
Abstract
In a recent past, transposable elements (TEs) were referred to as selfish genetic components only capable of copying themselves with the aim of increasing the odds of being inherited. Nonetheless, TEs have been initially proposed as positive control elements acting in synergy with the host. Nowadays, it is well known that TE movement into host genome comprises an important evolutionary mechanism capable of increasing the adaptive fitness. As insights into TE functioning are increasing day to day, the manipulation of transposition has raised an interesting possibility of setting the host functions, although the lack of appropriate genome engineering tools has unpaved it. Fortunately, the emergence of genome editing technologies based on programmable nucleases, and especially the arrival of a multipurpose RNA-guided Cas9 endonuclease system, has made it possible to reconsider this challenge. For such purpose, a particular type of transposons referred to as miniature inverted-repeat transposable elements (MITEs) has shown a series of interesting characteristics for designing functional drivers. Here, recent insights into MITE elements and versatile RNA-guided CRISPR/Cas9 genome engineering system are given to understand how to deploy the potential of TEs for control of the host transcriptional activity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Over the last decades, diverse genome engineering strategies manipulating the functioning of transposable elements (TE) have been intended to achieve site-specific integration of foreign DNA into host genomes. With this aim, the naturally occurring mechanism of transposition was exploited in animals to develop corresponding vectors that allow efficient gene transference and effective integration into genome (Szabo et al. 2003; Yant et al. 2007; Palazzoli et al. 2010). Moreover, the zinc-finger (ZF) and transcription activator-like effector (TALE) programmable nucleases, which can be efficiently redesigned to target specific genome sequences, have been also employed for the development of tools to efficiently achieve genome integration. ZF- and TALE-based constructs display important applications in genome engineering, reverse genetics, and targeting transgenic integration strategies (Walsh and Hochedlinger 2013; Bortesi and Fischer 2015; Cox et al. 2015). Finally, one RNA-guided system based on prokaryotic Type II clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated Cas9 endonuclease (CRISPR/Cas9) immune system has recently emerged as more efficient, easy to use, and low-cost genome editing tool able to produce desired genomic modifications (Cong et al. 2013; Ran et al. 2013; Lai et al. 2016) (Fig. 1).

CRISPR/Cas9 genome editing system. a CRISPR locus consists of one leader sequence (L) followed by the CRISPR array containing direct repeats (R) and spacers (S), and CRISPR-associated (Cas) genes. In natural type II CRISPR system, this locus is transcribed as a CRISPR RNA precursor (pre-crRNA) that is processed into mature CRISPR RNAs (crRNAs) molecules which are then bound to transacting CRISPR RNAs (tracrRNAs) to direct targeted Cas9-mediated Double Strand Breaks (DSBs) in complementary crRNA sequences. Thus, the natural CRISPR system is capable of defending the host against infection of matching crRNA pathogenic sequences. b In CRISPR/Cas9 genome engineering, a synthetic single-guide RNA (sgRNA) that combines the crRNA and tracrRNA is employed to mediate Cas9 DSB cleavage at targeted genomic sites which are subsequently repaired via non-homologous end-joining (NHEJ) or homology-directed (HR) competing cellular DNA repair pathways. By HR-mediated repair, it is possible to achieve targeted integration of desired templates and induce site-specific genome changes. The only requirement of the system is one short Cas9 recognition site known as Protospacer-Adjacent Motif (PAM). c Alternative Cas9-based chimeric fusion systems can be also engineered to achieve efficient transcriptional targeting, site-specific epigenetic modifications, cell imagining, etc
The CRISPR/Cas modules are adaptive host defense systems that protect archaea and bacteria against the invasion of viruses and plasmids (Jinek et al. 2012). The CRISPR locus was initially identified in Escherichia coli (Ishino et al. 1987) and the CRISPR/Cas modules were further characterized by Jansen et al. (2002), who identified CRISPR and CRISPR-associated (cas) genes in more than 40 species of prokaryotes. CRISPR/Cas systems can be divided into three main types (I, II, and III) and ten subtypes (Makarova et al. 2011). The CRISPR/Cas9 type II genome editing system uses a synthetic single-guide RNA (sgRNA) to produce targeted Cas9 DNA double strand breaks (DSBs) that are subsequently repaired by endogenous host mechanisms (Jinek et al. 2012; Doudna and Charpentier 2014). As a consequence of this, the DSBs generated are rejoined by either non-homologous end-joining (NHEJ) or homologous recombination (HR) pathways (see Fig. 1b). Depending on the repair mechanism activated, site-specific modifications involving gene disruption, gene replacement, or nucleotide substitution may be effectively generated. NHEJ is prone to produce indel mutations at target sites, while by use of DNA templates harboring desired sequences, it is possible to generate specific genome modifications by activation of HR-mediated repair (Sakuma and Yamamoto 2015). Alternative genome engineering strategies consisting of a nuclease-deactivated Cas9 (dCas9) fused with modular transcriptional domains (activators and/or repressors), chromatin remodelers, and fluorophores also enable efficient transcriptional control, site-specific chromatin modifications, and visualization of loci, respectively (Li et al. 2016; see Fig. 1c). The RNA-guided CRISPR/Cas9 system has dramatically improved our ability to edit the genome of many organisms, thereby being increasingly employed in biotechnology and therapeutics.
Transposable elements and the CRISPR/Cas9 genome engineering system: control of the host genome
The RNA-guided CRISPR/Cas9 system for design of homing endonuclease gene drives
Diverse endogenous genome elements including, among others, homing endonuclease genes (HEG), and transposable elements (TEs) are capable of exploiting the host machinery to increase the odds of being inherited. Such elements were previously referred to as “gene drives”, or parasitic genome players able to spread in the host genome by copying themselves into target sequences (Burt 2003; Adelman and Tu 2016). Since the beginning, it was believed that these elements might be used to design effective genome engineering strategies that allow coopting endogenous host molecular mechanisms. With this aim, Burt (2003) suggested an interesting, multi-talent, and promising procedure based on engineered HEG drives which would allow addressing the host cell machinery and control gene expression (Fig. 2); however, several technical constraints hindered effective design. Even though Burt’s proposal had not been implemented, his idea predicted the development of future applications. Fortunately, the emergence of the adaptable CRISPR/Cas9 genome editing system has overcome methodological restrictions, thereby Cas9-based drives were already successively engineered to bias inheritance in favor of particular fruit fly (Gantz and Bier 2015) and yeast (DiCarlo et al. 2015; see Fig. 2b) drive constructs which exhibited remarkable high transmission rates (97 and 99%, respectively). Recent genome engineering strategies based on CRISPR/Cas9-mediated gene drives have demonstrated having potential to address the evolution of natural populations (Hammond et al. 2016), whereby displaying the outstanding power that these emerging technologies possess and, besides, underlying an imperative need for public debate before each effective use (Esvelt et al. 2014).

Endonuclease-based gene drive systems. a Burt’s proposal (2003) based on targeted homing endonuclease gene (HEG) drives. Genome engineering HEG drives may spread in the genome host via targeted integration into recognition sites (light blue), thereby activating recombination while simultaneously preventing the cleavage of chromosomes that carry them. Following site-specific HEG insertion, HEG-based gene drives are expected to be able to modify host cellular functions (in this case, causing loss of function by gene disruption). b Biased inheritance of a CRISPR/Cas9 gene drive system in S. cerevisiae (Di Carlo et al. 2015). Gametes carrying a drive construct targeting the gene encoding the phosphoribosylaminoimidazole carboxylase (ADE2) enzyme mate with wild-type haploid cells and thus generate CRISPR/Cas9-based DSBs which are subsequently repaired by HR (gene replacement) or NHEJ (gene disruption) pathways. CRISPR/Cas9 genome editing can be monitored in the progeny by loss of function of ADE2. Blue box DNA sequence encoding sgRNA (single-guide RNA), Wt Chr wild-type chromosome, Gd Chr gene drive chromosome, HR homologous recombination, NHEJ non-homologous end-joining
CRISPR spacer acquisition and insertion of transposable elements: themes in common
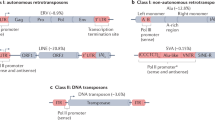
Several parallelisms between the CRISPR/Cas9 system and the eukaryotic RNA interference (RNAi) mechanism have been interestingly discovered (Mojica et al. 2005; Bolotin et al. 2005; Barrangou et al. 2015). In nature, CRISPR loci contain arrays of direct repeats (DR) associated with spacers sequences which likely derived from bacteriophages or plasmids (see Fig. 1a). The CRISPR DRs act as Cas9-mediated cleavage sites, whereas invasion-acquired spacers mediate immunologic responses to host reinvasion, thereby mimicking the eukaryote post-transcriptional gene silencing (PTGS) pathway mediated by small RNAs (sRNAs). Likewise CRISPR DRs, TEs also are flanked by cleavage sites recognized by particular enzymes (transposases) that mediate their own transposition. Essentially, there are two types of TE flanking sequences referred to as long terminal repeats (LTRs) or terminal inverted repeats (TIRs), belonging to elements which are classified into Class I and Class II, respectively. TEs are categorized according to their transposition intermediates into RNA-mediated (Class I) and DNA-mediated (Class II) elements, which are transposed through “copy-and-paste” and “cut-and-paste” mechanisms, respectively (for review, see Casacuberta and Santiago 2003). Remarkably, flanking CRISPR DRs derived from insertions of particular Class II TEs known as miniature inverted-repeat transposable elements (MITEs) have been discovered (Mai et al. 2016). MITEs are non-autonomous DNA (Class II) TEs usually composed of ~ 100–800 pb, flanked by TIRs and adjacent to target site duplications (TSDs) (Fig. 3). Resembling the functioning of CRISPR systems during bacteriophage infection, MITEs also can generate sRNAs that mediate the RNAi silencing pathway during stress responses and environmental (hormone) signals (Yan et al. 2011). Indeed, the existence of MITE-derived CRISPR DRs indicates that site-specific TE insertions have contributed to evolution of CRISPR arrays (Mai et al. 2016). Diverse types of TEs exhibit preference insertional targets, whereby hot spots for many TE families have been correspondingly characterized (Castilho and Casadaban 1991; Feng et al. 2013). In the case of plant MITEs, these elements often exhibit preferential transposition into both AT dinucleotide and ATT trinucleotide genome signatures (Jiang and Wessler 2001; Jiang et al. 2003).

Miniature inverted-repeat transposable elements (MITEs). MITE elements contain terminal inverted repeats (TIRs, triangles) adjacent to target site duplications (TSDs, arrows). It is believed that MITEs were originated from deletion derivatives of Class II TEs that lost the ability to drive transposition (i.e., transposases and functional enzymes), whereas maintaining the transpositional activity. MITE-derived sequences may not contain perfect TIRs or even lose the TDSs. An amplification burst of MITEs can dramatically increase the number of these elements in one host genome. E1 and E2 enzymes that mediate transposition, T transposase
The transposition mechanism by which TEs are scattered into host genome challenged the mistaken concept that considered the genome as one fixed, immutable entity (Mourier 2016). Nowadays, it is well known that transposition comprises an important adaptive mechanism capable of providing both hereditable and non-hereditable variability, source of critical phenotypic plasticity. Under particular environmental conditions, active transposases mediate transposition and this may positively affect host gene expression by providing promoter sequences (Jordan et al. 2003; Feschotte 2008), modifying gene expression patterns (Cowley and Oakey 2013; Kim et al. 2015), changing local chromatin structure (Hollister and Gaut 2009), etc., thereby potentially resulting in adaptive phenotypes. For example, Class I retrotransposons sequences are activated during human neuronal differentiation and consequent amplification triggers chromosome arrangements capable of conferring somatic plasticity (Singer et al. 2010; Baillie et al. 2011). In maize, Qüesta and colleagues (2010) proposed that UV-B-radiation induces the transposition via modulation of chromatin structure and thus generates variation in the genome. Moreover, in Arabidopsis, it has been showed that stress heat conditions induce the transposition of the ONSEN (Class I) copia-like retrotransposon and TE accumulation was encouraged in plant small intereference RNA (siRNA) deficient mutants (Matsunaga et al. 2012), whereby evidencing the critical role of RNAi during stress-mediated transposition. The eukaryotic RNAi machinery functions to epigenetically regulate the insertion of TEs by sequence-specific mechanisms and thus maintain genome stability (Buchon and Vaury 2006). Indeed, TE mobilization and epigenetic gene regulation mechanisms are deeply interconnected to each other, and nowadays, it is well known that transposition may trigger epigenetic modifications associated with more adaptable stress-tolerant phenotypes (Yang et al. 2005; Hou et al. 2012; Castelletti et al. 2014).
Control of host transcriptional activity by TE-based drives
The targeted insertional mutagenesis has emerged as an important strategy for deciphering the gene function by inducing large-scale mutations into genomic loci of interest. In cancer modeling, DNA Sleeping Beauty TE-based systems have been used to induce somatic-specific mutagenesis and, therefore, to identify essential genes involved in tumorgenesis (Molyneux et al. 2014). In plants, a transposon-tagging tool for genome-wide analysis based on the rice mPing MITEs was used in transgenic soybean (Hancock et al. 2011), showing this system preferential insertion for both nearby genes and AT-rich sequences. Hancock et al. (2011) observed an increased transpositional activity during specific developmental stages (cotyledon vs. globular stage), suggesting interestingly that insights into the developmental regulation pathways involved might be used to control transposition. Both results indicate how the genome fluidity can be efficiently manipulated via TE-based systems. In this sense, new molecular breeding strategies based on the induced activation of Class I retrotransposons have also already been suggested (Paszkowski 2015). TEs represent an important source of phenotype plasticity which is particularly interesting in view of the potentiality for addressing transposition through application of external stimuli. As a consequence of this, we are currently able to engineer TE-based strategies to induce transcriptional control of target genes (Fig. 4). For such purpose, the versatile CRISPR/Cas9 system can be used to mediate HR-based genome editing and thus to introduce transposition insertion sites upstream target genes whose transcriptional regulation is desired. Alternatively, RNA-guided genome engineering constructs based on the use of Cas9 fused with transposase enzymes might be likewise employed to direct the insertion of target sequences. In this regard, targeted transposition using vector delivery systems consisting of the piggyBac transposase fused with ZF/TALE nucleases has already been reported (Kettlun et al. 2011; Li et al. 2013; Owens et al. 2013).

MITE-based strategy to drive the transcription of target genes. The HR-mediated CRISPR/Cas9 genome editing system can be employed to generate MITE hotspots (in this case consisting of AT/ATT nucleotide signatures). Diverse environmental stimuli are capable of trigger selective amplification and movement of particular MITEs, which are thus accordingly integrated into CRISPR-targeted loci by taking advantage of the preferential insertion at genome-engineered hotspots. Such strategy makes use of the dual essential property of MITEs to either up or downregulate the transcription of nearby genes corresponding to loci in which they are hosted. References: open reading frame (ORF), yellow; MITE, light blue
Miniature inverted repeat transposable elements (MITEs) and their interactions with the host genome
To design effective TE-based genome engineering strategies for control of host cell functions, insights into the particular type of TE should be mandatory. In this work, MITE elements are described, since these Class II DNA transposons exhibit a series of characteristics that make them especially suitable for designing TE-based drives. (1) MITEs are abundant repeat elements in eukaryote genomes and they have played critical roles during genome evolution (Lu et al. 2012; Ye et al. 2016). For example, MITE elements represent the most usual type of TEs in rice genome (Jiang et al. 2004). (2) Insertion of MITEs can both upregulate and downregulate the expression of nearby genes (Chen et al. 2014). (3) Many active MITE families exist, among others, mPing and mGing in rice (Jiang et al. 2003; Dong et al. 2012), Stowaway in potato (Momose et al. 2010), and AhMITE1 in peanut (Shirasawa et al. 2012). (4) Particular MITE families have been shown to mobilize during cell differentiation under certain conditions (Dong et al. 2012). (5) MITEs are preferentially inserted close to genes (Lu et al. 2012; Wessler et al. 1995). For instance, the Tourist-like and Stowaway-like plant MITE superfamilies show target site preference for integration into specific gene sequences (Jiang et al. 2004). (6) Since MITEs do not possess the enzymes required for their own transposition, they are usually shorter than autonomous DNA TEs (Jiang and Wessler 2001), thereby being more easily manipulated for designing efficient TE-based genome engineering strategies.
Although different families of MITEs have been identified in plants and animals, their origins are still unclear. It has been proposed that MITEs might have derived from ancestral DNA transposons which gave rise to related autonomous elements whose activity eventually resulted in the generation of non-autonomous deletion derivatives elements. Subsequently, the amplification of these derivative elements would give rise to a collection of homogeneous non-autonomous TEs, e.g., a MITE subfamily (Feschotte et al. 2002a; see Fig. 3). This idea is supported by the sequence homology between MITEs and DNA transposons; however, most MITEs do not exhibit significant homology with previously characterized elements beyond the TIR sequences (Feschotte et al. 2002b). Similarly, mechanisms that trigger the temporal activation of MITEs via trans-acting transposases and/or “genome shock” phenomena such as, for example, cell culture or hybridization, remain to be elucidated (Lu et al. 2012).
In silico analyses show that MITE sequences are involved in RNA-based gene regulation, either by hairpin-like miRNA precursors (pre-miRNA) (Lorenzetti et al. 2016) or by siRNA biogenesis (Kuang et al. 2009). Interestingly, it has been recently demonstrated that sRNAs can be used by cell machinery for targeted gene expression control, not only downregulating but also upregulating the transcriptional activity (Li et al. 2006; Turner et al. 2014). Alternatively, insertion of MITEs can mediate regulation of gene expression by promoting the establishment of epigenetic modifications such as, for example, DNA and both histone methylation and acetylation marks. The currently active mPing MITEs represent a clear example of how TEs may act synergistically with the host modulating its functioning. The mPing family is present in high copies in the rice genome and exhibits preferential insertion into AT-gene-rich regions, avoiding exons while simultaneously choosing promoter regions. These elements are able to both upregulate or downregulate the expression of genes according to the localization of the insertion, although more than 80% of the mPing insertions did not exhibit detectable effects on the expression of nearby genes (Naito et al. 2009). Interestingly, Naito and colleagues (2014) proposed that active mPing elements should be benign to their hosts, because the consequent mPing amplification resulted in selective gene expression control, which is particularly useful in view of manipulating transcription of target genes. In crops, MITEs can modulate the transcriptional activity of essential genes and, therefore, MITE insertion represents an important source of genetic variation worthy to be considered for improvement of agronomic traits (Zhang et al. 2000; Patel et al. 2004; Yang et al. 2005; Li et al. 2014; Mao et al. 2015; Vaschetto 2016).
Concluding remarks
Most eukaryotic genomes are littered with TEs, and nowadays, it is well known that these elements have played critical roles in genome evolution (Casacuberta and Santiago 2003; Garfinkel et al. 2016). Although their mode of action resembles to selfish parasitic elements, TE dispersal may also be beneficial for hosts. Recent insights into the biology of TEs retake an initial position which postulated that environment-induced TE dispersion represent an important adaptive mechanism (McClintock 1984), and nowadays, it is recognized as a regulatory mechanism for both allocating gene motifs and genome sequences involved in the establishment of epigenetic profiles. It is worthy of note that amplification of TEs induced under certain conditions can offer significant benefits to the host by modulating expression of non-linked genes involved in the same gene regulatory pathways, even having potential for generating de novo regulatory networks (Feschotte 2008).
Transposon-based vectors are very useful and effective tools for achieving targeted mutagenesis (gene disruption/replacement). The emergence of versatile CRISPR/Cas9 genome editing system has enabled to overcome long-standing technical limitations to give the next step towards the design of more efficient TE-based targeting systems. Moreover, the preferential integration induced through application of particular stress/environmental treatments represents a key milestone to maximize the versatility of such systems, since it allows the exploitation/use at will of the natural potential that TEs have to control the host. As it is stated before, MITEs exhibit a series of well-defined characteristics that make them an appropriate tool for engineering of TE-based systems. In particular, the mPing MITE family from rice genome have shown to be useful for achieving this objective, since the movement of mPing elements can be activated by application of different treatments such as cell culture (Jiang et al. 2003; Kikuchi et al. 2003), hybridization (Shan et al. 2005), or pressurization (Lin et al. 2006). In addition, since it has been demonstrated that transgenic mPing-based transposon-tagging systems remain active in the soybean genome (Hancock et al. 2011), the functional characterization of mPing family would also allow designing effective genome engineering strategies involving not only endogenous but also exogenous control of the host cell machinery.
Diverse host functioning mechanisms are controlled by TEs, while efficient CRISPR/Cas9 technologies can be used to deploy the potential that these elements have to influence cellular activity of the host. In crops, the climatic fluctuations associated with global warming require the design of molecular breeding strategies tailored to optimize the plant development under such scenarios of change (Hou et al. 2012); thereby, it has already been suggested that TE amplification may represent a solution for generating functional genetic diversity in the face of ever-changing environments (Naito et al. 2009). Moreover, nowadays, we are able to engineer CRISPR/Cas9-targeted loci to efficiently achieve site-specific TE integration, thereby allowing regulation of target genes by exploiting intrinsic TE capabilities for controlling transcription. In addition, since it is well known that TEs are associated with control of different host genome mechanisms including telomere maintenance, chromosomal rearrangements, gene duplication, and epigenetic regulation, analogous strategies can be eventually developed to manipulate such important functions. Consequently, the emergence of versatile CRISPR/Cas9 genome editing tool raises TE-based strategies to endogenously coopt the host mechanisms, being therefore undoubtedly beneficial for the development of diverse biological applications.
References
Adelman ZN, Tu Z (2016) Control of mosquito-borne infectious diseases: sex and gene drive. Trends Parasitol 32:219–229
Baillie JK, Barnett MW, Upton KR, Gerhardt DJ, Richmond TA, De Sapio F, Talbot RT (2011) Somatic retrotransposition alters the genetic landscape of the human brain. Nature 479:534–537
Barrangou R, Birmingham A, Wiemann S, Beijersbergen RL, Hornung V, van Brabant Smith A (2015) Advances in CRISPR-Cas9 genome engineering: lessons learned from RNA interference. Nucleic Acids Res 43:3407–3419
Bolotin A, Quinquis B, Sorokin A, Ehrlich SD (2005) Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology 151:2551–2561
Bortesi L, Fischer R (2015) The CRISPR/Cas9 system for plant genome editing and beyond. Biotechnol Adv 33:41–52
Buchon N, Vaury C (2006) RNAi: a defensive RNA-silencing against viruses and transposable elements. Heredity 96:195
Burt A (2003) Site-specific selfish genes as tools for the control and genetic engineering of natural populations. Proc Biol Sci 270:921–928
Casacuberta JM, Santiago N (2003) Plant LTR-retrotransposons and MITEs: control of transposition and impact on the evolution of plant genes and genomes. Gene 311:1–11
Castelletti S, Tuberosa R, Pindo M, Salvi S (2014) A MITE transposon insertion is associated with differential methylation at the maize flowering time QTL Vgt1. G3 4:805–812
Castilho BA, Casadaban MJ (1991) Specificity of mini-Mu bacteriophage insertions in a small plasmid. J Bacteriol 173:1339–1343
Chen J, Hu Q, Lu C, Kuang H (2014) Evolutionary genomics of miniature inverted-repeat transposable elements (MITEs) in plants. In: Pontarotti P (ed) Evolutionary biology: genome evolution, speciation, coevolution and origin of life. Springer, Cham, pp 157–168
Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Zhang F (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339:819–823
Cowley M, Oakey RJ (2013) Transposable elements re-wire and fine-tune the transcriptome. PLoS Genet 9:e1003234
Cox DBT, Platt RJ, Zhang F (2015) Therapeutic genome editing: prospects and challenges. Nat Med 21:121–131
DiCarlo JE, Chavez A, Dietz SL, Esvelt KM, Church GM (2015) Safeguarding CRISPR-Cas9 gene drives in yeast. Nat Biotechnol. doi:10.1038/nbt.3412
Dong HT, Zhang L, Zheng KL, Yao HG, Chen J, Yu FC, Li DB (2012) A Gaijin-like miniature inverted repeat transposable element is mobilized in rice during cell differentiation. BMC Genom 13:135
Doudna JA, Charpentier E (2014) The new frontier of genome engineering with CRISPR-Cas9. Science 346:1258096
Esvelt KM, Smidler AL, Catteruccia F, Church GM (2014) Concerning RNA-guided gene drives for the alteration of wild populations. Elife 3:e03401
Feng G, Leem YE, Levin HL. 2013. Transposon integration enhances expression of stress response genes. Nucleic Acids Res 41:775–789
Feschotte C (2008) Transposable elements and the evolution of regulatory networks. Nat Rev Genet 9:397–405
Feschotte C, Jiang N, Wessler SR (2002a) Plant transposable elements: where genetics meets genomics. Nat Rev Genet 3:329
Feschotte C, Zhang X, Wessler S (2002b) Miniature inverted-repeat transposable elements (MITEs) and their relationship with established DNA transposons. In: Craig NL, Craigie R, Gellert M, Lambowitz AM (eds) Mobile DNA II, vol 35. American Society for Microbiology Press, Washington, DC, pp 1147–1158
Gantz VM, Bier E (2015) The mutagenic chain reaction: a method for converting heterozygous to homozygous mutations. Science 348:442–444
Garfinkel DJ, Tucker JM, Saha A, Nishida Y, Pachulska-Wieczorek K, Błaszczyk L, Purzycka KJ (2016) A self-encoded capsid derivative restricts Ty1 retrotransposition in Saccharomyces. Curr Genet 62:321–329
Hammond A, Galizi R, Kyrou K, Simoni A, Siniscalchi C, Katsanos D, …Burt A (2016) A CRISPR-Cas9 gene drive system targeting female reproduction in the malaria mosquito vector Anopheles gambiae. Nat Biotechnol 34:78–83
Hancock CN, Zhang F, Floyd K, Richardson AO, LaFayette P, Tucker D, Parrott WA (2011) The rice miniature inverted repeat transposable element mPing is an effective insertional mutagen in soybean. Plant Physiol 157:552–562
Hollister JD, Gaut BS (2009) Epigenetic silencing of transposable elements: a trade-off between reduced transposition and deleterious effects on neighboring gene expression. Genome Res 19:1419–1428
Hou J, Long Y, Raman H, Zou X, Wang J, Dai S, …Meng J (2012) A tourist-like MITE insertion in the upstream region of the BnFLC. A10 gene is associated with vernalization requirement in rapeseed (Brassica napus L.). BMC Plant Biol 12:1
Ishino Y, Shinagawa H, Makino K, Amemura M, Nakata A (1987) Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J Bacteriol 169:5429–5433
Jansen R, Embden J, Gaastra W, Schouls L (2002) Identification of genes that are associated with DNA repeats in prokaryotes. Mol Microbiol 43:1565–1575
Jiang N, Wessler SR (2001) Insertion preference of maize and rice miniature inverted repeat transposable elements as revealed by the analysis of nested elements. Plant Cell 13:2553–2564
Jiang N, Bao Z, Zhang X, Hirochika H, Eddy SR, McCouch SR, Wessler SR (2003) An active DNA transposon family in rice. Nature 421:163–167
Jiang N, Feschotte C, Zhang X, Wessler SR (2004) Using rice to understand the origin and amplification of miniature inverted repeat transposable elements (MITEs). Curr Opin Plant Biol 7:115–119
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337:816–821
Jordan IK, Rogozin IB, Glazko GV, Koonin EV (2003) Origin of a substantial fraction of human regulatory sequences from transposable elements. Trends Genet 19:68–72
Kettlun C, Galvan DL, George AL, Kaja A, Wilson MH (2011) Manipulating piggyBac transposon chromosomal integration site selection in human cells. Mol Ther 19:1636–1644
Kikuchi K, Terauchi K, Wada M, Hirano HY (2003) The plant MITE mPing is mobilized in anther culture. Nature 421:167–170
Kim KD, El Baidouri M, Abernathy B, Iwata-Otsubo A, Chavarro C, Gonzales M, Jackson SA (2015) A comparative epigenomic analysis of polyploidy-derived genes in soybean and common bean. Plant Physiol 168:1433–1447
Kuang H, Padmanabhan C, Li F, Kamei A, Bhaskar PB, Ouyang S, Baker B (2009) Identification of miniature inverted-repeat transposable elements (MITEs) and biogenesis of their siRNAs in the Solanaceae: new functional implications for MITEs. Genome Res 19:42–56
Lai WC, Sun HFS, Lin PH, Lin H, Shieh JC (2016) A new rapid and efficient system with dominant selection developed to inactivate and conditionally express genes in Candida albicans. Curr Genet 62:213–235
Li LC, Okino ST, Zhao H, Pookot D, Place RF, Urakami S, Enokida H, Dahiya R (2006) Small dsRNAs induce transcriptional activation in human cells. Proc Natl Acad Sci USA 103:17337–17342
Li X, Burnight ER, Cooney AL, Malani N, Brady T, Sander JD, Bushman FD (2013) piggyBac transposase tools for genome engineering. Proc Natl Acad Sci USA 110:E2279–E2287
Li J, Wang Z, Peng H, Liu Z (2014) A MITE insertion into the 3′-UTR regulates the transcription of TaHSP16. 9 in common wheat. Crop J 2:381–387
Li X, Wu R, Ventura A (2016) The present and future of genome editing in cancer research. Hum Genet 135:1083–1092
Lin X, Long L, Shan X, Zhang S, Shen S, Liu B (2006) In planta mobilization of mPing and its putative autonomous element Pong in rice by hydrostatic pressurization. J Exp Bot 57:2313–2323
Lorenzetti AP, de Antonio GY, Paschoal AR, Domingues DS (2016) PlanTE-MIR DB: a database for transposable element-related microRNAs in plant genomes. Funct Integr Genom 16:235–242
Lu C, Chen J, Zhang Y, Hu Q, Su W, Kuang H (2012) Miniature inverted-repeat transposable elements (MITEs) have been accumulated through amplification bursts and play important roles in gene expression and species diversity in Oryza sativa. Mol Biol Evol 29:1005–1017
Mai G, Ge R, Sun G, Meng Q, Zhou F (2016) A comprehensive curation shows the dynamic evolutionary patterns of prokaryotic CRISPRs. Biomed Res Int. doi:10.1155/2016/7237053
Makarova KS, Haft DH, Barrangou R, Brouns SJ, Charpentier E, Horvath P, Van Der Oost J (2011) Evolution and classification of the CRISPR-Cas systems. Nat Rev Microbiol 9:467
Mao H, Wang H, Liu S, Li Z, Yang X, Yan J, Qin F (2015) A transposable element in a NAC gene is associated with drought tolerance in maize seedlings. Nat Commun 6:8326
Matsunaga W, Kobayashi A, Kato A, Ito H (2012) The effects of heat induction and the siRNA biogenesis pathway on the transgenerational transposition of ONSEN, a copia-like retrotransposon in Arabidopsis thaliana. Plant Cell Physiol 53:824–833
McClintock B (1984) The significance of responses of the genome to challenge. Science 226:792–801
Mojica FJM, García-Martínez J, Soria E (2005) Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J Mol Evol 60:174–182
Molyneux SD, Waterhouse PD, Shelton D, Shao YW, Watling CM, Tang QL, Zhang X (2014) Human somatic cell mutagenesis creates genetically tractable sarcomas. Nat Genet 46:964–972
Momose M, Abe Y, Ozeki Y (2010) Miniature inverted-repeat transposable elements of Stowaway are active in potato. Genetics 186:59–66
Mourier T (2016) Potential movement of transposable elements through DNA circularization. Curr Genet 62:697–700
Naito K, Zhang F, Tsukiyama T, Saito H, Hancock CN, Richardson AO, Wessler SR (2009) Unexpected consequences of a sudden and massive transposon amplification on rice gene expression. Nature 461:1130–1134
Naito K, Monden Y, Yasuda K, Saito H, Okumoto Y (2014) mPing: the bursting transposon. Breeding Sci 64:109–114
Owens JB, Mauro D, Stoytchev I, Bhakta MS, Kim MS, Segal DJ, Moisyadi S (2013) Transcription activator like effector (TALE)-directed piggyBac transposition in human cells. Nucleic Acids Res 41:9197–9207
Palazzoli F, Testu FX, Merly F, Bigot Y (2010) Transposon tools: worldwide landscape of intellectual property and technological developments. Genetica 138:285–299
Paszkowski J (2015) Controlled activation of retrotransposition for plant breeding. Curr Opin Biotechnol 32:200–206
Patel M, Jung S, Moore K, Powell G, Ainsworth C, Abbott A (2004) High-oleate peanut mutants result from a MITE insertion into the FAD2 gene. Theor Appl Genet 108:1492–1502
Qüesta JI, Walbot V, Casati P (2010) Mutator transposon activation after UV-B involves chromatin remodeling. Epigenetics 5:352–363
Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, Zhang F (2013) Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154:1380–1389
Sakuma T, Yamamoto T (2015) CRISPR/Cas9: the leading edge of genome editing technology. Targeted genome editing using site-specific nucleases. Springer, Tokyo, pp 25–41
Shan X, Liu Z, Dong Z, Wang Y, Chen Y, Lin X, Liu B (2005) Mobilization of the active MITE transposons mPing and Pong in rice by introgression from wild rice (Zizania latifolia Griseb.). Mol Biol Evol 22:976–990
Shirasawa K, Hirakawa H, Tabata S, Hasegawa M, Kiyoshima H, Suzuki S, Isobe S (2012) Characterization of active miniature inverted-repeat transposable elements in the peanut genome. Theor Appl Genet 124:1429–1438
Singer T, McConnell MJ, Marchetto MC, Coufal NG, Gage FH (2010) LINE-1 retrotransposons: mediators of somatic variation in neuronal genomes? Trends Neurosci 33:345–354
Szabo M, Müller F, Kiss J, Balduf C, Strähle U, Olasz F (2003) Transposition and targeting of the prokaryotic mobile element IS30 in zebrafish. FEBS Lett 550:46–50
Turner M, Jiao A, Slack FJ (2014) Autoregulation of lin-4 microRNA transcription by RNA activation (RNAa) in C. elegans. Cell Cycle 13:772–781
Vaschetto LM (2016) Miniature inverted-repeat transposable elements (MITEs) and their effects on the regulation of major genes in cereal grass genomes. Mol Breed 36:1–4
Walsh RM, Hochedlinger K (2013) A variant CRISPR-Cas9 system adds versatility to genome engineering. Proc Natl Acad Sci USA 110:15514–15515
Wessler SR, Bureau TE, White SE (1995) LTR-retrotransposons and MITEs: important players in the evolution of plant genomes. Curr Opin Genet Dev 5:814–821
Yan Y, Zhang Y, Yang K, Sun Z, Fu Y, Chen X, Fang R (2011) Small RNAs from MITE-derived stem-loop precursors regulate abscisic acid signaling and abiotic stress responses in rice. Plant J 65:820–828
Yang G, Lee YH, Jiang Y, Shi X, Kertbundit S, Hall TC (2005) A two-edged role for the transposable element Kiddo in the rice ubiquitin2 promoter. Plant Cell 17:1559–1568
Yant SR, Huang Y, Akache B, Kay MA (2007) Site-directed transposon integration in human cells. Nucleic Acids Res 35:e50
Ye C, Ji G, Liang C (2016) detectMITE: a novel approach to detect miniature inverted repeat transposable elements in genomes. Sci Rep 6:19688
Zhang Q, Arbuckle J, Wessler SR (2000) Recent, extensive, and preferential insertion of members of the miniature inverted-repeat transposable element family Heartbreaker into genic regions of maize. Proc Natl Acad Sci USA 97:1160–1165
Acknowledgements
The author would like to thank Marcela Rivarola and Cristian Arellano for grammatical corrections. Also, I wish to thank Jimena Ingaramo who provided support in the preparation of the figures.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declares that has no conflict of interest.
Additional information
Communicated by M. Kupiec.
Rights and permissions
About this article

Cite this article
Vaschetto, L.M. Modulating signaling networks by CRISPR/Cas9-mediated transposable element insertion. Curr Genet 64, 405–412 (2018). https://doi.org/10.1007/s00294-017-0765-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00294-017-0765-9