
Abstract
The renin-angiotensin system (RAS) is a fundamental regulator of blood pressure and has emerged as an important player in the control of inflammatory processes. Accordingly, imbalance on RAS components either systemically or locally might trigger the development of inflammatory disorders by affecting immune cells. At the same time, alterations in the dopaminergic system have been consistently involved in the physiopathology of inflammatory disorders. Accordingly, the interaction between the RAS and the dopaminergic system has been studied in the context of inflammation of the central nervous system (CNS), kidney, and intestine, where they exert antagonistic actions in the regulation of the immune system. In this review, we summarized, integrated, and discussed the cross talk of the dopaminergic system and the RAS in the regulation of inflammatory pathologies, including neurodegenerative disorders, such as Parkinson’s disease. We analyzed the molecular mechanisms underlying the interaction between both systems in the CNS and in systemic pathologies. Moreover, we also analyzed the impact of the commensal microbiota in the regulation of RAS and dopaminergic system and how it is involved in inflammatory disorders. Furthermore, we summarized the therapeutic approaches that have yielded positive results in preclinical or clinical studies regarding the use of drugs targeting the RAS and dopaminergic system for the treatment of inflammatory conditions. Further understanding of the molecular and cellular regulation of the RAS-dopaminergic cross talk should allow the formulation of new therapies consisting of novel drugs and/or repurposing already existing drugs, alone or in combination, for the treatment of inflammatory disorders.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
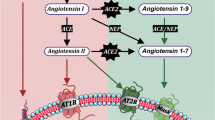
Inflammatory diseases include a huge number of pathologies that involve the perturbation immune homeostasis, causing systemic or local inflammation in the organism. These disorders represent a serious issue world-wide, involving life-long debilitating symptoms. Their incidence in the western hemisphere has enhanced in the last decade due to increased life span, environmental pollutants, lifestyle, and changes in diet, reaching about 5–7% among its inhabitants and affecting almost 24 million people in the USA [1]. Nevertheless, those statistics are based in a list of 80 pathologies classified as inflammatory disorders, which excludes many diseases with an inflammatory component, such as neurodegenerative and cardiovascular diseases. Importantly, genetic and environmental factors may contribute to the development and progression of inflammatory disorders, including the renin-angiotensin system (RAS) and the catecholaminergic system, among others [2,3,4,5,6]. The RAS is composed by a group of hormones and enzymes that together regulate both blood pressure and inflammation (Fig. 1). The system consists on a molecule called angiotensinogen, which is secreted by the liver into the blood stream. Angiotensinogen is then converted into angiotensin I (AngI) by the action of renin, and in turn, AngI might be converted into angiotensin II (AngII) in a reaction catalyzed by the angiotensin-converting enzyme 1 (ACE1). AngII has the ability to stimulate type 1 AngII receptors (ATR1) as well as type 2 AngII receptor (ATR2). The stimulation of ATR1, which is expressed in the kidneys, blood vessels, heart, and central nervous system (CNS), promotes increased blood pressure and favors the development of inflammation [7]. Conversely, stimulation of ATR2, which is expressed in kidney, adrenal gland, and the brain, induces anti-inflammatory effects and reduces blood pressure [7]. In addition, AngII might be further processed by angiotensin-converting enzyme 2 (ACE2) to yield a peptide called angiotensin (1–7) (Ang(1–7)). Ang(1–7) in turn may stimulate the Mas receptor (MasR), exerting anti-inflammatory and regenerative effects [8,9,10]. Importantly, in addition to the systemic RAS, several local RAS have been also identified in individual organs, where it plays a role not only in the homeostasis of blood pressure but also in the regulation of local immune response [11,12,13,14].

The renin-angiotensin system and its outcome in inflammation and blood pressure. The renin-angiotensin system (RAS) is composed by a number of enzymes and peptides that regulate blood vasodilation, electrolyte balance, and ultimately blood pressure in the organism. Nevertheless, this system also regulates inflammation. Systemic and local RAS have been identified. In the systemic RAS, pro-renin is converted to renin by the kidneys and released into the blood stream. Afterwards, renin catalyzes the cleavage of angiotensinogen, a protein released by the liver, yielding angiotensin I (AngI). Subsequently, AngI is further processed and converted into angiotensin II (AngII), a reaction catalyzed by the angiotensin-converting enzyme 1 (ACE1), which is produced by endothelial cells of the lungs. AngII exerts its effects by stimulating angiotensin receptor 1 (ATR1) on the cell surface, yielding an increase in blood pressure and pro-inflammatory responses. AngII can also stimulate ATR2 on target cells, reducing blood pressure and inflammation. ACE2 can further process AngII to a smaller peptide, angiotensin (1–7) (Ang(1–7)), which might stimulate the Mas receptor (MasR), dampening oxidative stress as well as promoting anti-inflammatory effects. Local RAS have been identified in several organs, which synthesize most or all the constituents of systemic RAS, such as the brain, the kidney, and the gut, among others
The immune system can also be modulated by the action of neurotransmitters, which have been extensively involved as mediators in neuro-immune communication. In particular, dopamine, a catecholaminergic neurotransmitter, has been consistently involved in the control of inflammation [15]. In this regard, dopamine plays an important role in the regulation of immune responses in the brain [16,17,18,19,20] and the gut [21, 22]. Dopaminergic regulation of immunity is exerted by the stimulation of dopamine receptors expressed on immune cells, including dendritic cells [23,24,25], T cells [26,27,28], and B cells among other cellular types [29, 30]. Dopaminergic effects are triggered by the stimulation of five different G protein-coupled receptors (GPCRs) expressed on the cell surface, which vary in their affinities for the ligand and are classified into two subtypes depending on their sequence homology, pharmacologic behavior, and signaling pathways coupled: type 1 dopamine receptors (including DRD1 and DRD5) and type 2 dopamine receptors (including DRD2, DRD3, and DRD4) [6]. It is noteworthy that high-affinity dopamine receptors (DRD3, DRD4, and DRD5), which are selectively stimulated by low dopamine concentrations (in the range of 20–500 nM), promote pro-inflammatory responses in the adaptive [20, 22, 27, 28, 31, 32] and innate immune cells [23,24,25, 33]. Conversely, the stimulation of low-affinity dopamine receptors (DRD1 and DRD2), which are exclusively stimulated by high dopamine levels (in the range of 1–10 μM), induces anti-inflammatory effects in the immune system [34, 35].
Anomalies in the dopaminergic regulation of these neuroimmune interaction have been associated to several autoimmune and neurodegenerative diseases [6, 19, 36]. Indeed, polymorphisms in some dopamine receptors have been identified as genetic risk factors for Parkinson’s disease [37, 38] and inflammatory bowel diseases [39] as well as increased fibrosis and kidney inflammation, independently of blood pressure [40, 41]. Furthermore, dopamine is also involved in the regulation of blood pressure, renal function, cognition, and voluntary movements [42, 43], thus suggesting a potential communication between dopaminergic signaling and either local or systemic RAS. Here, we analyzed the communication between RAS components, the immune system, and dopaminergic signaling in the context of homeostasis, inflammation, and neurodegenerative disorders.
Modulation of inflammation by dopamine and RAS
Dopamine is a catecholaminergic neurotransmitter with immunomodulatory effects in the CNS and in the periphery. Dysregulation in the dopaminergic system affects both innate and adaptive immunity, contributing to the development of autoimmune and neurodegenerative disorders [6]. In this regard, changes of dopamine levels have been associated with inflammatory conditions, including inflammatory bowel diseases [44, 45], Parkinson’s disease [46], and sepsis [34]. These changes in dopamine concentration are critical for the modulation of immune responses by promoting a shift in the type of dopamine receptors stimulated. Dopamine receptors are expressed on several immune cell populations [47] where they modulate fundamental processes required for the development of immune responses, such as antigen presentation [24, 33], effector cell differentiation [22, 27], regulatory T cell function [31, 48], and leukocyte migration [49, 50].
Inflammation is often mediated by the inflammasome, a group of proteins that becomes activated and assembled in response to both microbial inflammation and endogenous danger signals, such as tumor necrosis factor α (TNFα) and reactive oxygen species [51,52,53]. Its activation plays a critical role in the initiation and development of systemic immune responses by promoting the secretion of pro-inflammatory cytokines, including interleukin 1β (IL-1β) and IL-18 [54, 55]. The NLRP3 inflammasome, which is highly expressed in macrophages, is strongly regulated by dopamine. In this regard, the stimulation of the DRD1 triggers a rise of cAMP and the consequent ubiquitination and degradation of NLRP3 in macrophages, thus dampening inflammation [35]. Consequently, exercise-induced dopamine secretion by the vagus nerve ameliorates inflammation by reducing serum levels of TNFα in a mouse model of endotoxemia [56]. According to these findings, the in vitro stimulation of splenocytes with a dopaminergic agonist type 1 attenuated TNFα production. Conversely, TNFα production was increased by splenocytes upon treatment with a dopaminergic antagonist type 1. In the same direction, administration of fenoldopam, a type 1 dopaminergic agonist, ameliorated systemic inflammation and attenuated hyperglycemia in diabetic septic mice by inhibiting p65NF-kB phosphorylation and reducing serum TNFα [57]. Taken together, these findings suggest that stimulation of low-affinity dopamine receptors in immune cells attenuates systemic inflammation.
It is important to consider that other catecholamines, including epinephrine and norepinephrine, also play an important role in the regulation of systemic inflammation both in humans and animal models. In this regard, initial studies showed that antiviral function of alveolar macrophages was dampened after exercise in experimental animals, an effect that was abrogated by adrenalectomy or by the administration of propranolol, thus suggesting the involvement of β-adrenergic receptors (βAR) [58]. Similarly, a study performed with human samples showed a decrease in LPS-induced TNFα production and a concomitant increase in IL-10 secretion by mononuclear cells after an acute exposition to epinephrine [59]. Further studies have provided genetic and pharmacologic evidence indicating that sympathetic activity reduces inflammation [60, 61]. Accordingly, it has been shown that norepinephrine stimulates β2AR in macrophages, promoting IL-10 secretion and attenuating the production of TLR-induced pro-inflammatory cytokines by these cells [60]. In consequence, the genetic deficiency of Adrb2 (the gene encoding for β2AR) in experimental animals resulted in exacerbated susceptibility to dextran sodium sulfate (DSS)-induced inflammatory colitis and increased lethality to LPS-induced endotoxemia [60]. Despite the acute sympathetic activity exerts a β2AR-mediated immunosuppressive effect on inflammation, it has been shown that prolonged time of exercise training leads to increased IL-12 production by macrophages in response to LPS. It was due to that prolonged sympathetic stimulation (i.e., 3 weeks of exercise training) induces a downregulation of β2AR in macrophages [62]. Altogether these studies illustrate the important immunosuppressive role of sympathetic activity contributing to the control of inflammation.
On the other hand, systemic immunomodulatory effects have been also associated with global RAS activation. In this regard, preclinical and clinical studies have shown that both ATR1 signaling and ACE activity promote inflammatory responses through mechanisms that are independent of blood pressure. For instance, global RAS is activated during atherosclerosis and unstable angina, inducing myocardial damage mediated by T cells. Of note, ACE and ATR1 expression has been shown upregulated in T cells obtained from patients with unstable angina [63]. Furthermore, when T cells from these patients were cultured in the presence of AngII, ACE enzymatic activity was increased in the supernatant, indicating the activation of a positive feedback of the RAS in lymphocytes [63]. In the same direction, the systemic administration of an ACE inhibitor, quinapril, showed an anti-inflammatory effect, decreasing immune cell accumulation and reducing pro-inflammatory cytokine production in renal tubules in a rodent model of immune complex nephritis [64]. Another key process involved in inflammation and regulated by RAS is leukocyte migration. In this regard, it has been shown that AngII-mediated ATR1-stimulation promotes migration of naïve T cells from the bloodstream into the spleen. Mechanistic analyses showed that systemic administration of AngII induces the overexpression of CCR9 and CD62L in naïve T cells and the secretion of CCL25 and CCL19 in the spleen of mice [65]. Another study has shown that exogenous AngII induced upregulation of cell surface molecules involved in T cell proliferation and migration, including CD69, CD25, and CCR5 [66]. Moreover, the same authors showed that AngII produced by T cells upon TCR stimulation might act in an autocrine loop inducing NADPH-oxidase activity and the consequent production of superoxide, which ultimately promotes TNF-α production.
Interestingly, the RAS and the dopaminergic system have been described in the kidney, where dopaminergic stimulation of DRD1 plays an antagonistic role counteracting ATR1-mediated increase in blood pressure. In this regard, a number of studies have addressed the cross talk between DRD1 and ATR1 and ATR2 in animal models of chronic kidney diseases [67]. Regarding the effect of the RAS and the dopaminergic system on kidney inflammation, it was recently shown that chronic kidney disease progression in humans is associated with increasing concentrations of urinary AngII and decreased levels of urinary dopamine, thus indicating a positive association of renal AngII concentration and a negative association of renal dopamine levels with inflammation and kidney dysfunction [68]. In the same direction, a recent study conducted with Sprague-Dawley rats fed with an overload of fructose shows that kidney inflammation and renal dysfunction involved an increased urinary expression of AngII followed by a decrease in urinary levels of dopamine [69]. Interestingly, the pathogenic effects induced by the overload of fructose were reverted by the administration of Losartan, an ATR1 antagonist. The authors suggested a mechanism by which AngII inhibits DRD1 expression in renal tubules [69]. Consistently, another study using animals deficient in the catechol-O-methyltransferase (COMPT), the major enzyme catabolizing dopamine in kidneys, shows that increased levels of renal dopamine attenuate the detrimental effects induced in kidneys upon AngII exposure [70]. Mechanistic analyses of the antagonistic cross talk observed between renal ATR1 and DRD1 have suggested that both receptors might form a heteromeric complex which may regulate negatively and reciprocally the receptor-signaling exerted by each other in the organ [71].
Similarly, interactions between the RAS and the dopaminergic system have also been recently studied in the context of inflammatory bowel diseases (IBD). IBD are characterized by chronic inflammation of the gut mucosa, which is triggered by increased permeability due to epithelial disruption mediated by some pathobionts present in commensal bacteria [72,73,74]. Interestingly, it has been described that gut inflammation in IBD involves a sharp reduction of dopamine levels (up to 10-fold change in the colonic mucosa), which results from epithelial disruption, loss of some neurons of the enteric nervous system, and dysbiosis [44, 75]. In this context, using a mouse model of IBD induced by the adoptively transfer of naïve T cells into lymphopenic mice, it was shown that DRD3-signaling in T-lymphocytes promoted Th1 differentiation, inhibited the Th2 differentiation, and favored the expansion of Th17 cells in the gut mucosa [22]. Of note, reduced dopamine levels associated to gut inflammation (50–100 nM) are consistent with a selective stimulation of DRD3, which display the highest affinity for dopamine [15]. Conversely, DRD2 signaling, which is triggered by 1–5 μM dopamine and therefore is impaired upon gut inflammation, has been involved in promoting suppressive activity of regulatory T cells [76] and favoring epithelial permeability [77]. Accordingly, the administration of DRD2 agonists, such as cabergoline and quinpirole, has been shown to dampen gut inflammation in animal models [76, 77]. In the same direction, a DRD2 allele associated with lower DRD2 expression has been related to increased risk of Crohn’s diseases [39]. Thus, current evidence indicates that DRD3 signaling promotes gut inflammation, whereas DRD2 stimulation ameliorates IBD. Regarding the involvement of the RAS system in IBD, it has been shown that a genetic mouse model, which overexpresses renin in the liver, develops a more severe form of TNBS-induced inflammatory colitis than that of wild-type (WT) littermates. Accordingly, increased expression of renin promotes gut epithelium disruption and accumulation of Th1 and Th17 subsets of T cells in the intestinal lamina propria. In addition, administration of the ATR1 antagonist, losartan, reduced the severity of the disease manifestation, reaching levels similar to that of the WT mice [78]. Furthermore, the analysis of biopsies of IBD patients treated with losartan showed that ATR1 antagonism resulted in lower levels of pro-inflammatory cytokines in the colonic mucosa when compared with control IBD patients [78]. Another study analyzed the interaction between the RAS and the dopaminergic system in gut inflammation using mice deficient in genes encoding for some dopamine receptors and RAS components involved in inflammation. The results showed that genetic deficiency of ATR1 resulted in increased expression of either type 1 and 2 dopamine receptors in the proximal colon. Moreover, an increased ratio of ATR1 to ATR2 was observed in the gut upon genetic deficiency of either DRD1 or DRD2 [79]. Thus, this study confirms a reciprocal negative regulation between the pro-inflammatory receptor ATR1 and the anti-inflammatory receptors ATR2, DRD1, and DRD2. Interestingly, the same authors also demonstrated that aged rats, which are associated with a pro-inflammatory state on the gastrointestinal tract, display higher levels of ATR1 expression and lower expression of ATR2 and DRD2 in the colon, differences that could be partially reverted by the administration of the ATR1 antagonist candesartan [79]. Taken together, these results indicate that gut homeostasis involves a reciprocal regulation between some components of the RAS and the dopaminergic system, which becomes dysregulated upon inflammation associated to IBD and ageing, resulting in increased pro-inflammatory signaling mediated by ATR1 and DRD3, and impaired anti-inflammatory signaling mediated by ATR2, DRD1, and DRD2.
Interaction between RAS and dopaminergic system in neurogenerative disorders
Not only the kidney and the gut mucosa express local components of the RAS but also the brain, where it plays a key role regulating blood pressure and inflammation. In this regard, some components of the RAS have been identified in neurons and glial cells. Angiotensinogen and ACE activity have been found in astrocytes and neurons in particular areas of the brain [80, 81]. Although scarcely found in the brain, renin is sequestered by the pro-renin receptor, which seems to constitute a key component required to propagate AngII actions in the CNS [82]. After conversion of angiotensinogen into AngI and then in AngII, the latter might stimulate ATR1 expressed in microglia, triggering increased production of nitric oxide species and oxidative damage [83]. Conversely, the dopaminergic stimulation of DRD2 in astrocytes dampens the pro-inflammatory and oxidative effects exerted by local RAS, by downregulating the expression of microglial ATR1 and reducing the astrocytic production of angiotensinogen [83] (Fig. 2).

Interaction between RAS and dopaminergic system in the CNS. Astrocytes in the brain produce and secrete angiotensinogen to the parenchyma, which is processed by low concentrations of renin coming from the periphery yielding AngI. 1. AngI is subsequently converted to AngII by the action of ACE1 produced by astrocytes. 2. AngII might stimulate ATR1 on microglial cells, triggering the generation of nitric oxide and the production of the pro-inflammatory cytokines TNFα and IL1β. Furthermore, ATR1 signaling also induces the downregulation of dopamine receptors expression in astrocytes and microglia, thus rendering glial cells hyporeactive to this anti-inflammatory signal. This phenomenon promotes inflammation and vasoconstriction in the CNS. 3. AngII can also stimulate ATR2 on microglia, counteracting the response exerted by ATR1 stimulation. 4. AngII may be further degraded to generate the peptide Ang(1–7), which upon stimulation of MasR, inhibits secretion of pro-inflammatory cytokines, and promotes vasodilation. 5. The neurotransmitter dopamine can stimulate different dopamine receptors according their particular affinities. Low-affinity dopamine receptors are stimulated by high dopamine levels triggering increased expression of ATR2 in microglia and degradation of the NLRP3 inflammasome in glial cells. 6. DRD2 signaling induces increased expression of the anti-inflammatory protein αB-crystallin in astrocytes. 7. Stimulation of the high-affinity dopamine receptor DRD3 in astrocytes by low concentrations of dopamine inhibits the production of the anti-inflammatory mediator Fizz1 in microglial cells. 8. Given the close interaction between these systems, heteromeric complexes formed by dopamine and angiotensin receptors have been proposed by some researchers. Of note, heterodimerization of MasR and ATR2 in astrocytes has already been described, although its role in neuroinflammation remains pending to be explored
A huge number of studies performed in human and animals has indicated that neuroinflammation plays a major role in the development and progression of neurodegenerative disorders, including Parkinson’s disease, Alzheimer’s disease, and multiple sclerosis [84]. According to the regulatory role of the RAS in neuroinflammation, emerging evidence has shown a relevant role of this system in neurodegenerative disorders. In this regard, the inflammatory features associated to the mouse model of Parkinson’s disease induced by 6-hydroxydopamine (6-OHDA) were significantly dampened by the administration of candesartan, an ATR1 antagonist [85]. Patients with Parkinson’s disease often show gastrointestinal problems many years before the manifestation of the motor symptoms. Braak and colleagues proposed that Parkinson’s disease is initiated in the gut by the generation of alpha-synuclein aggregates in response to pathogenic bacteria, and that these aggregates migrate later to the brain through the vagus nerve, being accumulated in the substantia nigra and ultimately promoting degeneration of neurons of the nigrostriatal pathway [86, 87]. Of note, the nigrostriatal pathway is an area of the brain involved in the control of voluntary movements; thereby, a classic symptomatic manifestation in advanced Parkinson’s disease is the motor impairment. The Braak’s hypothesis has been the starting point for many studies addressing the mechanisms by which processes associated to the gut mucosa might trigger neuroinflammation. In this regard, a study addressing the role of the cross talk between the dopaminergic system and the RAS in the context of the gut-brain axis has demonstrated that gut inflammation induced in mice by the administration of DSS results in downregulation of DRD2 and ATR2 expression and increased expression of ATR1 in the nigrostriatal pathway. Accordingly, the subchronic administration of DSS resulted in significant neurodegeneration of dopaminergic neurons of the substantia nigra and overexpression of the pro-inflammatory cytokine IL-1β and of AngII [88]. Interestingly, vagotomy reverted the changes in the expression ratio of ATR1/ATR2 and partially recovered the expression levels of the nigrostriatal IL-1β [88]. Of note, these changes in the local CNS RAS were previously shown to be toxic for dopaminergic neurons [89,90,91]. On the other hand, when nigrostriatal pathway was directly lesioned by the stereotaxic administration of 6-OHDA, the ratio of ATR1 to ATR2 and IL-1β expression was increased in the colonic mucosa; nevertheless, dopamine levels were increased in this tissue. Moreover, vagotomy did not reversed the inflammatory features observed in the colonic mucosa upon nigrostriatal lesion [88]. Thereby, these results indicate a bidirectional communication of inflammatory components in the colonic mucosa and the nigrostriatal pathway, which is dependent in the vagus nerve only in the gut-to-brain direction but not in the opposite direction (Fig. 3). In addition, a number of studies have indicated that the initial reduction of dopamine levels in the nigrostriatal pathway induce pro-inflammatory responses in this area of the brain [19]. It is noteworthy that initial aggregation of α-synuclein in nigrostriatal neurons observed in Parkinson’s disease results in decreased secretion of dopamine, even before neurodegeneration [92]. Due to the early reduction of nigrostriatal dopamine, the role of DRD3 in neuroinflammation has been explored in the context of Parkinson’s disease. In this regard, it has been shown that DRD3 signaling in CD4+ T cells infiltrating the substantia nigra favors Th1 and Th17 effector function [17, 20]. In addition, DRD3 signaling in astrocytes has been also involved in neuroinflammation. In this regard, genetic and pharmacologic evidence has recently shown that DRD3 signaling in astrocytes attenuates the production of the anti-inflammatory factor Fizz1 by microglial cells, thus promoting neuroinflammation [93]. Accordingly, the systemic treatment of mice with a selective DRD3 antagonist, PG01037, exerted a significant therapeutic effect reducing neuroinflammation and neurodegeneration in two different mouse models of Parkinson’s disease [16]. In addition, another couple of studies have addressed the role of the low-affinity dopamine receptors in neuroinflammation associated to Parkinson’s disease. Using genetic and pharmacologic approaches, that study showed that DRD2 signaling in astrocytes induced the expression of the anti-inflammatory mediator αB-crystallin, which abolish the development of neuroinflammation and neurodegeneration in a mouse model of Parkinson’s disease induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) [94]. Another study has demonstrated that DRD1 signaling in either microglial cells or astrocytes promotes the degradation of the inflammasome NLRP3, attenuating neuroinflammation, and the consequent neurodegeneration associated to the treatment with MPTP in a mouse model [35]. Taken together these findings indicate that signaling through low-affinity dopamine receptors and ATR2 expressed in glial cells attenuates neuroinflammation, whereas DRD3 and ATR1 promote neuroinflammation. Furthermore, the cross talk between the RAS and dopaminergic system associated to Parkinson’s disease involves a reciprocal negative regulation between DRD1/DRD2 and ATR1/AngII (Fig. 2).

Interaction between RAS and the dopaminergic system in the context of gut-brain axis. Gut and brain local RAS mediate inflammatory responses in their corresponding organs. Communication between the gut and the brain involves a number of immune mediators. The loss of dopaminergic neurons in the brain induces an activation of the inflammatory arm of RAS in the gut by increasing ATR1/ATR2 balance. Consequently, expression of low-affinity dopamine receptors turns downregulated in the proximal colon and pro-inflammatory cytokine production is increased. Nevertheless, dopamine levels turn increased in the gut mucosa. Conversely, when intestinal inflammation is triggered, brain local RAS becomes activated preferentially by stimulation of ATR1. AngII levels and the ATR1/ATR2 ratio become higher, whereas DRD2 expression is downregulated in the brain. Subsequently, ATR1 signaling induces changes in the oxidative microenvironment through secretion of reactive oxygen species and the production of the pro-inflammatory cytokine TNFα, yielding dopaminergic neuronal death
Another example of neurodegenerative disease that is affected by the dopaminergic system and RAS is multiple sclerosis (MS). MS is a CD4+ T cell-driven autoimmune disease characterized by the destruction of the myelin sheath on the axons of motor neurons. Myelin is targeted by the adaptive and innate immune system once the blood-brain barrier becomes permeable leading to the infiltration of immune cells into the CNS [95,96,97]. Experimental autoimmune encephalomyelitis (EAE) is a mouse model of MS which is elicited by the immunization with a peptide derived from the sequence of myelin oligodendrocyte glycoprotein (MOG35-55) and recapitulates several immunologic and pathological aspects of MS. Addressing the role of RAS in EAE, it has been shown an upregulation of AngII, ACE1, and ATR1 either in CD4+ T-cells and monocytes infiltrating the CNS during the inflammatory process [98, 99]. Accordingly, the inhibition of ACE activity or ATR1 signaling promoted the accumulation of antigen-specific regulatory T cells and reduced infiltration of inflammatory Th1 and Th17 phenotypes in the CNS lesions [99]. In addition, the selective stimulation of DRD5 by low concentrations of dopamine in the CNS promotes antigen presentation and Th17- and Th1-mediated immunity [24, 100]. Moreover, the stimulation of MasR by Ang(1–7) promotes anti-inflammatory and regenerative effects in both the CNS and the periphery and is thought to counteract the action exerted by ATR1 signaling, thus reducing inflammation and oxidative stress [8,9,10]. According to the anti-inflammatory and pro-regenerative effects exerted by the Ang(1–7)-MasR axis, the administration of Ang(1–7) in mice undergoing EAE resulted in attenuated disease manifestation, and it was dependent on MasR signaling. This Ang(1–7)-induced EAE amelioration was correlated with decreased immune infiltration, reduced demyelination, and attenuated axonal loss compared with control mice, suggesting a role of the Ang(1–7)-MasR axis in the recruitment of immune cells into the CNS [101]. According to this idea, a previous work demonstrated that MasR signaling in monocytes/macrophages favored inflammatory responses in vivo by increasing monocytes/macrophage migration and infiltration into the spinal cord of EAE mice [102]. Thus, these studies indicate that the AngII-ATR1 axis as well as the selective stimulation of high-affinity dopamine receptors contribute to neuroinflammation associated to EAE.
Altogether, the current evidence suggests a dual role of RAS on neurodegenerative disorders, promoting neuroinflammation and oxidative damage by stimulating the ATR1 pathway and, conversely, dampening oxidative stress and neuroinflammation by action of MasR signaling. Although there is no direct evidence on the link of the RAS and dopaminergic system in the context of neurodegenerative disorders, it is clear that these pathologies are multifactorial and involve across talk between complex molecular networks that mediate disease development and progression.
Intestinal microbiota in the modulation of RAS and its role in inflammatory and metabolic diseases
More than 5000 different species of bacteria and more than 100 trillion individual microbial cells inhabit our intestine. The human body hosts more than 10-folds its own number of cells on intestinal microbiota, giving rise to the idea that it constitutes another organ of our body [103]. Intestinal microbiota interacts with the organism by synthesizing metabolites, such as γ-aminobutyric acid (GABA) or short-chain fatty acids (SCFA), which exerts their effects by stimulating specific receptors expressed on the host cells [104]. Of note, some of those metabolites, including SCFA, can only be obtained through gut microbiota-mediated food degradation. SCFA have acquired particular importance, as a relevant regulatory role in the development of inflammatory and neurodegenerative disorders has been consistently reported in the last 15 years [105,106,107,108]. Dietary fiber is mainly degraded into three different SCFA by the intestinal microbiota: acetate and propionate, which are mainly synthesized by the bacterial genus Bacteroidetes, and butyrate, produced by the Firmicutes genus [109]. Each of these molecules can act in the organism beyond the intestine by stimulating G protein-coupled receptors expressed in several type of cells in the host, including GPR41, GPR43, and GPR109a [75]. For instance, propionate has a protective role on the blood-brain barrier, promoting its integrity and reducing its permeability, thereby protecting the CNS from infections [110]. On the other hand, butyrate favors healing and promotes the structural integrity of the epithelial barrier of the gut mucosa, thus limiting bacterial translocation into the blood stream and thereby reducing the probability of gut inflammation [111]. At the same time, metabolites derived from commensal bacteria might stimulate their receptors expressed in innate and adaptive immune cells, strongly regulating the immune response of the host [104]. Thus, the presence of some commensal bacterial populations in the gut contributes to the regulation of homeostatic processes in the organism. In this regard, it has been shown that depletion of intestinal microbiota through the action of wide-spectrum antibiotics enhances the sensitivity to insulin, modifying the outcome of some metabolic disorders, which suggest the possibility that commensal bacteria are able to interact with intestinal RAS [112]. Indeed, the administration of sodium butyrate to uninephrectomyzed rats attenuated the increase on arterial pressure induced by high AngII and significantly reduced the production of inflammatory cytokine, including TNFα and IL-6 [113]. Moreover, the expression of pro-renin receptor, angiotensinogen, AngII, and ACE-I were all diminished in the kidney of rats treated with butyrate [113]. Another way by which gut microbiota might regulate host physiology is by degradation of proteins derived from the organism, yielding peptides with biological activity, including peptides derived from serum albumin, mucin, digestive enzymes, and debris protein derived from dying epithelial cells. Some of these peptides have shown to exert inhibitory effects on the activity of renin and ACE1 [114], thus modulating directly RAS in the gut and indirectly the dopaminergic intestinal system, as these systems are reciprocally regulated in the gut mucosa, as described above. In addition to the direct production of metabolites that might affect host physiology, intestinal microbiota can also influence the production of key regulators of immunity, including dopamine and norepinephrine [115, 116]. As previously mentioned in this review, either systemic or local dopamine levels have different effect in the modulation of the immune system, orchestrating either tolerogenic or inflammatory responses depending on the dopamine receptor that are preferentially being stimulated in adaptive or innate immune cells. However, further research is required to decipher how gut microbiota affects the different mechanisms involved in the interaction between the RAS and the dopaminergic system. The potential knowledge derived from these studies would give novel insight in the understanding of potential consequences of dysbiosis in the development of inflammatory affections, and in the design, combination or repurpose of dopaminergic drugs and RAS-targeting drugs as therapeutic approaches for inflammatory disorders
Conclusions and projections
Current evidence has shown a significant regulation of inflammation by RAS and dopaminergic system, as well as a complex interaction between both systems. Of note, the RAS and the dopaminergic system might trigger pro-inflammatory and anti-inflammatory outcomes systemically and in particular organs, including the kidney, the gut, and the brain. Whereas AngII-ATR1 axis exerts pro-inflammatory effects, the AngII-ATR2 and Ang(1–7)-MasR arm induces anti-inflammatory and regenerative processes. Similarly, DRD3 and DRD5 signaling promotes inflammatory effects, while DRD1 and DRD2 stimulation trigger anti-inflammatory effects. Furthermore, it has been shown that DRD1/DRD2 stimulation might exert inhibition of ATR1 signaling as well as attenuation of AngII synthesis. Conversely, ATR1 stimulation exerts downregulation of DRD1 expression. Thus, both systems regulate each other reciprocally. Further insight at the cellular and molecular level is needed in order to elucidate the detailed mechanisms of interaction by which changes on RAS or dopaminergic components control inflammation in the kidney, the gut, the brain, and systemically. Several preclinical studies have demonstrated the use of RAS inhibitors and dopaminergic drugs for the treatment of neurodegenerative and inflammatory diseases (Table 1). It is noteworthy that the lack of selectivity for dopaminergic drugs represents a challenge for the design of new drugs or other therapeutic approaches for targeting specific dopamine receptors as therapy for inflammatory disorders. Another interesting projection that arises from this study is that the combination of drugs targeting different components of the RAS and dopaminergic system could potentially reach synergistic outcomes as therapeutic strategies for the treatment of inflammation. Furthermore, the potential identification of heteromeric receptors conformed by protomers from different systems would open the possibility of development of new pharmacologic tools to be tested as therapeutic approaches for inflammatory disorders.
Data availability
Not applicable.
Abbreviations
- ACE:
-
Angiotensin-converting enzyme
- AngI:
-
Angiotensin I
- AngII:
-
Angiotensin II
- Ang(1–7):
-
Angiotensin (1–7)
- ATR1:
-
Type 1 AngII receptor
- ATR2:
-
Type 2 AngII receptor
- CNS:
-
Central nervous system
- COMPT:
-
Catechol-O-methyltransferase
- DRDn :
-
Dopamine receptor Dn
- DSS:
-
Dextran sodium sulfate
- IBD:
-
Inflammatory bowel diseases
- IL-n :
-
Interleukin n
- MasR:
-
Mas receptor
- MPTP:
-
1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- RAS:
-
Renin-angiotensin system
- Thn :
-
T-helper n
- TH:
-
Tyrosine hydroxylase
- TNF-α:
-
Tumor necrosis factor α
- WT:
-
Wild-type
- βAR:
-
β-adrenergic receptor
- β2AR:
-
β2-adrenergic receptor
- 6-OHDA:
-
6-hydroxydopamine
References
Pahwa R, Singh A, Jialal I (2020) Chronic inflammation. In: StatPearls. Treasure Island (FL),
Suzuki Y, Ruiz-Ortega M, Gomez-Guerrero C, Tomino Y, Egido J (2003) Angiotensin II, the immune system and renal diseases: another road for RAS? Nephrol Dial Transplant 18(8):1423–1426. https://doi.org/10.1093/ndt/gfg223
Capettini LS, Montecucco F, Mach F, Stergiopulos N, Santos RA, da Silva RF (2012) Role of renin-angiotensin system in inflammation, immunity and aging. Curr Pharm Des 18(7):963–970. https://doi.org/10.2174/138161212799436593
Satou R, Penrose H, Navar LG (2018) Inflammation as a regulator of the renin-angiotensin system and blood pressure. Curr Hypertens Rep 20(12):100. https://doi.org/10.1007/s11906-018-0900-0
Hammer A, Stegbauer J, Linker RA (2017) Macrophages in neuroinflammation: role of the renin-angiotensin-system. Pflugers Arch 469(3-4):431–444. https://doi.org/10.1007/s00424-017-1942-x
Vidal PM, Pacheco R (2019) Targeting the dopaminergic system in autoimmunity. J NeuroImmune Pharmacol 15:57–73. https://doi.org/10.1007/s11481-019-09834-5
Sparks MA, Crowley SD, Gurley SB, Mirotsou M, Coffman TM (2014) Classical renin-angiotensin system in kidney physiology. Compr Physiol 4(3):1201–1228. https://doi.org/10.1002/cphy.c130040
Cerrato BD, Frasch AP, Nakagawa P, Longo-Carbajosa N, Pena C, Hocht C, Gironacci MM (2012) Angiotensin-(1-7) upregulates central nitric oxide synthase in spontaneously hypertensive rats. Brain Res 1453:1–7. https://doi.org/10.1016/j.brainres.2012.03.022
Mecca AP, Regenhardt RW, O'Connor TE, Joseph JP, Raizada MK, Katovich MJ, Sumners C (2011) Cerebroprotection by angiotensin-(1-7) in endothelin-1-induced ischaemic stroke. Exp Physiol 96(10):1084–1096. https://doi.org/10.1113/expphysiol.2011.058578
Mordwinkin NM, Meeks CJ, Jadhav SS, Espinoza T, Roda N, diZerega GS, Louie SG, Rodgers KE (2012) Angiotensin-(1-7) administration reduces oxidative stress in diabetic bone marrow. Endocrinology 153(5):2189–2197. https://doi.org/10.1210/en.2011-2031
Liao Z, Chakrabarty A, Mu Y, Bhattacherjee A, Goestch M, Leclair CM, Smith PG (2017) A local inflammatory renin-angiotensin system drives sensory axon sprouting in provoked vestibulodynia. J Pain 18(5):511–525. https://doi.org/10.1016/j.jpain.2016.12.008
Labandeira-Garcia JL, Rodriguez-Perez AI, Garrido-Gil P, Rodriguez-Pallares J, Lanciego JL, Guerra MJ (2017) Brain renin-angiotensin system and microglial polarization: implications for aging and neurodegeneration. Front Aging Neurosci 9:129. https://doi.org/10.3389/fnagi.2017.00129
Santos CF, Morandini AC, Dionisio TJ, Faria FA, Lima MC, Figueiredo CM, Colombini-Ishikiriama BL, Sipert CR, Maciel RP, Akashi AP, Souza GP, Garlet GP, Rodini CO, Amaral SL, Becari C, Salgado MC, Oliveira EB, Matus I, Didier DN, Greene AS (2015) Functional local renin-angiotensin system in human and rat periodontal tissue. PLoS One 10(8):e0134601. https://doi.org/10.1371/journal.pone.0134601
He L, Du J, Chen Y, Liu C, Zhou M, Adhikari S, Rubin DT, Pekow J, Li YC (2019) Renin-angiotensin system promotes colonic inflammation by inducing TH17 activation via JAK2/STAT pathway. Am J Physiol Gastrointest Liver Physiol 316(6):G774–G784. https://doi.org/10.1152/ajpgi.00053.2019
Pacheco R (2017) Targeting dopamine receptor D3 signalling in inflammation. Oncotarget 8(5):7224–7225. https://doi.org/10.18632/oncotarget.14601
Elgueta D, Aymerich MS, Contreras F, Montoya A, Celorrio M, Rojo-Bustamante E, Riquelme E, Gonzalez H, Vasquez M, Franco R, Pacheco R (2017) Pharmacologic antagonism of dopamine receptor D3 attenuates neurodegeneration and motor impairment in a mouse model of Parkinson’s disease. Neuropharmacology 113(Pt A):110–123. https://doi.org/10.1016/j.neuropharm.2016.09.028
Elgueta D, Contreras F, Prado C, Montoya A, Ugalde V, Chovar O, Villagra R, Henriquez C, Abellanas MA, Aymerich MS, Franco R, Pacheco R (2019) Dopamine receptor d3 expression is altered in cd4+ t-cells from Parkinson’s disease patients and its pharmacologic inhibition attenuates the motor impairment in a mouse model. Front Immunol 10. https://doi.org/10.3389/fimmu.2019.00981
Arreola R, Alvarez-Herrera S, Perez-Sanchez G, Becerril-Villanueva E, Cruz-Fuentes C, Flores-Gutierrez EO, Garces-Alvarez ME, de la Cruz-Aguilera DL, Medina-Rivero E, Hurtado-Alvarado G, Quintero-Fabian S, Pavon L (2016) Immunomodulatory effects mediated by dopamine. J Immunol Res 2016:3160486–3160431. https://doi.org/10.1155/2016/3160486
Gonzalez H, Contreras F, Pacheco R (2015) Regulation of the neurodegenerative process associated to Parkinson’s disease by CD4+ T-cells. J NeuroImmune Pharmacol 10(4):561–575. https://doi.org/10.1007/s11481-015-9618-9
Gonzalez H, Contreras F, Prado C, Elgueta D, Franz D, Bernales S, Pacheco R (2013) Dopamine receptor d3 expressed on cd4+ t cells favors neurodegeneration of dopaminergic neurons during Parkinson’s disease. J Immunol 190(10):5048–5056. https://doi.org/10.4049/jimmunol.1203121
Pacheco R, Contreras F, Zouali M (2014) The dopaminergic system in autoimmune diseases. Front Immunol 5:117. https://doi.org/10.3389/fimmu.2014.00117
Contreras F, Prado C, Gonzalez H, Franz D, Osorio-Barrios F, Osorio F, Ugalde V, Lopez E, Elgueta D, Figueroa A, Lladser A, Pacheco R (2016) Dopamine receptor D3 signaling on CD4+ T cells favors Th1- and Th17-mediated immunity. J Immunol 196(10):4143–4149. https://doi.org/10.4049/jimmunol.1502420
Prado C, Bernales S, Pacheco R (2013) Modulation of T-cell mediated immunity by dopamine receptor d5. Endocr Metab Immune Disord Drug Targets 13(2):184–194
Prado C, Contreras F, Gonzalez H, Diaz P, Elgueta D, Barrientos M, Herrada AA, Lladser A, Bernales S, Pacheco R (2012) Stimulation of dopamine receptor D5 expressed on dendritic cells potentiates Th17-mediated immunity. J Immunol 188(7):3062–3070. https://doi.org/10.4049/jimmunol.1103096
Prado C, Gaiazzi M, Gonzalez H, Ugalde V, Figueroa A, Osorio-Barrios FJ, Lopez E, Lladser A, Rasini E, Marino F, Zaffaroni M, Cosentino M, Pacheco R (2018) Dopaminergic stimulation of myeloid antigen-presenting cells attenuates signal transducer and activator of transcription 3-activation favouring the development of experimental autoimmune encephalomyelitis. Front Immunol 9:571. https://doi.org/10.3389/fimmu.2018.00571
Cosentino M, Ferrari M, Kustrimovic N, Rasini E, Marino F (2015) Influence of dopamine receptor gene polymorphisms on circulating T lymphocytes: a pilot study in healthy subjects. Hum Immunol 76(10):747–752. https://doi.org/10.1016/j.humimm.2015.09.032
Franz D, Contreras F, Gonzalez H, Prado C, Elgueta D, Figueroa C, Pacheco R (2015) Dopamine receptors D3 and D5 regulate CD4(+)T-cell activation and differentiation by modulating ERK activation and cAMP production. J Neuroimmunol 284:18–29. https://doi.org/10.1016/j.jneuroim.2015.05.003
Osorio-Barrios F, Prado C, Contreras F, Pacheco R (2018) Dopamine receptor D5 signaling plays a dual role in experimental autoimmune encephalomyelitis potentiating Th17-mediated immunity and favoring suppressive activity of regulatory T-cells. Front Cell Neurosci 12:192. https://doi.org/10.3389/fncel.2018.00192
Papa I, Saliba D, Ponzoni M, Bustamante S, Canete PF, Gonzalez-Figueroa P, McNamara HA, Valvo S, Grimbaldeston M, Sweet RA, Vohra H, Cockburn IA, Meyer-Hermann M, Dustin ML, Doglioni C, Vinuesa CG (2017) TFH-derived dopamine accelerates productive synapses in germinal centres. Nature 547(7663):318–323. https://doi.org/10.1038/nature23013
Pacheco R, Prado CE, Barrientos MJ, Bernales S (2009) Role of dopamine in the physiology of T-cells and dendritic cells. J Neuroimmunol 216(1-2):8–19. https://doi.org/10.1016/j.jneuroim.2009.07.018
Cosentino M, Fietta AM, Ferrari M, Rasini E, Bombelli R, Carcano E, Saporiti F, Meloni F, Marino F, Lecchini S (2007) Human CD4 + CD25+ regulatory T cells selectively express tyrosine hydroxylase and contain endogenous catecholamines subserving an autocrine/paracrine inhibitory functional loop. Blood 109(2):632–642
Wang W, Cohen JA, Wallrapp A, Trieu KG, Barrios J, Shao F, Krishnamoorthy N, Kuchroo VK, Jones MR, Fine A, Bai Y, Ai X (2019) Age-related dopaminergic innervation augments T helper 2-type allergic inflammation in the postnatal lung. Immunity 51(6):1102–1118 e1107. https://doi.org/10.1016/j.immuni.2019.10.002
Figueroa C, Gálvez-Cancino F, Oyarce C, Contreras F, Prado C, Valeria C, Cruz S, Lladser A, Pacheco R (2017) Inhibition of dopamine receptor D3 signaling in dendritic cells increases antigen cross-presentation to CD8+ T-cells favoring anti-tumor immunity. J Neuroimmunol Accepted
Torres-Rosas R, Yehia G, Pena G, Mishra P, del Rocio T-BM, Moreno-Eutimio MA, Arriaga-Pizano LA, Isibasi A, Ulloa L (2014) Dopamine mediates vagal modulation of the immune system by electroacupuncture. Nat Med 20(3):291–295. https://doi.org/10.1038/nm.3479
Yan Y, Jiang W, Liu L, Wang X, Ding C, Tian Z, Zhou R (2015) Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell 160(1-2):62–73. https://doi.org/10.1016/j.cell.2014.11.047
Levite M, Marino F, Cosentino M (2017) Dopamine, T cells and multiple sclerosis (MS). J Neural Transm 124(5):525–542. https://doi.org/10.1007/s00702-016-1640-4
Comi C, Ferrari M, Marino F, Magistrelli L, Cantello R, Riboldazzi G, Bianchi ML, Bono G, Cosentino M (2017) Polymorphisms of dopamine receptor genes and risk of L-dopa-induced dyskinesia in Parkinson’s disease. Int J Mol Sci 18(2). https://doi.org/10.3390/ijms18020242
McGuire V, Van Den Eeden SK, Tanner CM, Kamel F, Umbach DM, Marder K, Mayeux R, Ritz B, Ross GW, Petrovitch H, Topol B, Popat RA, Costello S, Manthripragada AD, Southwick A, Myers RM, Nelson LM (2011) Association of DRD2 and DRD3 polymorphisms with Parkinson's disease in a multiethnic consortium. J Neurol Sci 307(1-2):22–29. https://doi.org/10.1016/j.jns.2011.05.031
Magro F, Cunha E, Araujo F, Meireles E, Pereira P, Dinis-Ribeiro M, Veloso FT, Medeiros R, Soares-da-Silva P (2006) Dopamine D2 receptor polymorphisms in inflammatory bowel disease and the refractory response to treatment. Dig Dis Sci 51(11):2039–2044. https://doi.org/10.1007/s10620-006-9168-3
Jiang X, Konkalmatt P, Yang Y, Gildea J, Jones JE, Cuevas S, Felder RA, Jose PA, Armando I (2014) Single-nucleotide polymorphisms of the dopamine D2 receptor increase inflammation and fibrosis in human renal proximal tubule cells. Hypertension 63(3):e74–e80. https://doi.org/10.1161/HYPERTENSIONAHA.113.02569
Zhang Y, Cuevas S, Asico LD, Escano C, Yang Y, Pascua AM, Wang X, Jones JE, Grandy D, Eisner G, Jose PA, Armando I (2012) Deficient dopamine D2 receptor function causes renal inflammation independently of high blood pressure. PLoS One 7(6):e38745. https://doi.org/10.1371/journal.pone.0038745
Pinoli M, Marino F, Cosentino M (2017) Dopaminergic regulation of innate immunity: a review. J Neuroimmune Pharm 12(4):602–623. https://doi.org/10.1007/s11481-017-9749-2
Beaulieu JM, Gainetdinov RR (2011) The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev 63(1):182–217. https://doi.org/10.1124/pr.110.002642
Magro F, Vieira-Coelho MA, Fraga S, Serrao MP, Veloso FT, Ribeiro T, Soares-da-Silva P (2002) Impaired synthesis or cellular storage of norepinephrine, dopamine, and 5-hydroxytryptamine in human inflammatory bowel disease. Dig Dis Sci 47(1):216–224
Magro F, Fraga S, Ribeiro T, Soares-da-Silva P (2004) Decreased availability of intestinal dopamine in transmural colitis may relate to inhibitory effects of interferon-gamma upon L-DOPA uptake. Acta Physiol Scand 180(4):379–386. https://doi.org/10.1111/j.1365-201X.2004.01260.x
Brochard V, Combadiere B, Prigent A, Laouar Y, Perrin A, Beray-Berthat V, Bonduelle O, Alvarez-Fischer D, Callebert J, Launay JM, Duyckaerts C, Flavell RA, Hirsch EC, Hunot S (2009) Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J Clin Invest 119(1):182–192
McKenna F, McLaughlin PJ, Lewis BJ, Sibbring GC, Cummerson JA, Bowen-Jones D, Moots RJ (2002) Dopamine receptor expression on human T- and B-lymphocytes, monocytes, neutrophils, eosinophils and NK cells: a flow cytometric study. J Neuroimmunol 132(1-2):34–40
Kipnis J, Cardon M, Avidan H, Lewitus GM, Mordechay S, Rolls A, Shani Y, Schwartz M (2004) Dopamine, through the extracellular signal-regulated kinase pathway, downregulates CD4 + CD25+ regulatory T-cell activity: implications for neurodegeneration. J Neurosci 24(27):6133–6143
Watanabe Y, Nakayama T, Nagakubo D, Hieshima K, Jin Z, Katou F, Hashimoto K, Yoshie O (2006) Dopamine selectively induces migration and homing of naive CD8+ T cells via dopamine receptor D3. J Immunol 176(2):848–856
Coley JS, Calderon TM, Gaskill PJ, Eugenin EA, Berman JW (2015) Dopamine increases CD14 + CD16+ monocyte migration and adhesion in the context of substance abuse and HIV neuropathogenesis. PLoS One 10(2):e0117450. https://doi.org/10.1371/journal.pone.0117450
Davis BK, Wen H, Ting JP (2011) The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol 29:707–735. https://doi.org/10.1146/annurev-immunol-031210-101405
Martinon F, Mayor A, Tschopp J (2009) The inflammasomes: guardians of the body. Annu Rev Immunol 27:229–265. https://doi.org/10.1146/annurev.immunol.021908.132715
Bauernfeind F, Niepmann S, Knolle PA, Hornung V (2016) Aging-associated TNF production primes inflammasome activation and NLRP3-related metabolic disturbances. J Immunol 197(7):2900–2908. https://doi.org/10.4049/jimmunol.1501336
Lamkanfi M, Dixit VM (2012) Inflammasomes and their roles in health and disease. Annu Rev Cell Dev Biol 28:137–161. https://doi.org/10.1146/annurev-cellbio-101011-155745
Tschopp J, Schroder K (2010) NLRP3 inflammasome activation: the convergence of multiple signalling pathways on ROS production? Nat Rev Immunol 10(3):210–215. https://doi.org/10.1038/nri2725
Shimojo G, Joseph B, Shah R, Consolim-Colombo FM, De Angelis K, Ulloa L (2019) Exercise activates vagal induction of dopamine and attenuates systemic inflammation. Brain Behav Immun 75:181–191. https://doi.org/10.1016/j.bbi.2018.10.005
Feketeova E, Li Z, Joseph B, Shah R, Spolarics Z, Ulloa L (2018) Dopaminergic control of inflammation and glycemia in sepsis and diabetes. Front Immunol 9:943. https://doi.org/10.3389/fimmu.2018.00943
Kohut ML, Davis JM, Jackson DA, Colbert LH, Strasner A, Essig DA, Pate RR, Ghaffar A, Mayer EP (1998) The role of stress hormones in exercise-induced suppression of alveolar macrophage antiviral function. J Neuroimmunol 81(1-2):193–200. https://doi.org/10.1016/s0165-5728(97)00179-3
van der Poll T, Coyle SM, Barbosa K, Braxton CC, Lowry SF (1996) Epinephrine inhibits tumor necrosis factor-alpha and potentiates interleukin 10 production during human endotoxemia. J Clin Invest 97(3):713–719. https://doi.org/10.1172/JCI118469
Agac D, Estrada LD, Maples R, Hooper LV, Farrar JD (2018) The beta2-adrenergic receptor controls inflammation by driving rapid IL-10 secretion. Brain Behav Immun 74:176–185. https://doi.org/10.1016/j.bbi.2018.09.004
Maisel AS, Knowlton KU, Fowler P, Rearden A, Ziegler MG, Motulsky HJ, Insel PA, Michel MC (1990) Adrenergic control of circulating lymphocyte subpopulations. Effects of congestive heart failure, dynamic exercise, and terbutaline treatment. J Clin Invest 85(2):462–467. https://doi.org/10.1172/JCI114460
Itoh CE, Kizaki T, Hitomi Y, Hanawa T, Kamiya S, Ookawara T, Suzuki K, Izawa T, Saitoh D, Haga S, Ohno H (2004) Down-regulation of beta2-adrenergic receptor expression by exercise training increases IL-12 production by macrophages following LPS stimulation. Biochem Biophys Res Commun 322(3):979–984. https://doi.org/10.1016/j.bbrc.2004.08.050
Coppo M, Bandinelli M, Chiostri M, Poggesi L, Boddi M (2017) Persistent and selective upregulation of renin-angiotensin system in circulating T lymphocytes in unstable angina. J Renin-Angiotensin-Aldosterone Syst 18(1):1470320317698849. https://doi.org/10.1177/1470320317698849
Ruiz-Ortega M, Bustos C, Hernandez-Presa MA, Lorenzo O, Plaza JJ, Egido J (1998) Angiotensin II participates in mononuclear cell recruitment in experimental immune complex nephritis through nuclear factor-kappa B activation and monocyte chemoattractant protein-1 synthesis. J Immunol 161(1):430–439
Silva-Filho JL, Souza MC, Henriques MG, Morrot A, Savino W, Caruso-Neves C, Pinheiro AA (2015) Renin-angiotensin system contributes to naive T-cell migration in vivo. Arch Biochem Biophys 573:1–13. https://doi.org/10.1016/j.abb.2015.02.035
Hoch NE, Guzik TJ, Chen W, Deans T, Maalouf SA, Gratze P, Weyand C, Harrison DG (2009) Regulation of T-cell function by endogenously produced angiotensin II. Am J Phys Regul Integr Comp Phys 296(2):R208–R216. https://doi.org/10.1152/ajpregu.90521.2008
Carey RM (2013) The intrarenal renin-angiotensin and dopaminergic systems: control of renal sodium excretion and blood pressure. Hypertension 61(3):673–680. https://doi.org/10.1161/HYPERTENSIONAHA.111.00241
Matsuyama T, Ohashi N, Ishigaki S, Isobe S, Tsuji N, Fujikura T, Tsuji T, Kato A, Miyajima H, Yasuda H (2018) The relationship between the intrarenal dopamine system and intrarenal renin-angiotensin system depending on the renal function. Intern Med 57(22):3241–3247. https://doi.org/10.2169/internalmedicine.0994-18
Rukavina Mikusic NL, Kouyoumdzian NM, Uceda A, Del Mauro JS, Pandolfo M, Gironacci MM, Puyo AM, Toblli JE, Fernandez BE, Choi MR (2018) Losartan prevents the imbalance between renal dopaminergic and renin angiotensin systems induced by fructose overload. l-Dopa/dopamine index as new potential biomarker of renal dysfunction. Metabolism 85:271–285. https://doi.org/10.1016/j.metabol.2018.04.010
Yang S, Yao B, Zhou Y, Yin H, Zhang MZ, Harris RC (2012) Intrarenal dopamine modulates progressive angiotensin II-mediated renal injury. Am J Physiol Ren Physiol 302(6):F742–F749. https://doi.org/10.1152/ajprenal.00583.2011
Khan F, Spicarova Z, Zelenin S, Holtback U, Scott L, Aperia A (2008) Negative reciprocity between angiotensin II type 1 and dopamine D1 receptors in rat renal proximal tubule cells. Am J Physiol Ren Physiol 295(4):F1110–F1116. https://doi.org/10.1152/ajprenal.90336.2008
Ihara S, Hirata Y, Koike K (2017) TGF-beta in inflammatory bowel disease: a key regulator of immune cells, epithelium, and the intestinal microbiota. J Gastroenterol 52(7):777–787. https://doi.org/10.1007/s00535-017-1350-1
Abraham BP, Ahmed T, Ali T (2017) Inflammatory bowel disease: pathophysiology and current therapeutic approaches. Handb Exp Pharmacol 239:115–146. https://doi.org/10.1007/164_2016_122
Azimi T, Nasiri MJ, Chirani AS, Pouriran R, Dabiri H (2018) The role of bacteria in the inflammatory bowel disease development: a narrative review. APMIS 126(4):275–283. https://doi.org/10.1111/apm.12814
Campos-Acuna J, Elgueta D, Pacheco R (2019) T-cell-driven inflammation as a mediator of the gut-brain axis involved in Parkinson’s disease. Front Immunol 10:239. https://doi.org/10.3389/fimmu.2019.00239
Wu W, Sun M, Zhang HP, Chen T, Wu R, Liu C, Yang G, Geng XR, Feng BS, Liu Z, Liu Z, Yang PC (2014) Prolactin mediates psychological stress-induced dysfunction of regulatory T cells to facilitate intestinal inflammation. Gut 63(12):1883–1892. https://doi.org/10.1136/gutjnl-2013-306083
Tolstanova G, Deng X, Ahluwalia A, Paunovic B, Prysiazhniuk A, Ostapchenko L, Tarnawski A, Sandor Z, Szabo S (2015) Role of dopamine and D2 dopamine receptor in the pathogenesis of inflammatory bowel disease. Dig Dis Sci 60(10):2963–2975. https://doi.org/10.1007/s10620-015-3698-5
Shi Y, Liu T, He L, Dougherty U, Chen L, Adhikari S, Alpert L, Zhou G, Liu W, Wang J, Deb DK, Hart J, Liu SQ, Kwon J, Pekow J, Rubin DT, Zhao Q, Bissonnette M, Li YC (2016) Activation of the renin-angiotensin system promotes colitis development. Sci Rep 6:27552. https://doi.org/10.1038/srep27552
Garrido-Gil P, Dominguez-Meijide A, Moratalla R, Guerra MJ, Labandeira-Garcia JL (2018) Aging-related dysregulation in enteric dopamine and angiotensin system interactions: implications for gastrointestinal dysfunction in the elderly. Oncotarget 9(13):10834–10846. https://doi.org/10.18632/oncotarget.24330
Yang G, Gray TS, Sigmund CD, Cassell MD (1999) The angiotensinogen gene is expressed in both astrocytes and neurons in murine central nervous system. Brain Res 817(1-2):123–131. https://doi.org/10.1016/s0006-8993(98)01236-0
Stornetta RL, Hawelu-Johnson CL, Guyenet PG, Lynch KR (1988) Astrocytes synthesize angiotensinogen in brain. Science 242(4884):1444–1446. https://doi.org/10.1126/science.3201232
Cuadra AE, Shan Z, Sumners C, Raizada MK (2010) A current view of brain renin-angiotensin system: Is the (pro)renin receptor the missing link? Pharmacol Ther 125(1):27–38. https://doi.org/10.1016/j.pharmthera.2009.07.007
Dominguez-Meijide A, Rodriguez-Perez AI, Diaz-Ruiz C, Guerra MJ, Labandeira-Garcia JL (2017) Dopamine modulates astroglial and microglial activity via glial renin-angiotensin system in cultures. Brain Behav Immun 62:277–290. https://doi.org/10.1016/j.bbi.2017.02.013
Gonzalez H, Pacheco R (2014) T-cell-mediated regulation of neuroinflammation involved in neurodegenerative diseases. J Neuroinflammation 11(1):201. https://doi.org/10.1186/s12974-014-0201-8
Munoz A, Garrido-Gil P, Dominguez-Meijide A, Labandeira-Garcia JL (2014) Angiotensin type 1 receptor blockage reduces l-dopa-induced dyskinesia in the 6-OHDA model of Parkinson’s disease. Involvement of vascular endothelial growth factor and interleukin-1beta. Exp Neurol 261:720–732. https://doi.org/10.1016/j.expneurol.2014.08.019
Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E (2003) Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 24(2):197–211. https://doi.org/10.1016/s0197-4580(02)00065-9
Braak H, Rub U, Gai WP, Del Tredici K (2003) Idiopathic Parkinson’s disease: possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J Neural Transm 110(5):517–536. https://doi.org/10.1007/s00702-002-0808-2
Garrido-Gil P, Rodriguez-Perez AI, Dominguez-Meijide A, Guerra MJ, Labandeira-Garcia JL (2018) Bidirectional neural interaction between central dopaminergic and gut lesions in Parkinson’s disease models. Mol Neurobiol 55:7297–7316. https://doi.org/10.1007/s12035-018-0937-8
Rodriguez-Pallares J, Rey P, Parga JA, Munoz A, Guerra MJ, Labandeira-Garcia JL (2008) Brain angiotensin enhances dopaminergic cell death via microglial activation and NADPH-derived ROS. Neurobiol Dis 31(1):58–73. https://doi.org/10.1016/j.nbd.2008.03.003
Joglar B, Rodriguez-Pallares J, Rodriguez-Perez AI, Rey P, Guerra MJ, Labandeira-Garcia JL (2009) The inflammatory response in the MPTP model of Parkinson’s disease is mediated by brain angiotensin: relevance to progression of the disease. J Neurochem 109(2):656–669. https://doi.org/10.1111/j.1471-4159.2009.05999.x
Labandeira-Garcia JL, Garrido-Gil P, Rodriguez-Pallares J, Valenzuela R, Borrajo A, Rodriguez-Perez AI (2014) Brain renin-angiotensin system and dopaminergic cell vulnerability. Front Neuroanat 8:67. https://doi.org/10.3389/fnana.2014.00067
Abeliovich A, Gitler AD (2016) Defects in trafficking bridge Parkinson’s disease pathology and genetics. Nature 539(7628):207–216. https://doi.org/10.1038/nature20414
Montoya A, Elgueta D, Campos J, Chovar O, Falcon P, Matus S, Alfaro I, Bono MR, Pacheco R (2019) Dopamine receptor D3 signalling in astrocytes promotes neuroinflammation. J Neuroinflammation 16(1):258. https://doi.org/10.1186/s12974-019-1652-8
Shao W, Zhang SZ, Tang M, Zhang XH, Zhou Z, Yin YQ, Zhou QB, Huang YY, Liu YJ, Wawrousek E, Chen T, Li SB, Xu M, Zhou JN, Hu G, Zhou JW (2013) Suppression of neuroinflammation by astrocytic dopamine D2 receptors via alphaB-crystallin. Nature 494(7435):90–94. https://doi.org/10.1038/nature11748
Fletcher JM, Lalor SJ, Sweeney CM, Tubridy N, Mills KH (2010) T cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Clin Exp Immunol 162(1):1–11. https://doi.org/10.1111/j.1365-2249.2010.04143.x
Haider L, Fischer MT, Frischer JM, Bauer J, Hoftberger R, Botond G, Esterbauer H, Binder CJ, Witztum JL, Lassmann H (2011) Oxidative damage in multiple sclerosis lesions. Brain J Neurol 134(Pt 7):1914–1924. https://doi.org/10.1093/brain/awr128
Smith KJ (2011) Newly lesioned tissue in multiple sclerosis--a role for oxidative damage? Brain J Neurol 134(Pt 7):1877–1881. https://doi.org/10.1093/brain/awr144
Kawajiri M, Mogi M, Higaki N, Matsuoka T, Ohyagi Y, Tsukuda K, Kohara K, Horiuchi M, Miki T, Kira JI (2009) Angiotensin-converting enzyme (ACE) and ACE2 levels in the cerebrospinal fluid of patients with multiple sclerosis. Mult Scler 15(2):262–265. https://doi.org/10.1177/1352458508097923
Platten M, Youssef S, Hur EM, Ho PP, Han MH, Lanz TV, Phillips LK, Goldstein MJ, Bhat R, Raine CS, Sobel RA, Steinman L (2009) Blocking angiotensin-converting enzyme induces potent regulatory T cells and modulates TH1- and TH17-mediated autoimmunity. Proc Natl Acad Sci U S A 106(35):14948–14953. https://doi.org/10.1073/pnas.0903958106
Kaasinen V, Joutsa J, Rissanen E, Airas L, Soilu-Hanninen M, Noponen T (2019) Progressive dopaminergic defect in a patient with primary progressive multiple sclerosis. Mult Scler Relat Disord 36:101385. https://doi.org/10.1016/j.msard.2019.101385
Lund BT, Stone R, Levy AM, Lee S, Amundson E, Kashani N, Rodgers KE, Kelland EE (2019) Reduced disease severity following therapeutic treatment with angiotensin 1-7 in a mouse model of multiple sclerosis. Neurobiol Dis 127:87–100. https://doi.org/10.1016/j.nbd.2019.02.018
Hammer A, Yang G, Friedrich J, Kovacs A, Lee DH, Grave K, Jorg S, Alenina N, Grosch J, Winkler J, Gold R, Bader M, Manzel A, Rump LC, Muller DN, Linker RA, Stegbauer J (2016) Role of the receptor Mas in macrophage-mediated inflammation in vivo. Proc Natl Acad Sci U S A 113(49):14109–14114. https://doi.org/10.1073/pnas.1612668113
Backhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI (2005) Host-bacterial mutualism in the human intestine. Science 307(5717):1915–1920. https://doi.org/10.1126/science.1104816
Pacheco R (2019) Cross-talk between T-cells and gut-microbiota in neurodegenerative disorders. Neural Regen Res 14(12):2091–2092. https://doi.org/10.4103/1673-5374.262582
Unger MM, Spiegel J, Dillmann KU, Grundmann D, Philippeit H, Burmann J, Fassbender K, Schwiertz A, Schafer KH (2016) Short chain fatty acids and gut microbiota differ between patients with Parkinson’s disease and age-matched controls. Parkinsonism Relat Disord 32:66–72. https://doi.org/10.1016/j.parkreldis.2016.08.019
Marino E, Richards JL, McLeod KH, Stanley D, Yap YA, Knight J, McKenzie C, Kranich J, Oliveira AC, Rossello FJ, Krishnamurthy B, Nefzger CM, Macia L, Thorburn A, Baxter AG, Morahan G, Wong LH, Polo JM, Moore RJ, Lockett TJ, Clarke JM, Topping DL, Harrison LC, Mackay CR (2017) Gut microbial metabolites limit the frequency of autoimmune T cells and protect against type 1 diabetes. Nat Immunol 18(5):552–562. https://doi.org/10.1038/ni.3713
Haase S, Haghikia A, Wilck N, Muller DN, Linker RA (2018) Impacts of microbiome metabolites on immune regulation and autoimmunity. Immunology 154(2):230–238. https://doi.org/10.1111/imm.12933
Hui W, Yu D, Cao Z, Zhao X (2019) Butyrate inhibit collagen-induced arthritis via Treg/IL-10/Th17 axis. Int Immunopharmacol 68:226–233. https://doi.org/10.1016/j.intimp.2019.01.018
Macfarlane S, Macfarlane GT (2003) Regulation of short-chain fatty acid production. Proc Nutr Soc 62(1):67–72. https://doi.org/10.1079/PNS2002207
Hoyles L, Snelling T, Umlai UK, Nicholson JK, Carding SR, Glen RC, McArthur S (2018) Microbiome-host systems interactions: protective effects of propionate upon the blood-brain barrier. Microbiome 6(1):55. https://doi.org/10.1186/s40168-018-0439-y
Morris G, Berk M, Carvalho A, Caso JR, Sanz Y, Walder K, Maes M (2017) The role of the microbial metabolites including tryptophan catabolites and short chain fatty acids in the pathophysiology of immune-inflammatory and neuroimmune disease. Mol Neurobiol 54(6):4432–4451. https://doi.org/10.1007/s12035-016-0004-2
Suarez-Zamorano N, Fabbiano S, Chevalier C, Stojanovic O, Colin DJ, Stevanovic A, Veyrat-Durebex C, Tarallo V, Rigo D, Germain S, Ilievska M, Montet X, Seimbille Y, Hapfelmeier S, Trajkovski M (2015) Microbiota depletion promotes browning of white adipose tissue and reduces obesity. Nat Med 21(12):1497–1501. https://doi.org/10.1038/nm.3994
Wang L, Zhu Q, Lu A, Liu X, Zhang L, Xu C, Liu X, Li H, Yang T (2017) Sodium butyrate suppresses angiotensin II-induced hypertension by inhibition of renal (pro)renin receptor and intrarenal renin-angiotensin system. J Hypertens 35(9):1899–1908. https://doi.org/10.1097/HJH.0000000000001378
Dave LA, Hayes M, Montoya CA, Rutherfurd SM, Moughan PJ (2016) Human gut endogenous proteins as a potential source of angiotensin-I-converting enzyme (ACE-I)-, renin inhibitory and antioxidant peptides. Peptides 76:30–44. https://doi.org/10.1016/j.peptides.2015.11.003
Asano Y, Hiramoto T, Nishino R, Aiba Y, Kimura T, Yoshihara K, Koga Y, Sudo N (2012) Critical role of gut microbiota in the production of biologically active, free catecholamines in the gut lumen of mice. Am J Physiol Gastrointest Liver Physiol 303(11):G1288–G1295. https://doi.org/10.1152/ajpgi.00341.2012
Diaz Heijtz R, Wang S, Anuar F, Qian Y, Bjorkholm B, Samuelsson A, Hibberd ML, Forssberg H, Pettersson S (2011) Normal gut microbiota modulates brain development and behavior. Proc Natl Acad Sci U S A 108(7):3047–3052. https://doi.org/10.1073/pnas.1010529108
Koronyo-Hamaoui M, Sheyn J, Hayden EY, Li S, Fuchs DT, Regis GC, Lopes DHJ, Black KL, Bernstein KE, Teplow DB, Fuchs S, Koronyo Y, Rentsendorj A (2020) Peripherally derived angiotensin converting enzyme-enhanced macrophages alleviate Alzheimer-related disease. Brain J Neurol 143(1):336–358. https://doi.org/10.1093/brain/awz364
Ongali B, Nicolakakis N, Tong XK, Lecrux C, Imboden H, Hamel E (2018) Transforming growth factor-beta1 induces cerebrovascular dysfunction and astrogliosis through angiotensin II type 1 receptor-mediated signaling pathways. Can J Physiol Pharmacol 96(5):527–534. https://doi.org/10.1139/cjpp-2017-0640
Singh N, Gurav A, Sivaprakasam S, Brady E, Padia R, Shi H, Thangaraju M, Prasad PD, Manicassamy S, Munn DH, Lee JR, Offermanns S, Ganapathy V (2014) Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity 40(1):128–139. https://doi.org/10.1016/j.immuni.2013.12.007
Gong X, Hu H, Qiao Y, Xu P, Yang M, Dang R, Han W, Guo Y, Chen D, Jiang P (2019) The involvement of renin-angiotensin system in lipopolysaccharide-induced behavioral changes, neuroinflammation, and disturbed insulin signaling. Front Pharmacol 10:318. https://doi.org/10.3389/fphar.2019.00318
Drews HJ, Yenkoyan K, Lourhmati A, Buadze M, Kabisch D, Verleysdonk S, Petschak S, Beer-Hammer S, Davtyan T, Frey WH 2nd, Gleiter CH, Schwab M, Danielyan L (2019) Intranasal losartan decreases perivascular beta amyloid, inflammation, and the decline of neurogenesis in hypertensive rats. Neurotherapeutics 16(3):725–740. https://doi.org/10.1007/s13311-019-00723-6
Trigiani LJ, Royea J, Lacalle-Aurioles M, Tong XK, Hamel E (2018) pleiotropic benefits of the angiotensin receptor blocker candesartan in a mouse model of Alzheimer disease. Hypertension 72(5):1217–1226. https://doi.org/10.1161/HYPERTENSIONAHA.118.11775
Rodriguez-Perez AI, Sucunza D, Pedrosa MA, Garrido-Gil P, Kulisevsky J, Lanciego JL, Labandeira-Garcia JL (2018) Angiotensin type 1 receptor antagonists protect against alpha-synuclein-induced neuroinflammation and dopaminergic neuron death. Neurotherapeutics 15(4):1063–1081. https://doi.org/10.1007/s13311-018-0646-z
Salmani H, Hosseini M, Beheshti F, Baghcheghi Y, Sadeghnia HR, Soukhtanloo M, Shafei MN, Khazaei M (2018) Angiotensin receptor blocker, losartan ameliorates neuroinflammation and behavioral consequences of lipopolysaccharide injection. Life Sci 203:161–170. https://doi.org/10.1016/j.lfs.2018.04.033
Khallaf WAI, Messiha BAS, Abo-Youssef AMH, El-Sayed NS (2017) Protective effects of telmisartan and tempol on lipopolysaccharide-induced cognitive impairment, neuroinflammation, and amyloidogenesis: possible role of brain-derived neurotrophic factor. Can J Physiol Pharmacol 95(7):850–860. https://doi.org/10.1139/cjpp-2017-0042
Ali MR, Abo-Youssef AM, Messiha BA, Khattab MM (2016) Tempol and perindopril protect against lipopolysaccharide-induced cognition impairment and amyloidogenesis by modulating brain-derived neurotropic factor, neuroinflammation and oxido-nitrosative stress. Naunyn Schmiedeberg's Arch Pharmacol 389(6):637–656. https://doi.org/10.1007/s00210-016-1234-6
Garrido-Gil P, Joglar B, Rodriguez-Perez AI, Guerra MJ, Labandeira-Garcia JL (2012) Involvement of PPAR-gamma in the neuroprotective and anti-inflammatory effects of angiotensin type 1 receptor inhibition: effects of the receptor antagonist telmisartan and receptor deletion in a mouse MPTP model of Parkinson's disease. J Neuroinflammation 9:38. https://doi.org/10.1186/1742-2094-9-38
Dominguez-Meijide A, Villar-Cheda B, Garrido-Gil P, Sierrra-Paredes G, Guerra MJ, Labandeira-Garcia JL (2014) Effect of chronic treatment with angiotensin type 1 receptor antagonists on striatal dopamine levels in normal rats and in a rat model of Parkinson's disease treated with L-DOPA. Neuropharmacology 76(Pt A):156–168. https://doi.org/10.1016/j.neuropharm.2013.07.016
Sonsalla PK, Coleman C, Wong LY, Harris SL, Richardson JR, Gadad BS, Li W, German DC (2013) The angiotensin converting enzyme inhibitor captopril protects nigrostriatal dopamine neurons in animal models of parkinsonism. Exp Neurol 250:376–383. https://doi.org/10.1016/j.expneurol.2013.10.014
Borrajo A, Rodriguez-Perez AI, Diaz-Ruiz C, Guerra MJ, Labandeira-Garcia JL (2014) Microglial TNF-alpha mediates enhancement of dopaminergic degeneration by brain angiotensin. Glia 62(1):145–157. https://doi.org/10.1002/glia.22595
Guo X, Namekata K, Kimura A, Harada C, Harada T (2017) The renin-angiotensin system regulates neurodegeneration in a mouse model of optic neuritis. Am J Pathol 187(12):2876–2885. https://doi.org/10.1016/j.ajpath.2017.08.012
Mori K, Yoshida K, Tani J, Nakagawa Y, Hoshikawa S, Ozaki H, Ito S (2008) Effects of angiotensin II blockade on the development of autoimmune thyroiditis in nonobese diabetic mice. Clin Immunol 126(1):97–103. https://doi.org/10.1016/j.clim.2007.08.018
Uzawa A, Mori M, Taniguchi J, Kuwabara S (2014) Modulation of the kallikrein/kinin system by the angiotensin-converting enzyme inhibitor alleviates experimental autoimmune encephalomyelitis. Clin Exp Immunol 178(2):245–252. https://doi.org/10.1111/cei.12413
Okunuki Y, Usui Y, Nagai N, Kezuka T, Ishida S, Takeuchi M, Goto H (2009) Suppression of experimental autoimmune uveitis by angiotensin II type 1 receptor blocker telmisartan. Invest Ophthalmol Vis Sci 50(5):2255–2261. https://doi.org/10.1167/iovs.08-2649
Miyazaki A, Kitaichi N, Ohgami K, Iwata D, Jin XH, Iwabuchi K, Morohashi T, Ohno S, Onoe K (2008) Anti-inflammatory effect of angiotensin type 1 receptor antagonist on endotoxin-induced uveitis in rats. Graefes Arch Clin Exp Ophthalmol 246(5):747–757. https://doi.org/10.1007/s00417-007-0730-2
Wang T, Nowrangi D, Yu L, Lu T, Tang J, Han B, Ding Y, Fu F, Zhang JH (2018) Activation of dopamine D1 receptor decreased NLRP3-mediated inflammation in intracerebral hemorrhage mice. J Neuroinflammation 15(1):2. https://doi.org/10.1186/s12974-017-1039-7
Zhu J, Hu Z, Han X, Wang D, Jiang Q, Ding J, Xiao M, Wang C, Lu M, Hu G (2018) Dopamine D2 receptor restricts astrocytic NLRP3 inflammasome activation via enhancing the interaction of beta-arrestin2 and NLRP3. Cell Death Differ 25(11):2037–2049. https://doi.org/10.1038/s41418-018-0127-2
Lieberknecht V, Junqueira SC, Cunha MP, Barbosa TA, de Souza LF, Coelho IS, Santos AR, Rodrigues AL, Dafre AL, Dutra RC (2017) Pramipexole, a dopamine D2/D3 receptor-preferring agonist, prevents experimental autoimmune encephalomyelitis development in mice. Mol Neurobiol 54(2):1033–1045. https://doi.org/10.1007/s12035-016-9717-5
Kawano M, Takagi R, Kaneko A, Matsushita S (2015) Berberine is a dopamine D1- and D2-like receptor antagonist and ameliorates experimentally induced colitis by suppressing innate and adaptive immune responses. J Neuroimmunol 289:43–55. https://doi.org/10.1016/j.jneuroim.2015.10.001
Du RH, Zhou Y, Xia ML, Lu M, Ding JH, Hu G (2018) alpha-Synuclein disrupts the anti-inflammatory role of Drd2 via interfering beta-arrestin2-TAB1 interaction in astrocytes. J Neuroinflammation 15(1):258. https://doi.org/10.1186/s12974-018-1302-6
Sadeghi H, Parishani M, Akbartabar Touri M, Ghavamzadeh M, Jafari Barmak M, Zarezade V, Delaviz H, Sadeghi H (2017) Pramipexole reduces inflammation in the experimental animal models of inflammation. Immunopharmacol Immunotoxicol 39(2):80–86. https://doi.org/10.1080/08923973.2017.1284230
Melnikov M, Belousova O, Murugin V, Pashenkov capital Em C, Boysmall ka CoCA (2016) The role of dopamine in modulation of Th-17 immune response in multiple sclerosis. J Neuroimmunol 292:97-101. doi:https://doi.org/10.1016/j.jneuroim.2016.01.020
Nakano K, Yamaoka K, Hanami K, Saito K, Sasaguri Y, Yanagihara N, Tanaka S, Katsuki I, Matsushita S, Tanaka Y (2011) Dopamine induces IL-6-dependent IL-17 production via D1-like receptor on CD4 naive T cells and D1-like receptor antagonist SCH-23390 inhibits cartilage destruction in a human rheumatoid arthritis/SCID mouse chimera model. J Immunol 186(6):3745–3752. https://doi.org/10.4049/jimmunol.1002475
Nakano K, Higashi T, Hashimoto K, Takagi R, Tanaka Y, Matsushita S (2008) Antagonizing dopamine D1-like receptor inhibits Th17 cell differentiation: preventive and therapeutic effects on experimental autoimmune encephalomyelitis. Biochem Biophys Res Commun 373(2):286–291
Funding
This work was supported by Programa de Apoyo a Centros con Financiamiento Basal AFB-170004 (to Fundación Ciencia & Vida) from “Comisión Nacional de Investigación Científica y Tecnológica de Chile (CONICYT)” and by grants FONDECYT-1170093 (to R.P.), from “Fondo Nacional de Desarrollo Científico y Tecnológico de Chile,” MJFF-10332.01, and MJFF-15076 (to R.P.) from Michael J. Fox Foundation for Parkinson Research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that they have no conflict of interest.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Code availability
Not applicable.
Additional information
This article is a contribution to the special issue on: Neuro-immune Interactions - Guest Editor: David Farrar
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Campos, J., Pacheco, R. Involvement of dopaminergic signaling in the cross talk between the renin-angiotensin system and inflammation. Semin Immunopathol 42, 681–696 (2020). https://doi.org/10.1007/s00281-020-00819-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-020-00819-8