
Abstract
The discovery of clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein (Cas) system, and its development into a set of powerful tools for manipulating the genome, has revolutionized genome editing. Precise, targeted CRISPR/Cas-based genome editing has become the most widely used platform in organisms ranging from plants to animals. The CRISPR/Cas system has been extensively modified to increase its efficiency and fidelity. In addition, the fusion of various protein motifs to Cas effector proteins has facilitated diverse set of genetic manipulations, such as base editing, transposition, recombination, and epigenetic regulation. The CRISPR/Cas system is undergoing continuous development to overcome current limitations, including off-target effects, narrow targeting scope, and issues associated with the delivery of CRISPR components for genome engineering and therapeutic approaches. Here, we review recent progress in a diverse array of CRISPR/Cas-based tools. We also describe limitations and concerns related to the use of CRISPR/Cas technologies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The progressive development of genome editing technologies, involving sequence-specific programmable nucleases, has enabled precise genome engineering. Four representative types of endonucleases, zinc-finger nucleases, engineered homing nucleases, transcription activator-like effector nucleases, and Cas nucleases, have been used for genome editing (Kim et al. 1996; Miller et al. 2007; Christian et al. 2010). However, endonuclease-guided target recognition has a major disadvantage, in that it is expensive and difficult to modify the nucleases so that they recognize the desired target sequences (Mushtaq et al. 2018). The discovery of the clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein (Cas) system, a form of prokaryotic immune system, enabled the far simpler RNA-guided genome editing, overcoming the limitations of the nuclease-guided genome editing tools (Marraffini and Sontheimer 2010). This system has been widely used in various forms of genome editing, such as gene knock-in/knock-out, functional genome screening, and the correction of disease-causing mutations (Liu et al. 2018a, b; Ryu et al. 2018). Further development of the CRISPR/Cas system to improve its editing efficiency through screening and engineering is currently in progress. Structural engineering and random mutagenesis of the Cas nucleases, as well as the creation of Cas fusion proteins, has expanded the utility, versatility, and target range of this system. In particular, Cas fusions with various functional motifs have made possible base editing, prime editing, CRISPR/Cas-mediated transposition, recombination, and epigenetic regulation. Limitations in these systems, such as the occurrence of unwanted genetic byproducts and off-target effects, must be resolved prior to therapeutic applications. We provide a various CRISPR/Cas technologies that have been used for genome editing.
Mechanism of actions: the CRISPR/Cas system
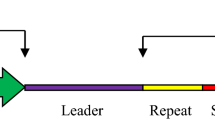
The CRISPR/Cas system is an evolved RNA-mediated adaptive defense system in bacteria and archaea that functions to protect cells from invading foreign phages and plasmids (Marraffini and Sontheimer 2010). This adaptive immune system is activated by the expression of the Cas gene operon and the CRISPR array, which consists of spacer sequences that exist between a series of direct repeats (Mohanraju et al. 2016). This defense system functions in three progressive phases (Fig. 1): the adaptive phase, expression phase, and interference phase (van der Oost et al. 2009; Jinek et al. 2012; Mohanraju et al. 2016; Rojo et al. 2018). In the adaptive phase, a short fragment of external genetic material (the protospacer) is integrated as a spacer in the CRISPR array (van der Oost et al. 2009; Jinek et al. 2012; Mohanraju et al. 2016; Rojo et al. 2018). When the same foreign material re-invades, the expression phase occurs, during which CRISPR array is transcribed and the precursor transcript is processed into a mature CRISPR RNA (crRNA) that corresponds to the foreign DNA (van der Oost et al. 2009; Jinek et al. 2012; Mohanraju et al. 2016; Rojo et al. 2018). In some systems, trans-activating RNA (tracrRNA) binds to a complementary region in pre-crRNAs to process them to their mature form. The mature crRNA is assembled with the Cas protein to form a CRISPR ribonucleoprotein (crRNP) complex (van der Oost et al. 2009; Jinek et al. 2012; Mohanraju et al. 2016; Rojo et al. 2018). Finally, in the interference phase, crRNPs are guided to specific sequences, complementary to the crRNA and adjacent to a protospacer adjacent motif (PAM) in the targeted DNA strand. The Cas protein then cleaves the foreign nucleic acid, eliminating it from cells (van der Oost et al. 2009; Jinek et al. 2012; Mohanraju et al. 2016; Rojo et al. 2018). As bacteria and phages co-evolved, phages have developed strategies for escaping CRISPR recognition. These strategies include spacer deletion, spacer mutation, and recombination between multiple phase species, making re-infection of previously infected, CRISPR-immune hosts possible (Han and Deem 2017; Westra and Levin 2020; Zhang et al. 2021). When the CRISPR/Cas system is used as a genome editing tool in cells, the CRISPR/Cas-mediated double stranded breaks (DSBs) are repaired using one of two endogenous intracellular DNA repair mechanisms: non-homologous end joining (NHEJ) or homology-directed repair (HDR). The NHEJ pathway results in the generation of small insertions/deletions (indels) at a target site during the repair of a Cas-induced DSB, which can be useful for gene knock-out but undesirable in other cases. Although NHEJ generally more efficient than HDR, it has a disadvantages in that it can cause non-specific gene disruptions such as insertions, deletions, and translocations, resulting in frameshift mutations or nonsense mutations (Zhang 2020). In contrast, the HDR pathway induces precise genome editing in the presence of a donor DNA template containing the desired sequence (Zhang 2020). However, as well as being less efficient than NHEJ, HDR is restricted to specific phases of the cell cycle (G2 and S phases) when sister chromatids are available to accept the template DNA (Chapman et al. 2012; Porto et al. 2020; Yang et al. 2020). Various attempts to increase the efficiency of HDR, such as chemical modulation to inhibit NHEJ (Chu et al. 2015), the use of linear repair templates, optimization of the lengths of the homology regions in the repair template, and the use of modified Cas protein, are underway (Liu et al. 2019).

Schematic summary of the CRISPR/Cas immune process. (i) Adaptive phase. When a virus or plasmid invades a bacterial cell, a short fragment of the foreign genetic material (the protospacer) is integrated as a spacer in the CRISPR array. (ii) Expression phase. When the same type of virus or plasmid subsequently re-invades the cell, the CRISPR array is transcribed and the precursor transcript is processed into a mature CRISPR RNA (crRNA) that corresponds to the foreign DNA. (iii) Interference phase. Finally, a RNP consisting of crRNA and Cas protein cleaves the foreign viral or plasmid DNA
Classification of CRISPR/Cas systems
CRISPR/Cas systems have been classified into 2 class, 6 types, and 33 subtypes, depending on the number of subunits constituting the Cas protein (the effector module), and the usage of tracrRNA for pre-crRNA processing (Makarova et al. 2015, 2018; Liu and Doudna 2020). The classification of CRISPR/Cas systems is briefly summarized in Table 1. Class I CRISPR/Cas systems, which include types I, III, and IV, are primary systems involving multi-subunit effector complexes (Makarova et al. 2015; Mohanraju et al. 2016; Rojo et al. 2018). In contrast, class II CRISPR/Cas systems, which include types II, V and VI, perform all functions with a single effector protein (Makarova et al. 2015; Rojo et al. 2018; Liu and Doudna 2020). This simplicity has resulted in type II CRISPR/Cas systems being the most widely used for genome editing. The class II Cas9 derived from Streptococcus pyogenes (SpCas9) is the most well characterized and frequently used Cas enzyme; it recognizes 5′-NGG-3′ PAMs located at the 3′ end of the target DNA sequence (Pyzocha and Chen 2018). A programmable, single-guide RNA (sgRNA), created by a synthetic fusion of crRNA and tracrRNA, is now the most widely used form for genome editing, given its simplicity and that its use results in similar or higher gene editing efficiencies compared to the two guide RNA system (Shapiro et al. 2020). When the sgRNA, target DNA, and Cas9 effector form a complex, the HNH and RuvC domains of Cas9 respectively cleave the complementary and noncomplementary DNA strands to generate DSBs at a site 3 bp upstream from the PAM (Fig. 2a) (Jinek et al. 2012). Although SpCas9 displays a high gene editing efficiency, it has several features that limit its usefulness for genome editing: (1) large size of Cas9 limits the choice of viral vectors for delivery, (2) the mismatch tolerance between the sgRNA and the target sequence may trigger off-target effects, and (3) pre-existing immunity against Cas9, which has been reported in humans, raises concerns for therapeutic applications. As an alternative to SpCas9, several Cas9 orthologs, including Staphylococcus aureus Cas9 (SaCas9) and Campylobacter jejuni Cas9 (CjCas9), can be used (Ran et al. 2015; Kim et al. 2017a; Dugar et al. 2018). Because they are smaller than SpCas9, the genes encoding these CRISPR components together with sequences encoding the appropriate sgRNA, can be packaged into small viral vector systems (Jo et al. 2019). Moreover, it has been reported that these nucleases exhibit reduced off-target nuclease activity compared to SpCas9 because they recognize longer PAMs, 5′-NNGRRT-3′ and 5′-NNNVRYAC-3′, respectively (Ran et al. 2015; Yamada et al. 2017; Dugar et al. 2018; Wang et al. 2019). In addition, Francisella novicida Cas9(FnCas9) (Hirano et al. 2016), Neisseria meningitidis Cas9 (Nme1Cas9, Nme2Cas9) (Zhang et al. 2013), Brevibacillus laterosporus Cas9 (BlatCas9) (Gao et al. 2020), Streptococcus thermophilus Cas9 (St1Cas9) (Deveau et al. 2008) and Staphylococcus auricularis Cas9 (SauriCas9) (Hu et al. 2020) are alternative options for genome editing.

Schematic summary of CRISPR/Cas tools used for genome editing. a The CRISPR/Cas9 system recognizes a PAM and generates DSBs at a site 3 bp upstream from the PAM. b CBE consists of an inactive or nickase form of Cas9 (dCas9 or nCas9) fused to a cytidine deaminase and UGI. After the dCas9 or nCas9 domain of a CBE recognizes a specific sequence, the cytosine deaminase deaminates C to generate U. Then, the G in the opposite strand is converted to A by cellular mismatch repair, and C is converted to T. UGI prevents the U from undergoing cellular base excision repair. c ABEs are constructed by fusion of nCas9 to E. coli tRNA adenosine deaminase, which deaminates A to generate I, which is then converted to G by DNA repair or replication. d Prime editors are constructed by fusion of an engineered reverse transcriptase domain to nCas9. A pegRNA binds to the 3′ end of the exposed target DNA strand, which was generated by nCas9. Then, the desired gene edit (which is contained in the pegRNA) is incorporated into the DNA by reverse transcriptase. e The Cas-transposon system was developed by fusing a transposase to Cas9. After the targeted sequence is recognized by Cas9 (dCas9, Cas12k or Cascades), the transpose inserts the desired sequence at the site. f Tools for CRISPR/Cas-mediated epigenetic regulation are constructed by conjugation of transcriptional repressors or activators to dCas9, generating CRISPRa and CRISPRi systems, respectively. In CRISPRa systems, transcriptional activators recruit RNA polymerase and transcription factors to a promoter and promote transcription. In CRISPRi systems, transcriptional repressors prevent binding of RNA polymerase (RNAP) to the promoter of interest
Type V (class II) CRISPR/Cas systems have some similarities with type II systems, but differ in key respects. Cas12a, which recognize T-rich PAMs at the 5′ end of the protospacer, is a representative example of type V CRISPR/Cas systems. This protein functions as a nuclease by using only a RuvC domain; it lacks an HNH domain (Zetsche et al. 2015). In addition, Cas12a is guided by a single, relatively compact crRNA and does not require a tracrRNA (Zetsche et al. 2015). The enzyme also exhibits RNase III activity, allowing it to process the precursor crRNA into the mature crRNA. These features have been exploited to allow multiplexed genome editing in which crRNAs for different targets are placed into an array, which is then processed by Cas12a to generate multiple mature crRNAs (Zetsche et al. 2015, 2017). It has been reported that Cas12a exhibits efficiency comparable to that of SpCas9, but higher specificity (Banakar et al. 2020). In contrast to Cas9 (which generates a blunt-ended DSB), Cas12a induces a staggered DSB, generating cuts in the non-targeted and targeted DNA strands at positions 18 and 23, respectively, in the protospacer (Zetsche et al. 2015). Advantageously, the gene encoding Cas12a is relatively short (3.6–3.9 kb), so it has been possible to incorporate it in adeno-associated viral vectors that were used as a gene delivery tool, resulting in efficient in vivo genome editing (Koo et al. 2018).
Some CRISPR/Cas systems protect bacteria from infections by targeting RNA. Cas13a, which belongs to the type VI system, cleaves RNA using two HEPN nuclease domains. Like other systems, the RNA editing process requires crRNA (unlike in the type II systems, tracrRNA is not required). The pre-crRNA undergoes maturation via the RNase function of the Helical-1 domain in the REC lobe (in LshCas13a) or the HEPN2 domain in the NUC lobe (in LbuCas13a). (Shmakov et al. 2015; Kingdom et al. 2017; Liu et al. 2017). The mature crRNA binds to the target RNA after recognition of the protospacer flanking sequence (PFS) located at the 3′ end of the protospacer (Abudayyeh et al. 2016; Burmistrz et al. 2020). After formation of a complex between crRNA-RNA and Cas13a, RNA degradation occurs (Knott et al. 2017). Representative Cas13a enzymes include LseCas13a, LwaCas13a, LshCas13a, LbuCa13a, and LbaCas13a, and the mechanisms through which their pre-crRNAs undergo maturation, the recognized PFSs, and the resulting RNA cleavage patterns are currently under investigation.
Engineered Cas9 and Cas12a variants
The CRISPR/Cas system has been rapidly developed by genetic engineering to expand its targeting scope and improve its specificity. Structure-guided and complimentary evolution-based engineering of Cas9 have led to increased PAM plasticity as summarized in Table 2. SpCas9-EQR, -VQR, -VRER and -VRQR were generated by modifying the Cas9 Arg1333 and Gln1335 residues, which respectively recognize the second and third guanine bases in the NGG PAM (Kleinstiver et al. 2015b, 2016; Anders et al. 2016). These Cas9 variants recognize 5′-NGNG-3′, 5′-NGAN-3′, 5′-NGCG-3′, and 5′-NGAH-3′ PAMs, respectively (Kleinstiver et al. 2015b, 2016; Anders et al. 2016). Through phage-assisted continuous evolution and selection of host cell containing evolved Cas9, xCas9 was developed to recognize 5′-NG-3′ PAMs (Hu et al. 2018). In addition, other SpCas9 variants (SpCas9-NRRH, SpCas9-NRCH and SpCas9-NRTH), which recognize most 5′-NR-3′ PAMs, have been generated by employing phage-assisted continuous and non-continuous evolution (Miller et al. 2020). Moreover, throughout use of a high-throughput PAM determination assay, engineered SpG and SpRY, which respectively recognize 5′-NGN-3′ and 5′-NRN-3′ PAMs were developed (Walton et al. 2020).
Furthermore, high-fidelity Cas variants, with increased targeting specificity and reduced off-target nuclease activity, have been developed. The general strategies for generating high-fidelity Cas9 variants are summarized in Table 2. As one example, structure-guided mutagenesis was used to neutralize the positively charged residues in the non-target strand binding groove in Cas9, leading to a requirement for more stringent base pairing between the sgRNA and the target DNA strand. With this strategy, eSpCas9 1.0 (K810A/K1003A/R1060A) and eSpCas9 1.1 (K848A/K1003A/R1060A) were generated, resulting in reduced off-target effects while maintaining on-target efficiency (Slaymaker et al. 2016). Similarly, SpCas9-HF1 (Kleinstiver et al. 2016), HypaCas9 (Chen et al. 2017), Sniper-Cas9 (Lee et al. 2018), and enAsCas12a-HF1 (Kleinstiver et al. 2019) also showed high-fidelity genome editing.
Cas9 orthologs have also been engineered to widen their targeting scope and increase their editing efficiency. For example, E782K/N968K/R1015H mutations were introduced into SaCas9 to create SaCas9-KKH, which recognizes 5′-NNNRRT-3′ PAMs (Kleinstiver et al. 2015a). RHA FnCas9 (E1369R/E1449H/R1556A), which recognizes 5′-YG-3′ PAMs, was generated by reducing the binding affinity of FnCas9 with the third guanine base in the 5′-NGG-3′ PAM (Hirano et al. 2016).
Cas12a, which recognizes T-rich PAMs, has a wider PAM window than other Cas variants. To take advantage of its wide targeting scope, mutations were introduced into Acidaminococcus Cas12a (AsCas12a) to generate the E174R/S542R/K548R variants, which recognize various PAMs, including 5′-TTYN-3′, 5′-VTTV-3′, and 5′-TRTV-3′ (Kleinstiver et al. 2019). To further expand the targeting scope of AsCas12a, additional variants were generated by modifying other residues (variant RR; S542R/K607R and variant RVR; S542R/K548V/N552R) that form hydrogen bonds with the PAM duplex; the RR and RVR variants recognize TYCV and 5′-TATV-3′ PAMs, respectively (Nishimasu et al. 2017; Kleinstiver et al. 2019). To reduce off-target effects of AsCas12a variants while maintaining their high levels of on-target activity, K949A was also introduced into the AsCas12a RR and RVR variants (Gao et al. 2017). In addition, G532R/K595R mutations were introduced into Lachnospiraceae bacterium Cas12a (LbCas12a) to generate LbCas12a-RR variant so that it recognizes 5′-TYCV-3′ PAMs, broadening its targeting scope (Li et al. 2018).
Base editors
Recently, several DNA base editing systems have been developed that allow single base conversions, also known as base editing, in cells and organisms in a guide RNA-dependent manner. By connecting a deaminase to a Cas9 protein, targeted base editing became possible, broadening the types of editing that could be achieved by CRISPR/Cas systems. Two general types of base editors (cytidine base editor (CBE), and adenine baes editor (ABE) have been developed, both with great potential for targeted base mutagenesis (Fig. 2b and 2c). CBE1, the first version of CBE, is a fusion between an inactive form of Cas9 (dead Cas9 or dCas9) and rat-derived cytosine deaminase apolipoprotein B mRNA editing enzyme catalytic subunit 1 (APOBEC1), a cytidine deaminase. CBE1 enables the direct conversion of a targeted C·G base pair to a T·A base pair in a process involving deamination of a C to create a U, which is then converted to T by the cell’s DNA mismatch repair process (Komor et al. 2016). CBE2 was generated by fusing a uracil DNA glycosylase inhibitor (UGI) to CBE1, which prevents the removal of the U by base excision repair involving uracil DNA glycosylase (Komor et al. 2016). Another form, CBE3, was created by exchanging dCas9 for Cas9 nickase (nCas9; D10A mutation). This version exhibits increased base editing efficiency compared to CBE1 and CBE2 (Komor et al. 2016). The development of CBE3 resulted in a six-fold increase in genome editing efficiency compared to CBE2, but it had a limitation in that window in which bases were converted was narrow (positions 4 to 8 in the protospacer, counting from the PAM distal base) (Komor et al. 2016). An improved version of CBE (CBE4) was generated by extending the length of linkers (rAPOBEC-nCas9 linker to 32 amino acids, nCas9-UGI linker to 9 amino acids) and attaching two UGIs to the C-terminus of the constructs. This modification in CBE4 resulted in a 1.5-fold increase in base editing efficiency and a 2.3-fold decrease in non-T product formation compared to CBE3 (Komor et al. 2017). As another base editing system, Target-activation-induced cytidine deaminase (Target-AID) was developed by fusing nCas9 (D10A) to a Petromyzon marinus cytidine deaminase 1 (PmCDA1). Target-AID exhibits a different targeting window (positions 1 to 5 in the protospacer) compared to CBEs and ability to edit methylated cytosines. In addition, several attempts have been made to expand the base editing window and increase its specificity as summarized in Table 3.
ABEs were constructed using an evolved version of Escherichia coli tRNA adenosine deaminase (TadA). Wild-type TadA can convert adenosine to inosine in tRNA. However, because no known natural adenosine deaminase recognizes DNA, TadA was evolved to exhibit this characteristic through bacterial selection methods, creating TadA* (Gaudelli et al. 2017). The first ABE (ABE1.2) was generated by fusing a mammalian codon-optimized TadA*, containing A106V and D108N mutations, to nCas9 (D10A) (Gaudelli et al. 2017). A later version, ABE7.10, which consists of a heterodimer of the wild-type TadA and TadA* fused with nCas9 (D10A) and exhibits improved base editing efficiency, was constructed through bacterial evolution and protein engineering (Gaudelli et al. 2017). ABE7.10 shows broad sequence compatibility, targeting a window spanning positions 4 to 7 in the protospacer. In addition, ABE7.10 exhibits highly efficient adenine base editing, in human cells (Gaudelli et al. 2017) and mice (Ryu et al. 2018). ABEs have been further engineered to exhibit even higher base editing efficiency as summarized in Table 3. For example, evolved TadA variants with increased deoxyadenosine deamination activity were used to generate several versions of ABE8e based on SpCas9, SaCas9, and LbCas12a, which exhibit higher efficiency and a wider activity window than ABE7.10 (Richter et al. 2020).
Glycosylase base editor (GBE) has also been developed by fusing uracil-DNA glycosylase to a CBE. The uracil-DNA glycosylase removes the U produced by the CBE and creates an apurinic/apyrimidinic site, which activates the DNA repair mechanism. GBE [that is, AID-nCas9-uracil DNA glycosylase] showed efficient C-to-A conversion in E. coli. Replacement of AID with rAPOBEC1 generated a GBE (APOBEC-nCas9- uracil DNA glycosylase), that enables C-to-G conversion in mammalian cells (Zhao et al. 2021).
A dual adenine and cytosine base editor (A&C-BE) has been developed that can simultaneously induce C-to-T and A-to-G base editing in the same allele. This dual editor consists of a human AID-TadA-TadA*-nCas9-UGIs fusion (Zhang et al. 2020). For A&C-BE, the A-to-G editing window remained consistent as positions 4–7, whereas the base editing window for C-to-T editing was extended to positions 2–17 in the protospacer (Zhang et al. 2020). Furthermore, synchronous programmable adenine and cytosine editors (SPACEs) were generated by a fusion of miniABEmax (which was generated by removing wild-type TadA domain from ABEmax and introducing V82G mutation) to target-AID (Grünewald et al. 2020). SPACEs can induce targeted A-to-G and C-to-T conversions at positions 4–7 and 2–7 in the protospacer, respectively, and exhibit reduced RNA off-target effects and comparable or lower DNA off-target effects compared to miniABEmax-V82G and Target-AID (Grünewald et al. 2020).
Prime editors
Prime editing is a recently developed genome editing technology that is capable of generating targeted insertions, deletions, and substitutions in a precise manner. A key feature of prime editing is that it does not require either a DSB or the HDR pathway for targeted editing. Prime editors are generated by fusing an engineered reverse transcriptase (RT) domain to nCas9 (H840A) (Fig. 2d) (Anzalone et al. 2019). This process requires an engineered prime editing guide RNA (pegRNA), which is similar to a sgRNA but also contains a primer binding site (PBS) and an RT template containing the desired edit. nCas9 first generates a nick in the DNA strand containing the PAM. Then, RT binds to the 3′ end of the exposed target DNA strand and performs reverse transcription, after which the non-edited protruding 5′-flap on the strand containing the PAM is degraded by cellular endonucleases. Finally, DNA ligation and repair occur to generate the desired DNA sequence (Anzalone et al. 2019).
Three versions of prime editors have been developed. Prime editor 1 (PE1) was generated by fusing nCas9 (H840A) to the wild-type Moloney murine leukemia virus-RT. The RT domain was further engineered in PE2, which exhibits increased editing efficiency (Anzalone et al. 2019). PE3 uses an additional sgRNA that leads to the generation of a nick in the non-edited strand by nCas9, resulting in a further two- to four-fold increase in editing efficiency. PE3b uses an sgRNA that targets only the edited strand to decrease the frequency of indels in the non-edited DNA strand (Anzalone et al. 2019). Various attempts to improve prime editing are actively underway; efficient genome editing has been demonstrated in plants and mouse models (Lin et al. 2020; Liu et al. 2020).
Prime editing efficiency is greatly influenced by structure of the pegRNA. Several factors such as SpCas9-induced indel frequencies, GC counts, and the PBS melting temperature must be considered to design the optimal pegRNA (Kim et al. 2020). Several prediction tools, including pegFinder (Chow et al. 2020) and PrimeDesign (Hsu et al. 2020) can be useful for designing pegRNAs and for predicting possible prime editing effects in the whole genome.
Transposase and recombinase fusions to Cas9
The CRISPR/Cas system has also been harnessed to enable target-specific insertions of longer DNA fragments. CRISPR/Cas-associated transposases that function in cyanobacterial cells have recently been reported. Here, a nuclease-deficient Cas effector directs transposition of a cargo gene in an RNA-guided manner (Strecker et al. 2019). Based on this system, a Cas-transposon system was developed by fusing a Tn7 transposase to type V-K Cas12k to facilitate target-specific transposition (Fig. 2e) (Strecker et al. 2019). Another type of reconstituted mariner-family transposase, Himar 1, conjugated to dCas9, resulted in site-specific insertion of DNA into the target TA motifs in the E.coli and mammalian cells (Chen and Wang 2019).
Genome editing tool that would enable programmable homologous recombination would also be a valuable addition to the CRISPR toolkit. Recombinase, which functions as a dimer, recognize a strict recombinase site, resulting in recombination; this process can cause the insertions, deletions, or inversions of specific sequences through cleavage, strand exchange, and re-ligation (Chaikind et al. 2016). Because recombinases do not induce cellular DNA repair procedures, recombination does not typically lead to byproducts of error-prone DNA repair such as indels. A fusion of dCas9 with an engineered recombinase, Ginβ, has been generated to overcome the sequence constraints of recombinase (Chaikind et al. 2016). Use of this system resulted in modest genome editing efficiency in mammalian cells, demonstrating its potential as an alternative genome editing tool (Cui et al. 2018).
Epigenetic regulators
CRISPR/Cas-mediated epigenetic regulation became possible by the conjugation of several epigenetic regulator proteins to dCas9, generating CRISPR interference (CRISPRi) and CRISPR activation (CRISPRa) systems (Fig. 2f). In CRISPRi systems, the Krüppel-associated box (KRAB) repressor, fused to dCas9, is commonly used as an effector (Gilbert et al. 2013; Thakore et al. 2015). KRAB interacts with heterochromatin-forming complexes, which can induce histone methylation and deacetylation to inhibit binding of RNA polymerases to enhancer or promoter regions, inactivating transcription (Gilbert et al. 2013; Thakore et al. 2015).
In contrast, CRISPRa systems promote transcription by using a transcriptional activator, such as the VP16 activation domain, which can activate transcription by interacting with the TATA-binding protein, TFIIB, and SAGA histone acetylase (Hall and Struhl 2002). Fusion of VP64 (4 copies of VP16) or VP192 (12 copies of VP16) to dCas9 led to transcriptional activation in vivo that was increased compared to that of VP16-conjutated dCas9 (Maeder et al. 2013; Balboa et al. 2015). In addition, fusions of SunTag (an array of repeating peptides that can mobilize multiple copies of an antibody-fusion protein) (Tanenbaum et al. 2014), VPR (composed of VP64, p65, and Rta and act as stronger activators than VP64) (Chavez et al. 2015), p300 (which promotes transcription through increased transactivation capacity) (Hilton et al. 2015), and TET (which performs demethylation by oxidizing methyl groups on proteins) (Liu et al. 2016) to dCas9 also promote transcription.
Challenges
The CRISPR/Cas system is certainly a useful gene editing tool, but there are several limitations to overcome before it can be used in various applications. Off-target effects (unwanted genome editing at unintended sites) are one of the major limitations. These effects can be caused by a tolerance for mismatches between the sgRNA and target DNA sequences (outside of the seed region located 1 to 5 nucleotides proximal to the PAM) (Fu et al. 2013). Furthermore, presence of non-canonical PAMs can decrease Cas specificity (Zhang et al. 2014). To minimize off-target effects, several bioinformatic tools such as Cas-OFFinder (Bae et al. 2014), CCTop (Stemmer et al. 2015), and CT-Finder (Zhu et al. 2016) can be useful for designing appropriate sgRNAs.
To apply the CRISPR/Cas system therapeutically, immunity against CRISPR components must also be resolved. Preexisting adaptive immune responses against Cas9 (anti-SpCas9 and anti-SaCas9 antibodies and Cas9-specific T-cells) have been detected in the majority of human serum samples tested, because the bacterial species that are the sources of these components regularly infect humans (Charlesworth et al. 2019). Another report supports the idea that CRISPR RNAs elicit innate immune responses in human cells (Kim et al. 2018). Recently, a phase 1 clinical trial involving the CRISPR/Cas system has been conducted in lung cancer patients, who were infused with CRISPR-PD 1-edited T cells (Lu et al. 2020). The safety and feasibility of this approach were confirmed without any detectable side effects in this study, but more extensive investigations of safety of CRISPR/Cas-modified cells should be conducted. In addition to the CRISPR/Cas trial in cancer therapy, a range of therapeutic approaches for treating various diseases are being tested in vitro and in vivo, and continuous developments of the CRISPR/Cas system are underway to make these therapeutic applications more feasible.
Conclusions
The development of CRISPR/Cas technology has revolutionized the field of genome engineering. CRISPR/Cas-based tools are the most sophisticated and versatile editing tools to be used in areas ranging from basic research to medical therapy development. In addition, CRISPR/Cas technology allows researchers to perform genome-wide screens to study the impact of changes in gene expression on cell function and link genotypes and phenotypes. As one example, CRISPR/Cas knockout screening was used to search for therapeutic targets in pancreatic cancer cells, leading to the finding that such cells were sensitive to MEK inhibitors (Kanarek et al. 2018; Szlachta et al. 2018; Behan et al. 2019).
Applications of the CRISPR/Cas system include transcriptional regulation made possible by conjugation of functional proteins to Cas9, disease modeling, and gene therapies. Nevertheless, many challenges remain; these include off-target effects, bystander effects, the development of efficient delivery methods, and immunity against CRISPR components, all of which need to be fully addressed. Rapid improvements in CRISPR/Cas-based tools are ongoing to overcome these limitations. These tools should reach their full potential in various applications in the near future.
References
Abudayyeh OO, Gootenberg JS, Konermann S, Joung J, Slaymaker IM, Cox DBT, Shmakov S, Makarova KS, Semenova E, Minakhin L, Severinov K, Regev A, Lander ES, Koonin EV, Zhang F (2016) C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science. https://doi.org/10.1126/science.aaf5573
Anders C, Bargsten K, Jinek M (2016) Structural plasticity of PAM recognition by engineered variants of the RNA-guided endonuclease Cas9. Mol Cell 61:895–902. https://doi.org/10.1016/j.molcel.2016.02.020
Anzalone AV, Randolph PB, Davis JR, Sousa AA, Koblan LW, Levy JM, Chen PJ, Wilson C, Newby GA, Raguram A, Liu DR (2019) Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576:149–157. https://doi.org/10.1038/s41586-019-1711-4
Bae S, Park J, Kim JS (2014) Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 30:1473–1475. https://doi.org/10.1093/bioinformatics/btu048
Balboa D, Weltner J, Eurola S, Trokovic R, Wartiovaara K, Otonkoski T (2015) Conditionally stabilized dCas9 activator for controlling gene expression in human cell reprogramming and differentiation. Stem Cell Rep 5:448–459. https://doi.org/10.1016/j.stemcr.2015.08.001
Banakar R, Schubert M, Collingwood M, Vakulskas C, Eggenberger AL, Wang K (2020) Comparison of CRISPR-Cas9/Cas12a ribonucleoprotein complexes for genome editing efficiency in the rice phytoene desaturase (OsPDS) gene. Rice 13:4. https://doi.org/10.1186/s12284-019-0365-z
Behan FM, Iorio F, Picco G, Gonçalves E, Beaver CM, Migliardi G, Santos R, Rao Y, Sassi F, Pinnelli M, Ansari R, Harper S, Jackson DA, McRae R, Pooley R, Wilkinson P, van der Meer D, Dow D, Buser-Doepner C, Bertotti A, Trusolino L, Stronach EA, Saez-Rodriguez J, Yusa K, Garnett MJ (2019) Prioritization of cancer therapeutic targets using CRISPR–Cas9 screens. Nature 568:511–516. https://doi.org/10.1038/s41586-019-1103-9
Burmistrz M, Krakowski K, Krawczyk-Balska A (2020) RNA-targeting CRISPR–Cas systems and their applications. Int J Mol Sci. https://doi.org/10.3390/ijms21031122
Chaikind B, Bessen JL, Thompson DB, Hu JH, Liu DR (2016) A programmable Cas9-serine recombinase fusion protein that operates on DNA sequences in mammalian cells. Nucleic Acids Res 44:9758–9770. https://doi.org/10.1093/nar/gkw707
Chapman JR, Taylor MRG, Boulton SJ (2012) Playing the end game: DNA double-strand break repair pathway choice. Mol Cell 47:497–510. https://doi.org/10.1016/j.molcel.2012.07.029
Charlesworth CT, Deshpande PS, Dever DP, Camarena J, Lemgart VT, Cromer MK, Vakulskas CA, Collingwood MA, Zhang L, Bode NM, Behlke MA, Dejene B, Cieniewicz B, Romano R, Lesch BJ, Gomez-Ospina N, Mantri S, Pavel-Dinu M, Weinberg KI, Porteus MH (2019) Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nat Med 25:249–254. https://doi.org/10.1038/s41591-018-0326-x
Chatterjee P, Lee J, Nip L, Koseki SRT, Tysinger E, Sontheimer EJ, Jacobson JM, Jakimo N (2020) A Cas9 with PAM recognition for adenine dinucleotides. Nat Commun. https://doi.org/10.1038/s41467-020-16117-8
Chavez A, Scheiman J, Vora S, Pruitt BW, Tuttle M, Iyer PR, Lin E, Kiani S, Guzman S, Wiegand CD, Ter-Ovanesyan DJ, Braff D, Davidsohn JL, Housden N, Perrimon BE, Weiss N, Aach R, Collins J, Church JJ (2015) Highly efficient Cas9-mediated transcriptional programming. Nat Methods 12:326–328. https://doi.org/10.1038/nmeth.3312
Chen JS, Dagdas YS, Kleinstiver BP, Welch MM, Sousa AA, Harrington LB, Sternberg SH, Joung JK, Yildiz A, Doudna JA (2017) Enhanced proofreading governs CRISPR-Cas9 targeting accuracy. Nature 550:407–410. https://doi.org/10.1038/nature24268
Chen SP, Wang HH (2019) An engineered Cas-transposon system for programmable and site-directed DNA transpositions. Cris J 2:376–394. https://doi.org/10.1089/crispr.2019.0030
Chow RD, Chen JS, Shen J, Chen S (2020) A web tool for the design of prime-editing guide RNAs. Nat Biomed Eng 5:190–194. https://doi.org/10.1038/s41551-020-00622-8
Christian M, Cermak T, Doyle EL, Schmidt C, Zhang F, Hummel A, Bogdanove AJ, Voytas DF (2010) Targeting DNA double-strand breaks with TAL effector nucleases. Genetics 186:757–761. https://doi.org/10.1534/genetics.110.120717
Chu VT, Weber T, Wefers B, Wurst W, Sander S, Rajewsky K, Kühn R (2015) Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat Biotechnol 33:543–548. https://doi.org/10.1038/nbt.3198
Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F (2013) Multiplex genome engineering using CRISPR/Cas systems. Science. https://doi.org/10.1126/science.1231143
Cui Y, Niu Y, Zhou J, Chen Y, Cheng Y, Li S, Ai Z, Chu C, Wang H, Zheng B, Chen X, Sha J, Guo X, Huang X, Ji W (2018) Generation of a precise Oct4-hrGFP knockin cynomolgus monkey model via CRISPR/Cas9-assisted homologous recombination. Cell Res 28:383–386. https://doi.org/10.1038/cr.2018.10
Deveau H, Barrangou R, Garneau JE, Labonté J, Fremaux C, Boyaval P, Romero DA, Horvath P, Moineau S (2008) Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J Bacteriol 190:1390–1400. https://doi.org/10.1128/JB.01412-07
Dugar G, Leenay RT, Eisenbart SK, Bischler T, Aul BU, Beisel CL, Sharma CM (2018) CRISPR RNA-dependent binding and cleavage of endogenous RNAs by the Campylobacter jejuni Cas9. Mol Cell 69:839–905. https://doi.org/10.1016/j.molcel.2018.01.032
Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, Sander JD (2013) High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol 31:822–826. https://doi.org/10.1038/nbt.2623
Gao L, Cox DBT, Yan WX, Manteiga JC, Schneider MW, Yamano T, Nishimasu H, Nureki O, Crosetto N, Zhang F (2017) Engineered Cpf1 variants with altered PAM specificities increase genome targeting range. Nat Biotechnol 35:789–792. https://doi.org/10.1038/nbt.3900.Engineered
Gao N, Zhang C, Hu Z, Li M, Wei J, Wang Y, Liu H (2020) Characterization of Brevibacillus laterosporus Cas9 (BlatCas9) for mammalian genome editing. Front Cell Dev Biol. https://doi.org/10.3389/fcell.2020.583164
Gaudelli NM, Komor AC, Rees HA, Packer MS, Badran AH, Bryson DI, Liu DR (2017) Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 551:464–471. https://doi.org/10.1038/nature24644
Gaudelli NM, Lam DK, Rees HA, Solá-Esteves NM, Barrera LA, Born DA, Edwards A, Gehrke JM, Lee SJ, Liquori AJ, Murray R, Packer MS, Rinaldi C, Slaymaker IM, Yen J, Young LE, Ciaramella G (2020) Directed evolution of adenine base editors with increased activity and therapeutic application. Nat Biotechnol 38:892–900. https://doi.org/10.1038/s41587-020-0491-6
Gehrke JM, Cervantes O, Clement MK, Wu Y, Zeng J, Bauer DE, Pinello L, Joung JK (2018) An APOBEC3A-Cas9 base editor with minimized bystander and off-target activities. Nat Biotechnol 36:977–982. https://doi.org/10.1038/nbt.4199
Gilbert LA, Larson MH, Leonardo M, Zairan L, Brar GA, Torress SE, Stern-Ginossar N, Onn B, Doudna JA (2013) CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154:442–451
Grünewald J, Zhou R, Garcia SP, Iyer S, Lareau CA, Aryee MJ, Joung JK (2019a) Transcriptome-wide off-target RNA editing induced by CRISPR-guided DNA base editors. Nature 569:433–437. https://doi.org/10.1038/s41586-019-1161-z
Grünewald J, Zhou R, Iyer S, Lareau CA, Garcia SP, Aryee MJ, Joung JK (2019b) CRISPR DNA base editors with reduced RNA off-target and self-editing activities. Nat Biotechnol 37:1041–1048. https://doi.org/10.1038/s41587-019-0236-6
Grünewald J, Zhou R, Lareau CA, Garcia SP, Miller BR, Langner LM, Hsu JY, Aryee MJ, Joung K (2020) A dual-deaminase CRISPR base editor enables concurrent adenine and cytosine editing. 38:861–864. https://doi.org/10.1038/s41587-020-0535-y.A
Hall DB, Struhl K (2002) The VP16 activation domain interacts with multiple transcriptional components as determined by protein-protein cross-linking in vivo. J Biol Chem 277:46043–46050. https://doi.org/10.1074/jbc.M208911200
Han P, Deem MW (2017) Non-classical phase diagram for virus bacterial coevolution mediated by clustered regularly interspaced short palindromic repeats. J R Soc Interface. https://doi.org/10.1098/rsif.2016.0905
Hilton IB, D'Ippolito AM, Vockley CM, Thakore PI, Crawford GE, Reddy TE, Gersbach CA (2015) Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat Biotechnol 33(5):510–517. https://doi.org/10.1038/nbt.3199
Hirano H, Gootenberg, Jonathan S, Horii T, Abudayyeh OO, Kimura M, Hsu PD, Nakane T, Ishitani R, Hatada I, Zhang F, Nishimasu H, Nureki O (2016) Structure and engineering of Francisella novicida Cas9. Cell 164:277–294. https://doi.org/10.1016/j.cell.2016.01.039.Structure
Hsu JY, Anzalone AV, Grünewald J, Lam KC, Shen MW, Liu DR, Joung JK, Pinello L (2020) PrimeDesign software for rapid and simplified design of prime editing guide RNAs. BioRxiv. https://doi.org/10.1101/2020.05.04.077750
Hu JH, Miller SM, Geurts MH, Tang W, Chen L, Sun N, Zeina CM, Gao X, Rees HA, Lin Z, Liu DR (2018) Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature 556:57–63. https://doi.org/10.1038/nature26155
Hu Z, Wang S, Zhang C, Gao N, Li M, Wang D, Wang D, Liu D, Liu H, Ong SG, Wang H, Wang Y (2020) A compact cas9 ortholog from staphylococcus auricularis (sauricas9) expands the DNA targeting scope. PLoS Biol 18:e3000686. https://doi.org/10.1371/JOURNAL.PBIO.3000686
Jiang W, Feng S, Huang S, Yu W, Li G, Yang G, Liu Y, Zhang Y, Zhang L, Hou Y, Chen J, Chen J, Huang X (2018) BE-PLUS: a new base editing tool with broadened editing window and enhanced fidelity. Cell Res 28:855–861. https://doi.org/10.1038/s41422-018-0052-4
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E (2012) A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science 337:816–822
Jo DH, Koo T, Cho CS, Kim JH, Kim JS, Kim JH (2019) Long-term effects of in vivo genome editing in the mouse retina using Campylobacter jejuni Cas9 expressed via adeno-associated virus. Mol Ther 27:130–136. https://doi.org/10.1016/j.ymthe.2018.10.009
Kanarek N, Keys HR, Cantor JR, Lewis CA, Chan SH, Kunchok T, Abu-Remaileh M, Freinkman E, Schweitzer LD, Sabatini DM (2018) Histidine catabolism is a major determinant of methotrexate sensitivity. Nature 559:632–636. https://doi.org/10.1038/s41586-018-0316-7
Kim YG, Cha J, Chandrasegaran S (1996) Hybrid restriction enzymes: Zinc finger fusions to Fok I cleavage domain. Proc Natl Acad Sci USA 93:1156–1160. https://doi.org/10.1073/pnas.93.3.1156
Kim E, Koo T, Park SW, Kim D, Kim K, Cho HY, Song DW, Lee KJ, Jung MH, Kim S, Kim JH, Kim JH, Kim JS (2017a) In vivo genome editing with a small Cas9 orthologue derived from Campylobacter jejuni. Nat Commun 8(1). https://doi.org/10.1038/ncomms14500
Kim YB, Komor AC, Levy JM, Packer MS, Zhao KT, Liu DR (2017b) Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nat Biotechnol 35:371–376. https://doi.org/10.1038/nbt.3803
Kim S, Koo T, Jee HG, Cho HY, Lee G, Lim DG, Shin HS, Kim JS (2018) CRISPR RNAs trigger innate immune responses in human cells. Genom Res 28:367–373. https://doi.org/10.1101/gr.231936.117
Kim HK, Yu G, Park J, Min S, Lee S, Yoon S, Kim HH (2020) Predicting the efficiency of prime editing guide RNAs in human cells. Nat Biotechnol 39:198–206. https://doi.org/10.1038/s41587-020-0677-y
Kingdom U, Kingdom U, London F, Health N, Foundation S, Kingdom U, Kingdom U (2017) Two distinct RNase activities of CRISPR-C2c2 enable guide RNA processing and RNA detection. Nature 21:129–139. https://doi.org/10.1038/nature19802.Two
Kleinstiver BP, Prew MS, Tsai SQ, Nguyen NT, Topkar VV, Zheng Z, Joung JK (2015a) Broadening the targeting range of Staphylococcus aureus CRISPR-Cas9 by modifying PAM recognition. Nat Biotechnol 33:1293–1298. https://doi.org/10.1038/nbt.3404
Kleinstiver BP, Prew MS, Tsai SQ, Topkar VV, Nguyen NT, Zheng Z, Gonzales APW, Li Z, Peterson RT, Yeh JRJ, Aryee MJ, Joung JK (2015b) Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature 523:481–485. https://doi.org/10.1038/nature14592
Kleinstiver BP, Pattanayak V, Prew MS, Tsai SQ, Nguyen NT, Zheng Z, Joung JK (2016) High-fidelity CRISPR-Cas9 nucleases with undetectable genome-wide off-target effects. Nature 529:490–495. https://doi.org/10.1038/nature16526
Kleinstiver BP, Sousa AA, Walton RT, Tak YE, Hsu JY, Clement K, Welch MM, Horng JE, Malagon-Lopez J, Scarfò I, Maus MV, Pinello L, Aryee MJ, Joung JK (2019) Engineered CRISPR–Cas12a variants with increased activities and improved targeting ranges for gene, epigenetic and base editing. Nat Biotechnol 37:276–282. https://doi.org/10.1038/s41587-018-0011-0
Knott GJ, East-Seletsky A, Cofsky JC, Holton JM, Charles E, O’Connell MR, Doudna JA (2017) Guide-bound structures of an RNA-targeting A-cleaving CRISPR-Cas13a enzyme. Nat Struct Mol Biol 24:825–833. https://doi.org/10.1038/nsmb.3466
Koblan LW, Doman JL, Wilson C, Levy JM, Tay T, Newby GA, Maianti JP, Raguram A, Liu DR (2018) Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction. Nat Biotechnol 36:843–848. https://doi.org/10.1038/nbt.4172
Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR (2016) Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533:420–424. https://doi.org/10.1038/nature17946
Komor AC, Zhao KT, Packer MS, Gaudelli NM, Waterbury AL, Koblan LW, Kim YB, Badran AH, Liu DR (2017) Improved base excision repair inhibition and bacteriophage Mu Gam protein yields C:G-to-T:A base editors with higher efficiency and product purity. Sci Adv 3:1–9. https://doi.org/10.1126/sciadv.aao4774
Koo T, Park SW, Jo DH, Kim D, Kim JH, Cho HY, Kim J, Kim JH, Kim JS (2018) CRISPR-LbCpf1 prevents choroidal neovascularization in a mouse model of age-related macular degeneration. Nat Commun 9:1855. https://doi.org/10.1038/s41467-018-04175-y
Koonin EV, Makarova KS, Zhang F (2017) Diversity, classification and evolution of CRISPR-Cas systems. Curr Opin Microbiol 37:67–78. https://doi.org/10.1016/j.mib.2017.05.008
Lee JK, Jeong E, Lee J, Jung M, Shin E, Kim Y, Lee K, Jung I, Kim D, Kim S, Kim JS (2018) Directed evolution of CRISPR-Cas9 to increase its specificity. Nat Commun 9:3048. https://doi.org/10.1038/s41467-018-05477-x
Li S, Zhang X, Wang W, Guo X, Wu Z, Du W, Zhao Y, Xia L (2018) Expanding the scope of CRISPR/Cpf1-mediated genome editing in rice. Mol Plant 11:995–998. https://doi.org/10.1016/j.molp.2018.03.009
Lin Q, Zong Y, Xue C, Wang S, Jin S, Zhu Z, Wang Y, Anzalone AV, Raguram A, Doman JL, Liu DR, Gao C (2020) Prime genome editing in rice and wheat. Nat Biotechnol 38:582–585. https://doi.org/10.1038/s41587-020-0455-x
Liu XS, Wu H, Ji X, Stelzer Y, Wu X, Czauderna S, Shu J, Dadon D, Young RA, Jaenisch R (2016) Editing DNA methylation in the mammalian genome. Cell 167:233-247.e17. https://doi.org/10.1016/j.cell.2016.08.056
Liu L, Li X, Ma J, Li Z, You L, Wang J, Wang M, Zhang X, Wang Y (2017) The molecular architecture for RNA-guided RNA cleavage by Cas13a. Cell 170:714-726.e10. https://doi.org/10.1016/j.cell.2017.06.050
Liu Z, Chen M, Chen S, Deng J, Song Y, Lai L, Li Z (2018a) Highly efficient RNA-guided base editing in rabbit. Nat Commun. https://doi.org/10.1038/s41467-018-05232-2
Liu Z, Lu Z, Yang G, Huang S, Li G, Feng S, Liu Y, Li J, Yu W, Zhang Y, Chen J, Sun Q, Huang X (2018b) Efficient generation of mouse models of human diseases via ABE- and BE-mediated base editing. Nat Commun. https://doi.org/10.1038/s41467-018-04768-7
Liu M, Rehman S, Tang X, Gu K, Fan Q, Chen D, Ma W (2019) Methodologies for improving HDR efficiency. Front Genet 9:691. https://doi.org/10.3389/fgene.2018.00691
Liu TY, Doudna JA (2020) Chemistry of Class 1 CRISPR-Cas effectors: binding, editing, and regulation. J Biol Chem 295:14473–14487. https://doi.org/10.1074/jbc.REV120.007034
Liu Y, Li X, He S, Huang S, Li C, Chen Y, Liu Z, Huang X, Wang X (2020) Efficient generation of mouse models with the prime editing system. Cell Discov. https://doi.org/10.1038/s41421-020-0165-z
Lu Y, Xue J, Deng T, Zhou X, Yu K, Deng L, Huang M, Yi X, Liang M, Wang Y, Shen H, Tong R, Wang W, Li L, Song J, Li J, Su X, Ding Z, Gong Y, Zhu J, Wang Y, Zou B, Zhang Y, Li Y, Zhou L, Liu Y, Yu M, Wang Y, Zhang X, Yin L, Xia X, Zeng Y, Zhou Q, Ying B, Chen C, Wei Y, Li W, Mok T (2020) Safety and feasibility of CRISPR-edited T cells in patients with refractory non-small-cell lung cancer. Nat Med 26:732–740. https://doi.org/10.1038/s41591-020-0840-5
Maeder ML, Linder SJ, Cascio VM, Fu Y, Ho QH, Joung JK (2013) CRISPR RNA-guided activation of endogenous human genes. Nat Methods 10:977–979. https://doi.org/10.1038/nmeth.2598
Makarova KS, Koonin EV (2015) Annotation and classification of CRISPR-Cas systems. Methods Mol Biol 1311:47–75. https://doi.org/10.1007/978-1-4939-2687-9_4
Makarova KS, Wolf YI, Alkhnbashi OS, Costa F, Shah SA, Saunders SJ, Barrangou R, Brouns SJJ, Charpentier E, Haft DH, Horvath P, Moineau S, Mojica FJM, Terns RM, Terns MP, White MF, Yakunin AF, Garrett RA, Van Der Oost J, Backofen R, Koonin EV (2015) An updated evolutionary classification of CRISPR-Cas systems. Nat Rev Microbiol 13:722–736. https://doi.org/10.1038/nrmicro3569
Makarova KS, Wolf YI, Koonin EV (2018) Classification and nomenclature of CRISPR-Cas systems: where from here? Cris J 1:325–336. https://doi.org/10.1089/crispr.2018.0033
Mao Z, Bozzella M, Seluanov A, Gorbunova V (2008) Comparison of nonhomologous end joining and homologous recombination in human cells. DNA Repair 7:1765–1771. https://doi.org/10.1016/j.dnarep.2008.06.018
Marraffini LA, Sontheimer EJ (2010) CRISPR interference: RNA-directed adaptive immunity in bacteria and archaea. Nat Rev Genet. https://doi.org/10.1038/nrg2749
Miller JC, Holmes MC, Wang J, Guschin DY, Lee YL, Rupniewski I, Beausejour CM, Waite AJ, Wang NS, Kim KA, Gregory PD, Pabo CO, Rebar EJ (2007) An improved zinc-finger nuclease architecture for highly specific genome editing. Nat Biotechnol 25:778–785. https://doi.org/10.1038/nbt1319
Miller SM, Wang T, Randolph PB, Arbab M, Shen MW, Huang TP, Matuszek Z, Newby GA, Rees HA, Liu DR (2020) Continuous evolution of SpCas9 variants compatible with non-G PAMs. Nat Biotechnol 38:471–481. https://doi.org/10.1038/s41587-020-0412-8
Mohanraju P, Makarova KS, Zetsche B, Zhang F, Koonin EV, Van Der Oost J (2016) Diverse evolutionary roots and mechanistic variations of the CRISPR-Cas systems. Science. https://doi.org/10.1126/science.aad5147
Mushtaq M, Bhat JA, Mir ZA, Sakina A, Ali S, Singh AK, Tyagi A, Salgotra RK, Dar AA, Bhat R (2018) CRISPR/Cas approach: a new way of looking at plant-abiotic interactions. J Plant Physiol 224–225:156–162. https://doi.org/10.1016/j.jplph.2018.04.001
Nishida K, Arazoe T, Yachie N, Banno S, Kakimoto M, Tabata M, Mochizuki M, Miyabe A, Araki M, Hara KY, Shimatani Z, Kondo A (2016) Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science. https://doi.org/10.1126/science.aaf8729
Nishimasu H, Yamano T, Gao L, Zhang F, Ishitani R, Nureki O (2017) Structural basis for the altered PAM recognition by engineered CRISPR-Cpf1. Mol Cell 67(1):139–147.e2. https://doi.org/10.1016/j.molcel.2017.04.019
Pinilla-Redondo R, Mayo-Muñoz D, Russel J, Garrett RA, Randau L, Sørensen SJ, Shah SA (2020) Type IV CRISPR-Cas systems are highly diverse and involved in competition between plasmids. Nucleic Acids Res 48:2000–2012. https://doi.org/10.1093/nar/gkz1197
Porto EM, Komor AC, Slaymaker IM, Yeo GW (2020) Base editing: advances and therapeutic opportunities. Nat Rev Drug Discov 19:839–859. https://doi.org/10.1038/s41573-020-0084-6
Pyzocha NK, Chen S (2018) Diverse Class 2 CRISPR-Cas effector proteins for genome engineering applications. ACS Chem Biol 13:347–356
Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, Kriz AJ, Zetsche B, Shalem O, Wu X, Makarova KS, Koonin EV, Sharp PA, Zhang F (2015) In vivo genome editing using Staphylococcus aureus Cas9. Nature 520:186–191. https://doi.org/10.1038/nature14299
Rees HA, Wilson C, Doman JL, Liu DR (2019) Analysis and minimization of cellular RNA editing by DNA adenine base editors. Sci Adv. https://doi.org/10.1126/sciadv.aax5717
Richter MF, Zhao KT, Eton E, Lapinaite A, Newby GA, Thuronyi BW, Wilson C, Koblan LW, Zeng J, Bauer DE, Doudna JA, Liu DR (2020) Phage-assisted evolution of an adenine base editor with improved Cas domain compatibility and activity. Nat Biotechnol 38:883–891. https://doi.org/10.1038/s41587-020-0453-z
Rojo FP, Nyman RKM, Johnson AAT, Navarro MP, Ryan MH, Erskine W, Kaur P (2018) CRISPR-cas systems: ushering in the new genome editing era. Bioengineered 9:214–221. https://doi.org/10.1080/21655979.2018.1470720
Ryu SM, Koo T, Kim K, Lim K, Baek G, Kim ST, Kim HS, Kim D, Lee H, Chung E, Kim JS (2018) Adenine base editing in mouse embryos and an adult mouse model of Duchenne muscular dystrophy. Nat Biotechnol 36(6):536–539. https://doi.org/10.1038/nbt.4148
Shapiro J, Iancu O, Jacobi AM, McNeill MS, Turk R, Rettig GR, Amit I, Tovin-Recht A, Yakhini Z, Behlke MA, Hendel A (2020) Increasing CRISPR efficiency and measuring its specificity in HSPCs using a clinically relevant system. Mol Ther - Methods Clin Dev 17:1097–1107. https://doi.org/10.1016/j.omtm.2020.04.027
Shmakov S, Abudayyeh OO, Makarova KS, Wolf YI, Gootenberg JS, Semenova E, Minakhin L, Joung J, Konermann S, Severinov K, Zhang F, Koonin EV (2015) Discovery and functional characterization of diverse class 2 CRISPR-Cas systems. Mol Cell 60:358–397. https://doi.org/10.1016/j.molcel.2015.10.008
Slaymaker IM, Gao L, Zetsche B, Scott DA, Yan WX, Zhang F (2016) Rationally engineered Cas9 nucleases with improved specificity. Science 351:84–88. https://doi.org/10.1126/science.aad5227
Stemmer M, Thumberger T, Del Sol Keyer M, Wittbrodt J, Mateo JL (2015) CCTop: an intuitive, flexible and reliable CRISPR/Cas9 target prediction tool. PLoS One 10:e0124633. https://doi.org/10.1371/journal.pone.0124633
Strecker J, Ladha A, Gardner Z, Schmid-Burgk JL, Makarova KS, Koonin EV, Zhang F (2019) RNA-guided DNA insertion with CRISPR-associated transposases. Science 364:48–53. https://doi.org/10.1126/science.aax9181
Szlachta K, Kuscu C, Tufan T, Adair SJ, Shang S, Michaels AD, Mullen MG, Fischer NL, Yang J, Liu L, Trivedi P, Stelow EB, Stukenberg PT, Parsons JT, Bauer TW, Adli M (2018) CRISPR knockout screening identifies combinatorial drug targets in pancreatic cancer and models cellular drug response. Nat Commun. https://doi.org/10.1038/s41467-018-06676-2
Tanenbaum ME, Gilbert LA, Qi LS, Weissman JS, Vale RD (2014) A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell 159:635–646. https://doi.org/10.1016/j.cell.2014.09.039
Thakore PI, D’Ippolito AM, Song L, Safi A, Shivakumar NK, Kabadi AM, Reddy TE, Crawford GE, Gersbach CA (2015) Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat Methods 12:1143–1149. https://doi.org/10.1038/nmeth.3630
Thuronyi BW, Koblan LW, Levy JM, Yeh WH, Zheng C, Newby GA, Wilson C, Bhaumik M, Shubina-Oleinik O, Holt JR, Liu DR (2019) Continuous evolution of base editors with expanded target compatibility and improved activity. Nat Biotechnol. https://doi.org/10.1038/s41587-019-0193-0
van der Oost J, Jore MM, Westra ER, Lundgren M, Brouns SJJ (2009) CRISPR-based adaptive and heritable immunity in prokaryotes. Trends Biochem Sci 34:401–407. https://doi.org/10.1016/j.tibs.2009.05.002
Walton RT, Christie KA, Whittaker MN, Kleinstiver BP (2020) Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants. Science 368:290–296. https://doi.org/10.1126/science.aba8853
Wang Y, Wang B, Xie H, Ren Q, Liu X, Li F, Lv X, He X, Cheng C, Deng R, Li J, Zhao J, Song Z, Gu F (2019) Efficient human genome editing using SaCas9 ribonucleoprotein complexes. Biotechnol J 14:e1800689. https://doi.org/10.1002/biot.201800689
Wang J, Zhang C, Feng B (2020) The rapidly advancing Class 2 CRISPR-Cas technologies: a customizable toolbox for molecular manipulations. J Cell Mol Med 24:3256–3270. https://doi.org/10.1111/jcmm.15039
Westra ER, Levin BR (2020) It is unclear how important CRISPR-Cas systems are for protecting natural populations of bacteria against infections by mobile genetic elements. Proc Natl Acad Sci USA 117:27777–27785
Xu Z, Li Y, Li M, Xiang H, Yan A (2021) Harnessing the type I CRISPR-Cas systems for genome editing in prokaryotes. Environ Microbiol 23:542–558. https://doi.org/10.1111/1462-2920.15116
Yamada M, Watanabe Y, Gootenberg JS, Hirano H, Ran FA, Nakane T, Ishitani R, Zhang F, Nishimasu H, Nureki O (2017) Crystal structure of the minimal Cas9 from Campylobacter jejuni reveals the molecular diversity in the CRISPR-Cas9 systems. Mol Cell 65:1109-1121.e3. https://doi.org/10.1016/j.molcel.2017.02.007
Yang H, Ren S, Yu S, Pan H, Li T, Ge S, Zhang J, Xia N (2020) Methods favoring homology-directed repair choice in response to CRISPR/Cas9 induced-double strand breaks. Int J Mol Sci 21:6461. https://doi.org/10.3390/ijms21186461
Zetsche B, Gootenberg JS, Abudayyeh OO, Slaymaker IM, Makarova KS, Essletzbichler P, Volz SE, Joung J, Van Der Oost J, Regev A, Koonin EV, Zhang F (2015) Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 163:759–771. https://doi.org/10.1016/j.cell.2015.09.038
Zetsche B, Heidenreich M, Mohanraju P, Fedorova I, Kneppers J, DeGennaro EM, Winblad N, Choudhury SR, Abudayyeh OO, Gootenberg JS, Wu WY, Scott DA, Severinov K, van der Oost J, Zhang F (2017) Multiplex gene editing by CRISPR–Cpf1 using a single crRNA array. Nat Biotechnol 35(1):31–34. https://doi.org/10.1038/nbt.3737
Zhang B (2020) CRISPR/Cas gene therapy. J Cell Physiol 236:2459–2481. https://doi.org/10.1002/jcp.30064
Zhang Y, Heidrich N, Ampattu BJ, Gunderson CW, Seifert HS, Schoen C, Vogel J, Sontheimer EJ (2013) Processing-independent CRISPR RNAs limit natural transformation in Neisseria meningitidis. Mol Cell 50:488–503. https://doi.org/10.1016/j.molcel.2013.05.001
Zhang Y, Ge X, Yang F, Zhang L, Zheng J, Tan X, Jin ZB, Qu J, Gu F (2014) Comparison of non-canonical PAMs for CRISPR/Cas9-mediated DNA cleavage in human cells. Sci Rep 4:5405. https://doi.org/10.1038/srep05405
Zhang X, Zhu B, Chen L, Xie L, Yu W, Wang Y, Li L, Yin S, Yang L, Hu H, Han H, Li Y, Wang L, Chen G, Ma X, Geng H, Huang W, Pang X, Yang Z, Wu Y, Siwko S, Kurita R, Nakamura Y, Yang L, Liu M, Li D (2020) Dual base editor catalyzes both cytosine and adenine base conversions in human cells. Nat Biotechnol 38:856–860. https://doi.org/10.1038/s41587-020-0527-y
Zhang M, Eshraghian EA, Jammal O, Al Zhang Z, Zhu X (2021) CRISPR technology: the engine that drives cancer therapy. Biomed Pharmacother. https://doi.org/10.1016/j.biopha.2020.111007
Zhao D, Li J, Li S, Xin X, Hu M, Price MA, Rosser SJ, Bi C, Zhang X (2021) Glycosylase base editors enable C-to-A and C-to-G base changes. Nat Biotechnol 39:35–40. https://doi.org/10.1038/s41587-020-0592-2
Zhou C, Sun Y, Yan R, Liu Y, Zuo E, Gu C, Han L, Wei Y, Hu X, Zeng R, Li Y, Zhou H, Guo F, Yang H (2019) Off-target RNA mutation induced by DNA base editing and its elimination by mutagenesis. Nature 571:275–278. https://doi.org/10.1038/s41586-019-1314-0
Zhu H, Misel L, Graham M, Robinson ML, Liang C (2016) CT-Finder: a web service for CRISPR optimal target prediction and visualization. Sci Rep 6:25516. https://doi.org/10.1038/srep25516
Acknowledgements
This work was supported by grants from the National Research Foundation of Korea [2021R1C1C1005851; T Koo] and the Ministry of Trade, Industry & Energy, South Korea [20012445; T Koo].
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original article was revised as the Fig. 2d was erroneously published. The original article has been corrected.
Rights and permissions
About this article

Cite this article
Song, M., Koo, T. Recent advances in CRISPR technologies for genome editing. Arch. Pharm. Res. 44, 537–552 (2021). https://doi.org/10.1007/s12272-021-01336-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12272-021-01336-4