Cao Y, Deng Y, Chen X et al (2012) Efficient targeted gene disruption in Xenopus embryos using engineered transcription activator-like effector nucleases (TALENs). Proc Natl Acad Sci 109:17484–17489. https://doi.org/10.1073/pnas.1215421109
Levy A et al (2015) CRISPR adaptation biases explain preference for acquisition of foreign DNA. Nature 520(7548):505–510. https://doi.org/10.1038/nature14302
Li L, Wu LP, Chandrasegaran S (1992) Functional domains in Fok I restriction endonuclease. Proc Natl Acad Sci 89:4275–4279. https://doi.org/10.1073/pnas.89.10.4275
Li T, Huang S, Zhao X, Wright DA, Carpenter S, Spalding MH et al (2011) Modularly assembled designer TAL effector nucleases for targeted gene knockout and gene replacement in eukaryotes. Nucleic Acids Res 39:6315–6325. https://doi.org/10.1093/nar/gkr188
Li T, Liu B, Spalding MH, Weeks DP, Yang B (2012) High-efficiency TALEN-based gene editing produces disease-resistant rice. Nat Biotechnol 30:390. https://doi.org/10.1038/nbt.2199
Li D, Zhou H, Zeng X (2019) Battling CRISPR-Cas9 off-target genome editing. Springer. https://doi.org/10.1007/s10565-019-09485-5
Liu Q, Segal DJ, Ghiara JB, Barbas CF (1997) Design of polydactyl zinc-finger proteins for unique addressing within complex genomes. Proc Natl Acad Sci 94:5525–5530. https://doi.org/10.1073/pnas.94.11.5525
Liu P-Q, Rebar EJ, Zhang L, Liu Q, Jamieson AC, Liang Y et al (2001) Regulation of an endogenous locus using a panel of designed zinc finger proteins targeted to accessible chromatin regions: activation of vascular endothelial growth factor A. J Biol Chem 276:11323–11334. https://doi.org/10.1074/jbc.M011172200
Liu J, Li C, Yu Z, Huang P, Wu H, Wei C et al (2012) Efficient and specific modifications of the Drosophila genome by means of an easy TALEN strategy. J Genetic Genomic 39:209–215. https://doi.org/10.1016/j.jgg.2012.04.003
Liu, W., Rudis, M. R., Cheplick, M. H., Millwood, R. J., Yang, J. P., Ondzighi-Assoume, C. A., ... & Stewart, C. N. (2020). Lipofection-mediated genome editing using DNA-free delivery of the Cas9/gRNA ribonucleoprotein into plant cells. Plant Cell Rep 39(2):245–257.
Lloyd A, Plaisier CL, Carroll D, Drews GN (2005) Targeted mutagenesis using zinc-finger nucleases in Arabidopsis. Proc Natl Acad Sci 102:2232–2237. https://doi.org/10.1073/pnas.0409339102
Ma S, Zhang S, Wang F, Liu Y, Liu Y, Xu H et al (2012) Highly efficient and specific genome editing in silkworm using custom TALENs. PLoS One 7:e45035. https://doi.org/10.1371/journal.pone.0045035
Maeder ML, Thibodeau-Beganny S, Osiak A, Wright DA, Anthony RM, Eichtinger M et al (2008) Rapid “open-source” engineering of customized zinc-finger nucleases for highly efficient gene modification. Mol Cell 31:294–301. https://doi.org/10.1016/j.molcel.2008.06.016
Makarova KS, Haft DH, Barrangou R, Brouns SJ, Charpentier E, Horvath P et al (2011) Evolution and classification of the CRISPR–Cas systems. Nat Rev Microbiol 9:467–477. https://doi.org/10.1038/nrmicro2577
Mali P, Aach J, Stranges PB, Esvelt KM, Moosburner M, Kosuri S et al (2013) CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat Biotechnol 31:833–838. https://doi.org/10.1038/nbt.2675
Maruyama T, Dougan SK, Truttmann MC, Bilate AM, Ingram JR, Ploegh HL (2015) Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat Biotechnol 33:538–542. https://doi.org/10.1038/nbt.3190
Mashimo T, Takizawa A, Voigt B, Yoshimi K, Hiai H, Kuramoto T, Serikawa T (2010) Generation of knockout rats with X-linked severe combined immunodeficiency (X-SCID) using zinc-finger nucleases. PLoS One 5:e8870. https://doi.org/10.1371/journal.pone.0008870
Matsoukas IG (2020) Prime editing: genome editing for rare genetic diseases without double-strand breaks or donor DNA. Front Genet 11:528. https://doi.org/10.3389/fgene.2020.00528
McNamara AR, Hurd PJ, Smith AE, Ford KG (2002) Characterisation of site-biased DNA methyltransferases: specificity, affinity and subsite relationships. Nucleic Acids Res 30:3818–3830. https://doi.org/10.1093/nar/gkf501
Meng X, Thibodeau-Beganny S, Jiang T, Joung JK, Wolfe SA (2007) Profiling the DNA-binding specificities of engineered Cys2His2 zinc finger domains using a rapid cell-based method. Nucleic Acids Res 35:e81. https://doi.org/10.1093/nar/gkm385
Meng X, Noyes MB, Zhu LJ, Lawson ND, Wolfe SA (2008) Targeted gene inactivation in zebrafish using engineered zinc-finger nucleases. Nat Biotechnol 26:695–701. https://doi.org/10.1038/nbt1398
Meyer RR, Laine PS (1990) The single-stranded DNA-binding protein of Escherichia coli. Microbiol Mol Biol Rev 54:342–380
Miao ZH, Lam E (1995) Targeted disruption of the TGA3 locus in Arabidopsis thaliana. Plant J 7:359–365. https://doi.org/10.1046/j.1365-313X.1995.7020359.x
Miller JC, Tan S, Qiao G, Barlow KA, Wang J, Xia DF et al (2011) A TALE nuclease architecture for efficient genome editing. Nat Biotechnol 29:143–148. https://doi.org/10.1038/nbt.1755
Moore M, Klug A, Choo Y (2001) Improved DNA binding specificity from polyzinc finger peptides by using strings of two-finger units. Proc Natl Acad Sci 98:1437–1441. https://doi.org/10.1073/pnas.98.4.1437
Morbitzer R, Römer P, Boch J, Lahaye T (2010) Regulation of selected genome loci using de novo-engineered transcription activator-like effector (TALE)-type transcription factors. Proc Natl Acad Sci 107:21617–21622. https://doi.org/10.1073/pnas.1013133107
Morton J, Davis MW, Jorgensen EM, Carroll D (2006) Induction and repair of zinc-finger nuclease-targeted double-strand breaks in Caenorhabditis elegans somatic cells. Proc Natl Acad Sci 103:16370–16375. https://doi.org/10.1073/pnas.0605633103
Moscou MJ, Bogdanove AJ (2009) A simple cipher governs DNA recognition by TAL effectors. Science 326:1501–1501. https://doi.org/10.1126/science.1178817
Müller AE, Kamisugi Y, Grüneberg R, Niedenhof I, Hörold RJ, Meyer P (1999) Palindromic sequences and A+T-rich DNA elements promote illegitimate recombination in Nicotiana tabacum. J Mol Biol 291:29–46. https://doi.org/10.1006/jmbi.1999.2957
Nelson CE, Gersbach CA (2016) Engineering delivery vehicles for genome editing. Ann Rev Chem Biomol Eng 7:637–662. https://doi.org/10.1146/annurev-chembioeng-080615-034711
Newton MD, Taylor BJ, Driessen RP, Roos L, Cvetesic N, Allyjaun S et al (2019) DNA stretching induces Cas9 off-target activity. Nat Struct Mol Biol 26:185–192. https://doi.org/10.1038/s41594-019-0188-z
Nishida K, et al (2016) Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science 353(6305). See all authors and affiliations. https://doi.org/10.1126/science.aaf8729
Ochiai H, Fujita K, Suzuki KI, Nishikawa M, Shibata T, Sakamoto N, Yamamoto T (2010) Targeted mutagenesis in the sea urchin embryo using zinc-finger nucleases. Genes Cells 15:875–885. https://doi.org/10.1111/j.1365-2443.2010.01425.x
Offringa R, de Groot MJ, Haagsman HJ, Does MP, van den Elzen PJ, Hooykaas PJ (1990) Extrachromosomal homologous recombination and gene targeting in plant cells after Agrobacterium mediated transformation. EMBO J 9:3077–3084. https://doi.org/10.1002/j.1460-2075.1990.tb07504.x
Osakabe K, Osakabe Y, Toki S (2010) Site-directed mutagenesis in Arabidopsis using custom-designed zinc finger nucleases. Proc Natl Acad Sci 107:12034–12039. https://doi.org/10.1073/pnas.1000234107
Pabo CO, Peisach E, Grant RA (2001) Design and selection of novel Cys2His2 zinc finger proteins. Annu Rev Biochem 70:313–340. https://doi.org/10.1146/annurev.biochem.70.1.313
Pâques F, Haber JE (1999) Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol Mol Biol Rev 63:349–404. https://doi.org/10.1128/MMBR.63.2.349-404.1999
Paris Ü et al (2015) NHEJ enzymes LigD and Ku participate in stationary-phase mutagenesis in Pseudomonas putida. DNA Repair 31:11–18. https://doi.org/10.1016/j.dnarep.2015.04.005
Pastink A, Eeken JC, Lohman PH (2001) Genomic integrity and the repair of double-strand DNA breaks. Mutat Res Fundam Mol Mech Mutagen 480:37–50. https://doi.org/10.1016/S0027-5107(01)00167-1
Paszkowski J, Baur M, Bogucki A, Potrykus I (1988) Gene targeting in plants. Embo J 7:4021–4026. https://doi.org/10.1002/j.1460-2075.1988.tb03295.x
Pavletich NP, Pabo CO (1991) Zinc finger-DNA recognition: crystal structure of a Zif268-DNA complex at 2.1 A. Science 252:809–817. https://doi.org/10.1126/science.2028256
Petes TD (1991) Recombination in yeast. In: The molecular and cellular biology of the yeast Saccharomyces: genome dynamics, protein synthesis and energetics. Cold Spring Harbor Laboratory Press, New York, pp 407–521
Porteus MH, Baltimore D (2003) Chimeric nucleases stimulate gene targeting in human cells. Science 300:763. https://doi.org/10.1126/science.1078395
Potrykus I, Spangenberg G (2013) Gene transfer to plants. Springer, Tokyo
Puchta H (1999) Double-strand break-induced recombination between ectopic homologous sequences in somatic plant cells. Genetics 152:1173–1181. https://doi.org/10.1093/genetics/152.3.1173
Puchta H (2002) Gene replacement by homologous recombination in plants. In: Functional genomics. Springer, Dordrecht, pp 173–182. https://doi.org/10.1007/978-94-010-0448-0_12
Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, Lim WA (2013) Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152:1173–1183. https://doi.org/10.1016/j.cell.2013.02.022
Ramakrishna S, Dad A-BK, Beloor J, Gopalappa R, Lee S-K, Kim H (2014) Gene disruption by cell-penetrating peptide-mediated delivery of Cas9 protein and guide RNA. Genome Res 24:1020–1027. https://doi.org/10.1101/gr.171264.113
Ramirez CL, Foley JE, Wright DA, Müller-Lerch F, Rahman SH, Cornu TI et al (2008) Unexpected failure rates for modular assembly of engineered zinc fingers. Nat Methods 5:374–375. https://doi.org/10.1038/nmeth0508-374
Ran FA, Hsu PD, Lin C-Y, Gootenberg JS, Konermann S, Trevino AE et al (2013a) Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154:1380–1389. https://doi.org/10.1016/j.cell.2013.08.021
Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F (2013b) Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8:2281–2308. https://doi.org/10.1038/nprot.2013.143
Ran, Y., Liang, Z., & Gao, C. (2017). Current and future editing reagent delivery systems for plant genome editing. Sci China Life Sci 60(5):490–505.
Rebar EJ (2004) Development of pro-angiogenic engineered transcription factors for the treatment of cardiovascular disease. Expert Opin Investig Drugs 13:829–839. https://doi.org/10.1517/13543784.13.7.829
Rebar EJ, Huang Y, Hickey R, Nath AK, Meoli D, Nath S et al (2002) Induction of angiogenesis in a mouse model using engineered transcription factors. Nat Med 8:1427–1432. https://doi.org/10.1038/nm1202-795
Rees HA, Liu DR (2018) Base editing: precision chemistry on the genome and transcriptome of living cells. Nat Rev Genet 19(12):770–788. https://doi.org/10.1038/s41576-018-0059-1
Reisch CR, Prather KL (2015) The no-SCAR (S carless C as9 A ssisted R ecombineering) system for genome editing in Escherichia coli. Sci Rep 5:1–12. https://doi.org/10.1038/srep15096
Rémy S, Tesson L, Ménoret S, Usal C, Scharenberg AM, Anegon I (2010) Zinc-finger nucleases: a powerful tool for genetic engineering of animals. Transgenic Res 19:363–371. https://doi.org/10.1007/s11248-009-9323-7
Ren D, Collingwood TN, Rebar EJ, Wolffe AP, Camp HS (2002) PPARγ knockdown by engineered transcription factors: exogenous PPARγ2 but not PPARγ1 reactivates adipogenesis. Genes Dev 16:27–32. https://doi.org/10.1101/gad.953802
Ren, B., Yan, F., Kuang, Y., Li, N., Zhang, D., Zhou, X., ... & Zhou, H. (2018). Improved base editor for efficiently inducing genetic variations in rice with CRISPR/Cas9-guided hyperactive hAID mutant. Mol Plant 11(4):623–626.
Reski R (1999) Molecular genetics of Physcomitrella. Planta 208:301–309. https://doi.org/10.1007/s004250050563
Reyon D, Tsai SQ, Khayter C, Foden JA, Sander JD, Joung JK (2012) FLASH assembly of TALENs for high-throughput genome editing. Nat Biotechnol 30:460–465. https://doi.org/10.1038/nbt.2170
Roca AI, Cox MM (1997) RecA protein: structure, function, and role in recombinational DNA repair. Prog Nucleic Acid Res Mol Biol 56:129–223. https://doi.org/10.1016/s0079-6603(08)61005-3
Rusche LN, Kirchmaier AL, Rine J (2003) The establishment, inheritance, and function of silenced chromatin in Saccharomyces cerevisiae. Annu Rev Biochem 72:481–516. https://doi.org/10.1146/annurev.biochem.72.121801.161547
Ryan DE, Taussig D, Steinfeld I, Phadnis SM, Lunstad BD, Singh M et al (2018) Improving CRISPR–Cas specificity with chemical modifications in single-guide RNAs. Nucleic Acids Res 46:792–803. https://doi.org/10.1093/nar/gkx1199
San Filippo J, Sung P, Klein H (2008) Mechanism of eukaryotic homologous recombination. Annu Rev Biochem 77:229–257. https://doi.org/10.1146/annurev.biochem.77.061306.125255
Sander JD, Cade L, Khayter C, Reyon D, Peterson RT, Joung JK, Yeh J-RJ (2011) Targeted gene disruption in somatic zebrafish cells using engineered TALENs. Nat Biotechnol 29:697–698. https://doi.org/10.1038/nbt.1934
Schaefer DG (2001) Gene targeting in Physcomitrella patens. Curr Opin Plant Biol 4:143–150. https://doi.org/10.1016/S1369-5266(00)00150-3
Sedivy JM, Dutriaux A (1999) Gene targeting and somatic cell genetics–a rebirth or a coming of age? Trends Genet 15:88–90. https://doi.org/10.1016/s0168-9525(98)01689-8
Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS et al (2014) Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343:84–87. https://doi.org/10.1126/science.1247005
Sharma S et al (2017) Recent advances in CRISPR/Cas mediated genome editing for crop improvement. Plant Biotechnol Rep 11(4):193–207. https://doi.org/10.1007/s11816-017-0446-7
Shen B, Zhang J, Wu H, Wang J, Ma K, Li Z et al (2013) Generation of gene-modified mice via Cas9/RNA-mediated gene targeting. Cell Res 23:720–723. https://doi.org/10.1038/cr.2013.46
Shi Y, Berg JM (1995) A direct comparison of the properties of natural and designed zinc-finger proteins. Chem Biol 2:83–89. https://doi.org/10.1016/1074-5521(95)90280-5
Shimatani Z et al (2017) Targeted base editing in rice and tomato using a CRISPR-Cas9 cytidine deaminase fusion. Nat Biotechnol 35(5):441–443. https://doi.org/10.1038/nbt.3833
Shimizu Y, Bhakta MS, Segal DJ (2009) Restricted spacer tolerance of a zinc finger nuclease with a six amino acid linker. Bioorg Med Chem Lett 19:3970–3972. https://doi.org/10.1016/j.bmcl.2009.02.109
Shukla VK, Doyon Y, Miller JC, DeKelver RC, Moehle EA, Worden SE et al (2009) Precise genome modification in the crop species Zea mays using zinc-finger nucleases. Nature 459:437–441. https://doi.org/10.1038/nature07992
Siede W, Friedl AA, Dianova I, Eckardt-Schupp F, Friedberg EC (1996) The Saccharomyces cerevisiae Ku autoantigen homologue affects radiosensitivity only in the absence of homologous recombination. Genetics 142:91–102. https://doi.org/10.1093/genetics/142.1.91
Silva G et al (2011) Meganucleases and other tools for targeted genome engineering: perspectives and challenges for gene therapy. Curr Gene Ther 11(1):11–27. https://doi.org/10.2174/156652311794520111
Singh V, Braddick D, Dhar PK (2017) Exploring the potential of genome editing CRISPR-Cas9 technology. Gene 599:1–18. https://doi.org/10.1016/j.gene.2016.11.008
Smart EJ, Selman BR (1991) Isolation and characterization of a Chlamydomonas reinhardtii mutant lacking the gamma-subunit of chloroplast coupling factor 1 (CF1). Mol Cell Biol 11:5053–5058. https://doi.org/10.1128/mcb.11.10.5053
Smithies O (1987) Targeted correction of a mutant HPRT gene in mouse ES cells. Nature 330:576–578. https://doi.org/10.1038/330576a0
Snowden AW, Zhang L, Urnov F, Dent C, Jouvenot Y, Zhong X et al (2003) Repression of vascular endothelial growth factor A in glioblastoma cells using engineered zinc finger transcription factors. Cancer Res 63:8968–8976
Sodeinde OA, Kindle KL (1993) Homologous recombination in the nuclear genome of Chlamydomonas reinhardtii. Proc Natl Acad Sci 90:9199–9203. https://doi.org/10.1073/pnas.90.19.9199
Song J, Yang D, Xu J, Zhu T, Chen YE, Zhang J (2016) RS-1 enhances CRISPR/Cas9-and TALEN-mediated knock-in efficiency. Nat Commun 7:1–7. https://doi.org/10.1038/ncomms10548
Standage-Beier K, Zhang Q, Wang X (2015) Targeted large-scale deletion of bacterial genomes using CRISPR-nickases. ACS Synth Biol 4:1217–1225. https://doi.org/10.1021/acssynbio.5b00132
Swarthout JT, Raisinghani M, Cui X (2011) Zinc Finger nucleases: a new era for transgenic animals. Ann Neurosci 18:25–28. https://doi.org/10.5214/ans.0972.7531.1118109
Symington LS (2002) Role of RAD52 epistasis group genes in homologous recombination and double-strand break repair. Microbiol Mol Biol Rev 66:630–670. https://doi.org/10.1128/mmbr.66.4.630-670.2002
Terada N, Hamazaki T, Oka M, Hoki M, Mastalerz DM, Nakano Y et al (2002) Bone marrow cells adopt the phenotype of other cells by spontaneous cell fusion. Nature 416:542–545. https://doi.org/10.1038/nature730
Tesson L, Usal C, Ménoret S, Leung E, Niles BJ, Remy S et al (2011) Knockout rats generated by embryo microinjection of TALENs. Nat Biotechnol 29:695–696. https://doi.org/10.1038/nbt.1940
Thykjær T, Finnemann J, Schauser L, Christensen L, Poulsen C, Stougaard J (1997) Gene targeting approaches using positive-negative selection and large flanking regions. Plant Mol Biol 35:523–530. https://doi.org/10.1023/A:1005865600319
Tian S et al (2018) Engineering herbicide-resistant watermelon variety through CRISPR/Cas9-mediated base-editing. Plant Cell Rep 37(9):1353–1356. https://doi.org/10.1007/s00299-018-2299-0
Townsend JA, Wright DA, Winfrey RJ, Fu F, Maeder ML, Joung JK, Voytas DF (2009) High-frequency modification of plant genes using engineered zinc-finger nucleases. Nature 459:442–445. https://doi.org/10.1038/nature07845
Tsai SQ, Wyvekens N, Khayter C, Foden JA, Thapar V, Reyon D et al (2014) Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat Biotechnol 32:569–576. https://doi.org/10.1038/nbt.2908
Upadhyay SK, Kumar J, Alok A, Tuli R (2013) RNA-guided genome editing for target gene mutations in wheat. G3: Genes. Genomes Genet 3:2233–2238. https://doi.org/10.1534/g3.113.008847
Urnov FD, Miller JC, Lee Y-L, Beausejour CM, Rock JM, Augustus S et al (2005) Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature 435:646–651. https://doi.org/10.1038/nature03556
van den Bosch M, Lohman PH, Pastink A (2002) DNA double-strand break repair by homologous recombination. Biol Chem 383:873–892. https://doi.org/10.1515/bc.2002.095
Vartak SV, Raghavan SC (2015) Inhibition of nonhomologous end joining to increase the specificity of CRISPR/Cas9 genome editing. FEBS J 282:4289–4294. https://doi.org/10.1111/febs.13416
Vento JM, Crook N, Beisel CL (2019) Barriers to genome editing with CRISPR in bacteria. J Ind Microbiol Biotechnol 46:1327–1341. https://doi.org/10.1007/s10295-019-02195-1
Vogel P, Stafforst T (2019) Critical review on engineering deaminases for site-directed RNA editing. Curr Opin Biotechnol 55:74–80. https://doi.org/10.1016/j.copbio.2018.08.006
Wang X et al (2018) Efficient base editing in methylated regions with a human APOBEC3A-Cas9 fusion. Nat Biotechnol 36(10):946–949. https://doi.org/10.1038/nbt.4198
Watanabe T, Ochiai H, Sakuma T, Horch HW, Hamaguchi N, Nakamura T et al (2012) Non-transgenic genome modifications in a hemimetabolous insect using zinc-finger and TAL effector nucleases. Nat Commun 3:1–8. https://doi.org/10.1038/ncomms2020
Weber E, Gruetzner R, Werner S, Engler C, Marillonnet S (2011) Assembly of designer TAL effectors by Golden Gate cloning. PLoS One 6:e19722. https://doi.org/10.1371/journal.pone.0019722
Weber T, Wefers B, Wurst W, Sander S, Rajewsky K, Kühn R (2015) Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat Biotechnol 33:543–548. https://doi.org/10.1038/nbt.3198
Weinstock DM, Jasin M (2006) Alternative pathways for the repair of RAG-induced DNA breaks. Mol Cell Biol 26:131–139. https://doi.org/10.1128/MCB.26.1.131-139.2006
West SC (1996) The RuvABC proteins and Holliday junction processing in Escherichia coli. J Bacteriol 178:1237–1241. https://doi.org/10.1128/jb.178.5.1237-1241.1996
Whitby MC, Lloyd RG (1998) Targeting Holliday junctions by the RecG branch migration protein of Escherichia coli. J Biol Chem 273:19729–19739. https://doi.org/10.1074/jbc.273.31.19729
Wood AJ, Lo T-W, Zeitler B, Pickle CS, Ralston EJ, Lee AH et al (2011) Targeted genome editing across species using ZFNs and TALENs. Science 333:307–307. https://doi.org/10.1126/science.1207773
Wright DA, Townsend JA, Winfrey RJ Jr, Irwin PA, Rajagopal J, Lonosky PM et al (2005) High-frequency homologous recombination in plants mediated by zinc-finger nucleases. Plant J 44:693–705. https://doi.org/10.1111/j.1365-313X.2005.02551.x
Wright DA et al (2014) TALEN-mediated genome editing: prospects and perspectives. Biochem J 462(1):15–24. https://doi.org/10.1042/BJ20140295
Wu J et al (2020) Engineering herbicide-resistant oilseed rape by CRISPR/Cas9-mediated cytosine base-editing. Plant Biotechnol J 18(9):1857–1859. https://doi.org/10.1038/s41598-021-87669-y
Wyman C, Kanaar R (2006) DNA double-strand break repair: all’s well that ends well. Annu Rev Genet 40:363–383. https://doi.org/10.1146/annurev.genet.40.110405.090451
Xie K, Minkenberg B, Yang Y (2015) Boosting CRISPR/Cas9 multiplex editing capability with the endogenous tRNA-processing system. Proc Natl Acad Sci 112:3570–3575. https://doi.org/10.1073/pnas.1420294112
Xu G-L, Bestor TH (1997) Cytosine methylation targetted to pre-determined sequences. Nat Genet 17:376–378. https://doi.org/10.1038/ng1297-376
Xu R et al (2020) Development of plant prime-editing systems for precise genome editing. Plant Commun 1(3):100043. https://doi.org/10.1016/j.xplc.2020.100043
Yang L, Guell M, Byrne S, Yang JL, De Los Angeles A, Mali P et al (2013) Optimization of scarless human stem cell genome editing. Nucleic Acids Res 41:9049–9061. https://doi.org/10.1093/nar/gkt555
Yin K et al (2017) Progress and prospects in plant genome editing. Nat Plant 3(8):1–6. https://doi.org/10.1038/nplants.2017.107
Young JJ, Cherone JM, Doyon Y, Ankoudinova I, Faraji FM, Lee AH et al (2011) Efficient targeted gene disruption in the soma and germ line of the frog Xenopus tropicalis using engineered zinc-finger nucleases. Proc Natl Acad Sci 108:7052–7057. https://doi.org/10.1073/pnas.1102030108
Yu C, Liu Y, Ma T, Liu K, Xu S, Zhang Y et al (2015) Small molecules enhance CRISPR genome editing in pluripotent stem cells. Cell Stem Cell 16:142–147. https://doi.org/10.1016/j.stem.2015.01.003
Zaman QU et al (2019) Genome editing opens a new era of genetic improvement in polyploid crops. Crop J 7(2):141–150. https://doi.org/10.1016/j.cj.2018.07.004
Zhan X et al (2020) Genome editing for plant research and crop improvement. J Integr Plant Biol 63(I1):3–33. https://doi.org/10.1111/jipb.13063
Zhang L, Spratt SK, Liu Q, Johnstone B, Qi H, Raschke EE et al (2000) Synthetic zinc finger transcription factor action at an endogenous chromosomal site: activation of the human erythropoietin gene. J Biol Chem 275:33850–33860. https://doi.org/10.1074/jbc.M005341200
Zhang F, Maeder ML, Unger-Wallace E, Hoshaw JP, Reyon D, Christian M et al (2010) High frequency targeted mutagenesis in Arabidopsis thaliana using zinc finger nucleases. Proc Natl Acad Sci 107:12028–12033. https://doi.org/10.1073/pnas.0914991107
Zhang F, Wen Y, Guo X (2014) CRISPR/Cas9 for genome editing: progress, implications and challenges. Hum Mol Genet 23:R40–R46. https://doi.org/10.1093/hmg/ddu125
Zhang Y et al (2019) The emerging and uncultivated potential of CRISPR technology in plant science. Nat Plant 5(8):778–794. https://doi.org/10.1038/s41477-019-0461-5
Zhang Y et al (2020) A CRISPR way for accelerating improvement of food crops. Nat Food 1(4):200–205
Zhao L et al (2007) The restriction fold turns to the dark side: a bacterial homing endonuclease with a PD-(D/E)-XK motif. EMBO J 26(9):2432–2442. https://doi.org/10.1038/sj.emboj.7601672
Zupan J, Muth TR, Draper O, Zambryski P (2000) The transfer of DNA from Agrobacterium tumefaciens into plants: a feast of fundamental insights. Plant J 23:11–28. https://doi.org/10.1046/j.1365-313x.2000.00808.x
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Sharma, D., Kaur, H., Kapoor, H.K., Sharma, R., Kaur, H., Kyum, M. (2022). Genome Editing: A Review of the Challenges and Approaches. In: Wani, S.H., Hensel, G. (eds) Genome Editing. Springer, Cham. https://doi.org/10.1007/978-3-031-08072-2_4
Download citation
DOI: https://doi.org/10.1007/978-3-031-08072-2_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-08071-5
Online ISBN: 978-3-031-08072-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)