
Abstract
Purpose
Genetic variation in the activation of the prodrug cyclophosphamide (CP) by cytochrome P450 (CYP) enzymes has been shown to influence outcomes. However, CYP are also subject to phenoconversion due to either the effects of comedications or cancer associated down-regulation of expression. The aim of this study was to assess the relationship between CP bioactivation with CYP2B6 and CYP2C19 genotype, as well as CYP2C19 phenotype, in breast cancer patients.
Methods
CP and the active metabolite levels were assessed in breast cancer patients (n = 34) at cycle 1 and cycle 3 of treatment. Patients were genotyped for a series of SNP known to affect CYP2B6 and CYP2C19 function. The activity of CYP2C19 was also assessed using a probe drug.
Results
We found a significant linear gene-dose relationship with CYP2B6 coding SNP and formation of 4-hydroxycyclophosphamide. A possible association with CYP2C19 null genotype at cycle 1 was obscured at cycle 3 due to the substantial intra-individual change in CP bioactivation on subsequent dosing.
Conclusion
Comedications may be the cause for this inter-occasion variation in bioactivation of cyclophosphamide and the ensuing phenoconversion may account for the conflicting reports in the literature about the relationship between CYP2C19 genotype and CP bioactivation pharmacokinetics. Trial registration ANZCTR363222 (6/11/2012, retrospectively registered).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Cyclophosphamide (CP) is an alkylating agent used in the treatment of solid and haematological malignancies. It is also used as an immunosuppressive agent in bone marrow transplantation (stem cell mobilization and conditioning regimens), as prophylaxis against post-transplantation Graft-versus Host Disease, and for lymphodepletion in chimeric antigen receptor T-cell (CAR-T) therapy, in addition to its use in the treatment of autoimmune disorders such as lupus nephritis.
As a prodrug cyclophosphamide is dependent on bioactivation by hepatic cytochrome P450 (CYP) enzymes to elicit its therapeutic effect. This results in the formation of 4-hydroxycyclophosphamide (4OHCP), which equilibrates with its tautomer aldophosphamide in the systemic circulation and undergoes intracellular hydrolysis to form the DNA alkylating compound phosphoramide mustard (Supplementary Fig. 1). Since the rate limiting step in the activation of cyclophosphamide (CP) is formation of 4OHCP, any variability in activity or expression of the CYP enzymes involved in this hydroxylation could influence therapeutic outcomes. Numerous CYP have been reported to catalyse this reaction including CYP2B6, CYP2C9, CYP2C19, CYP3A4, CYP3A5 and CYP2J2 [summarised in 1]. However, CYP2B6 and CYP2C19 are the hepatic enzymes with the highest activation of cyclophosphamide (intrinsic clearance 56.9 and 5 µL/min mg, respectively) [2]. Importantly both these enzymes have approximately threefold higher activity than CYP3A4 when assessed for formation of 4OHCP and subsequent DNA damage [3].
Both CYP2B6 and CYP2C19 display substantial inherited variation in expression and activity due common single nucleotide polymorphisms (SNP) [4, 5]. Individuals who inherit two loss of function variant alleles for CYP2C19 have null enzyme function and are termed ‘poor metabolisers’ of drug substrates for this enzyme. The prevalence of these individuals varies globally, with ~ 3% of people of European and ~ 20% of people of Japanese ancestry having this phenotype. CYP2B6 pharmacogenetics are complex since the common SNP exist in various combinations. Of these, the *6 allele (rs2279343 + rs3745274 haplotype) is most prevalent in populations with European ancestry. Moreover, the coding region SNP in CYP2B6 appear to alter activity in a substrate-dependent manner.
Whilst there is substantial evidence for the role of CYP2C19 and CYP2B6 germline pharmacogenetic variation in both plasma pharmacokinetics and the clinical outcomes of cyclophosphamide, in contexts as diverse as haematological malignancy, breast cancer, systemic lupus erythematosus and myeloablation [reviewed in 1], there is often a lack of assessment of both of these CYP in many of these studies.
In addition, to pharmacogenetic variation, CYP enzyme activity is also subject to phenoconversion. This is where there is either induction or inhibition of the enzyme by comedications. Phenoconversion can also occur due to disease-associated down-regulation of CYP expression [6]. It has previously been demonstrated that there is a high prevalence of genotype–phenotype discordance for CYP2C19 probe drugs in cancer patients [7,8,9,10]. It is not clear whether this additional phenoconversion influences the relationship between CYP pharmacogenetics and CP bioactivation.
The aim of this study was to assess the relationship between CP bioactivation with CYP2B6 and CYP2C19 genotype, as well as CYP2C19 phenotype, in breast cancer patients.
Methods
This study received approval from the New Zealand Heath and Disability Northern X Regional Ethics committee NTX/12/06/052 and was registered (ANZCTR363222). Patients were eligible for the study if they were diagnosed with carcinoma of the breast and scheduled to receive cyclophosphamide treatment. Patients had to be at least 18 years of age and able to give informed written consent. Only patients with good ECOG performance status (0–2) were eligible. Patients with poor liver and kidney function were not eligible for the study (i.e. serum creatinine > 1.5 × ULN; AST, ALT > 2.5 × ULN; ALP > 5 × ULN; Bilirubin > ULN). Patients with any active infection or concurrent chronic inflammatory condition were excluded from the study.
To minimise any drug–drug interactions with the CYP2C19 phenotyping test patients were not eligible for the study if receiving a CYP2C19 inducer drug required for other concurrent medical conditions when a washout period of 5 days was not clinically feasible. Administration of known CYP2C19 inhibitor drugs, especially omeprazole, were suspended for a washout period of at least 24 h prior to phenotyping.
Following written informed consent whole blood (8.5 mL) was collected into PAXgene blood DNA tubes (Qiagen, Hilden, Germany) and stored at − 20 °C prior to analysis. DNA was extracted using the PAXgene Blood DNA kit (Qiagen, Hilden, Germany) and analysed for CYP2C19*2 (rs4244285), CYP2C19*3 (rs49486893), CYP2C19*17 (rs12248560) and CYP2B6 − 2320 T > C (rs7254579) alleles using Sequenom MASSarray (Grafton Clinical Genomics, Auckland). Primer sequences are given in Supplementary Table 1. CYP2B6 genotypes were determined using RFLP-PCR as previously reported [11].
The plasma pharmacokinetics of 4OHCP are formation-rate limited and directly correlate with plasma CP concentrations. Since patients receive individualised CP dosage (mg/m2) based on body size, data are reported as the ratio of 4OHCP/CP to indicate the fraction of drug bioactivated. However, this assumes that the competing dechloroethylation pathway catalysed by CYP3A4 is minimal (Supplementary Fig. 1), hence the data are also shown as formation of 4OHCP. To minimise patient clinic time, we chose to use limited sampling, with blood samples collected at 15 min and 30 min after completion of 1 h infusion. These post-infusion timepoints approximate to Tmax and there is a previously characterised direct relationship between the prodrug and 4OHCP metabolite exposure over time [12]. The mean values from these two timepoints are reported as the bioactivation ratio. Plasma concentrations of CP and 4OHCP (immediately stabilised upon blood collection) were determined as previously reported [2].
The CYP2C19 probe substrate proguanil (200 mg, PO) was administered 1 day prior to scheduled cycle of CP treatment on two separate test occasions (at cycle 1 and cycle 3 of treatment). Proguanil (PG) and the CYP2C19 catalysed metabolite cycloguanil (CG) were quantified in the 3 h plasma sample as previously reported and a PG/CG cut-point of > 10 used to identify phenotypic poor metabolisers [9, 10].
C-reactive protein was analysed by the local clinical laboratory service.
Statistical analyses were performed in GraphPad PRISM (version 6, GraphPad Software Inc., USA). Normality was determined using D’Agostino-Pearson test. Normally distributed continuous data are reported as mean and standard deviation (SD). Non-normally distributed data are reported as median and interquartile range (IQR). Group comparisons for non-parametric data used Wilcoxon matched-pairs signed rank test, and for parametric data either an un-paired T test or the ANOVA linear trend test was used, as appropriate. Values p < 0.05 were considered to be statistically significant.
Results
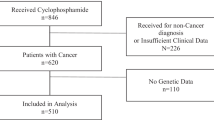
Forty-three female patients were registered in the study over a 2-year period (2012–2014). Of these, three patients were ineligible or declined the trial prior to start and one patient declined further chemotherapy after two cycles following febrile neutropenia and infection. Bioanalysis data were incomplete or invalid for a further five patients, hence, the relationship between cyclophosphamide bioactivation at cycle 1 and cycle 3 with pharmacogenetics was only assessable in 34 patients.
Patients received treatment for breast cancer based on clinical best practice and a number of different cyclophosphamide regimens were used (Table 1). The majority of the patients received adjuvant treatment. Dosages were typically 500–600 mg/m2, more than half of the patients (56%) had a BMI > 30 kg/m2.
Plasma concentrations of 4OHCP ranged from 0 to 0.57 µg/mL (15–30 min after end of 1 h infusion). Plasma CP concentrations ranged from 54.36 to 134.8 µg/mL at these time points. These values are similar to those previously reported in pharmacokinetic studies at the end of 1 h infusion at dosages < 1000 mg/m2. The bioactivation ratio for cyclophosphamide (4OHCP/CP) ranged from 0.00023 to 0.00366 and was not normally distributed. There was substantial inter-occasion variation in bioactivation ratios between treatment cycles 1 and 3, ranging from 30 to 1591% of the cycle 1 values. The bioactivation at cycle 3 compared to cycle 1 increased (> 150%) in 11 patients and decreased (< 65%) in six patients. There was minimal change (± 20%) in 16 patients; 8 of these individuals had almost identical values (± 5%) at both cycles 1 and 3. However, there was no significant difference (p < 0.56) in the values between cycles across the n = 34 patients. There was no simple relationship between regimen and these inter-occasion changes in bioactivation ratio (Fig. 1).

Bioactivation of cyclophosphamide in the patient cohort at cycle 1 compared with cycle 3. Plasma concentrations of 4-hydroxycyclophosphamide (4OHCP) relative to cyclophosphamide (CP) are reported. At cycle 1 across all CP regimens the median (IQR) ratio was 0.00105 (0.0006–0.00165) versus 0.00119 (0.00029–0.0019) at cycle 3. This was not significantly different between cycles (P = 0.20). Data are grouped based on the type of cyclophosphamide regimen received. FEC 5-FU, epirubicin and cyclophosphamide (+ docetaxel); TC docetaxel (taxotere) and cyclophosphamide; AC adriamycin and cyclophosphamide. Samples with no detectable 4OHCP were given a nominal value (lower limit of detection of 0.01 µg/mL) to calculate the ratio
All SNP assessed were in Hardy–Weinberg equilibrium, and the minor allele frequencies were consistent with that expected for the mixed ancestry (European, Māori, Pacific Peoples, Asian) of the cohort (Supplementary Table 2). At cycle 1, patients (n = 3) with a homozygous null function genotype (*2/*2) for CYP2C19 had a significantly lower (p = 0.043) bioactivation ratio (mean ± SD, 0.00051 ± 0.00028) compared with individuals who were not carriers of CYP2C19 null variants (mean ± SD, 0.00135 ± 0.0006). Whilst there appeared to be a gene-dose trend at cycle 1 this was not statistically significant (p = 0.148), and no associations were observed at cycle 3 (Fig. 2).

Relationship between CYP2C19 null function genotype or CYP2B6 SNP variant genotype and cyclophosphamide bioactivation ratio. Data are shown as scatter plots with mean values for each genotype group. CYP2C19 null function subjects (homozygous *2/*2) have significantly lower bioactivation compared to wildtype (WT) individuals (p = 0.043) at cycle 1. The *17 carriers are shown as grey symbols. ANOVA linear trend test indicates no significant relationship for CYP2C19 *2 genotype and bioactivation at cycle 1 (p = 0.148) and cycle 3 (p = 0.844). Individuals homozygous variant for CYP2B6 A785G or G516T allele did not have a significantly lower bioactivation compared with individuals wildtype at these loci (p = 0.329 and 0.328, respectively, at cycle 1; p = 0.340 and 0.390 at cycle 3). There was an apparent (non-significant) trend towards a gene-dose relationship for A785G and G516T at cycle 1 and/or cycle 3 (p values shown on graphs)
The relationships for each CYP2B6 coding SNP variant are also shown in Fig. 2. One individual was homozygous variant at both the A785G and G516T SNP (i.e. GG, TT). These two SNP are often co-inherited and form a haplotype termed the *6 allele. Another individual was homozygous variant at C1459T (i.e. TT, the *5 allele). Whilst there appeared to be a gene-dose trend for both A785G and G516T SNP at cycle 1, this was not significant (p = 0.0841 and p = 0.0572, respectively). Unlike the CYP2C19 null function genotype, individuals who were homozygous variant for either A785G or G516T did not have a significantly lower bioactivation ratio compared with those individuals who were wildtype at these loci (p = 0.329 and 0.328, respectively, at cycle 1). No gene-dose association was observed for the C1459T variant at either cycle (p > 0.50). The promoter region variants (− 750 T > C and − 2320 T > C) also had no association with bioactivation ratio (p > 0.50) at either cycle of treatment (Supplementary Fig. 2).
The bioactivation ratio assumes minimal conversion of CP via the competing pathway to form dechlorethylcyclophosphamide, which is catalysed by CYP3A4. The association between CYP2C19 or CYP2B6 genotype and formation of 4OHCP is shown (Fig. 3). There was lower 4OHCP formation at 15 min post-infusion in the three individuals who were homozygous null (*2/*2) for CYP2C19 and this was significantly lower than that observed in individuals who were not carriers of null function alleles (p < 0.05). However, this association was not observed at cycle 3. There was a significant gene-dose linear trend for both CYP2B6 A785G and G516T and 4OHCP formation at cycle 1 (p < 0.05) and this trend remained significant for the A785G SNP at cycle 3 (p < 0.001). This same significant gene-dose effect for CYP2B6 was observed for 4OHCP formation at 30 min post-infusion (Supplementary Fig. 3).

Relationship between CYP2C19 null function genotype or CYP2B6 SNP variant genotype and 4-hydroxycylophosphamide (4OHCP) formation. Data are shown as scatter plots with mean values for each genotype group. CYP2C19 null function subjects have significantly lower 4OHCP formation 15 min after end of infusion compared to wildtype (WT) individuals (p = 0.029) at cycle 1. The *17 carriers are shown as grey symbols. ANOVA linear trend test indicates no significant relationship for CYP2C19 genotype and 4OHCP at cycle 1 (p = 0.238) and cycle 3 (p = 0.929). There was a significant gene-dose trend for A785G and G516T at cycle 1 and for A785G at cycle 3 (p values shown on graphs)
CYP2C19 activity was probed and a PG/CG > 10 used as a cut-point to detect poor metaboliser phenotype. Three individuals were confirmed as homozygous null genotype (*2/*2) and had values above this cut-point (Fig. 4). Previous studies have shown that phenotype–genotype discordance can occur in a proportion of cancer patients (i.e. poor metaboliser CYP2C19 activity in the presence of at least one functional allele). At test 1, phenotype–genotype discordance was observed in one patient. At test 2 (prior to cycle 3), genotype–phenotype discordance was apparent in six patients. Notably the CYP2C19 activity had significantly declined (p = 0.0012) across the whole cohort between test 1 and test 2 (i.e. PG/CG ratio increased). The median (IQR) value for PG/CG across the patients at test 1 was 1.71 (0.8–5.3) versus 3.46 (1.08–14.81) at test 2. There was no simple relationship between tumour burden (metastatic disease vs adjuvant) and decreased CYP2C19 activity. CRP ranged between 0 and 108 mg/L at test 1 and 0–32 mg/L at test 2, however, the median (IQR) was identical at 3 (1–6) mg/L on the two test occasions. In those individuals who were not CYP2C19 null genotype, there was a linear relationship between CRP and PG/CG ratio at test 2 (slope 0.248, r2 = 0.819). There were no relationships between either CRP or the PG/CG ratios with CP bioactivation at either test occasion.

CYP2C19 activity in patients at two test occasions, prior to cycle 1 and cycle 3 of chemotherapy. Proguanil (PG) concentrations relative to cycloguanil (CG) are shown as scatter plots for each CYP2C19 genotype category. CG was not detectable in some samples and to calculate the PG/CG ratio a nominal value (lower limit of detection, 1 ng/ml was used). Squares are patients with metastatic disease. The cut-point previously used to identify null CYP2C19 activity (PG/CG > 10) is shown as a dotted line. The one individual with *17/*2 genotype is shown as a filled circle. A high PG/CG ratio indicates low CYP2C19 activity
Discussion
The role of CYP2C19 and CYP2B6 pharmacogenes in the inter-individual variability of 4OHCP formation has been widely but inconsistently studied. More than 20 studies have demonstrated that CYP2B6 and/or CYP2C19 loss of function SNP variants appear to influence bioactivation pharmacokinetics or therapeutic outcomes [1]. In this small study of 34 breast cancer patients, using a limited sampling technique, we have shown that the coding region SNP (A785G and G516T) in CYP2B6 (*6 allele) demonstrate a significant gene-dose trend for decreased formation of 4OHCP. A number of previous studies have demonstrated associations with these individual SNP (or the *6 allele) for bioactivation and clinical outcomes in cancer patients [13,14,15,16,17,18,19,20]. Whilst, CYP2C19 homozygous null function also appears to influence 4OHCP formation, this was weak compared with CYP2B6. This confirms previous reports [14, 15, 21, 22] and re-iterates the importance of inclusion of CYP2C19 null function genotype in CP pharmacokinetic bioactivation studies, since this pharmacogene is often overlooked. Whilst recombinant CYP2B6 has higher intrinsic activity than CYP2C19 for CP hydroxylation [2], SNP coding region variants of CYP2B6 lead to only partially altered activity (due to lower protein expression and/or changes in catalytic function), rather than the total loss of functional protein caused by the CYP2C19*2 and *3 variants. Hence these CYP2C19 null function variants may have more clinical impact on the hydroxylation of CP than would be expected based solely on intrinsic activity. Assessment of CYP2J2*7 (rs890293) should be considered in future studies since this SNP increases the activity of this extrahepatic enzyme, which can catalyse formation of 4OHCP albeit with relatively low intrinsic activity [23].
Importantly, the possible association between decreased 4OHCP formation and CYP2C19 null function, as well as the gene-dose trend for CYP2B6 G516T, was only observed at the first cycle of treatment. This may have been driven by the substantial change in apparent CP bioactivation of each individual between cycle 1 and cycle 3. Notably, many of the previous studies which have shown associations between either of these pharmacogenes and plasma pharmacokinetics of 4OHCP formation relative to CP have assessed patients at the first dose or first treatment cycle.
Inter-occasion variability in CP pharmacokinetics was initially reported in 1980 [24]. In the present study, we noted that whilst almost half (47%) of the patients had no substantive change between cycles, in approximately one-third of patients bioactivation ratio increased, whereas in about 15% of the patients bioactivation ratio declined between cycles. These proportions and extent of inter-occasion variability are almost identical to that previously reported for CP clearance in breast cancer patients over two or three cycles of treatment [25, 26].
The factors influencing this intra-occasion variability likely include gene expression changes (induction or down-regulation) and drug–drug inhibition due to comedications. Using a probe drug, we demonstrated that there was a significant decline in CYP2C19 activity across the cohort of patients prior to cycle 3 compared with the initial test values. Numerous studies have suggested that enzymes such as CYP2C19 and CYP3A4 may be particularly sensitive to down-regulation during periods of inflammation [27, 28]. Down-regulation of CYP2C19 can lead to a phenomenon known as phenotype-genotype discordance [7,8,9,10]. Whilst discordance was only observed in one patient at test 1 (naïve patients), discordance was observed in five patients at test 2 (after two cycles of chemotherapy). There was no obvious relationship between these additional phenotypic poor metaboliser individuals and CP bioactivation at cycle 3. Indeed, the impact of decreased CYP2C19 activity in these patients is likely to be minor compared with changes in CYP2B6 expression, particularly since in contrast to CYP2C19, CYP2B6 has been shown to be up-regulated by inflammation [29].
There was no association with decreased CYP2C19 function and tumour burden (metastatic vs adjuvant), however, a possible association with the circulating biomarker C-reactive protein was observed. This general decline in CYP2C19 function could be due to localised hepatic rather than systemic inflammation, since hepatoxicity can be observed following CP treatment in breast cancer patients [30]. Doxorubicin, which is often given in combination with CP, can also cause hepatic damage and decreased Cyp2c mRNA expression [31].
An additional cause of decreased CP bioactivation in some patients could be direct inhibition of CYP2C19 or CYP2B6 catalysed 4-hydroxylation of CP by a comedication. A number of anti-infective medications are known to inhibit CP metabolism, including fluconazole, chloramphenicol and sulphaphenazole [32]. Moreover, ciprofloxacin has been shown in rats to not only inhibit the formation of 4OHCP but to down-regulate the expression of cyp2b, cyp2c and cyp3a genes [33]. At least one patient was prescribed ciprofloxacin immediately prior to cycle 3 of treatment, and this could have influenced CP bioactivation.
Whilst the general decline in CYP2C19 activity, and or the use of comedications, may explain the decreased bioactivation ratio at cycle 3 (observed in ~ 15% of patients), bioactivation ratio increased at cycle 3 in a substantial number of individuals (~ 30%). Cyclophosphamide is known to induce (up-regulate) CYP2B6 and CYP3A4 protein expression in human liver [34]. When administered continuously over multiple consecutive days CP is a known autoinducer (up-regulation) of its own metabolism in humans [32, 35]. However, the ability of CP to autoinduce its own clearance when given as a single dose in monthly treatment cycles (21 days between doses) is less clear. However, a number of comedications, particularly anti-emetics, could also induce CP metabolism.
It has been suggested that concomitant aprepitant may alter the bioactivation of cyclophosphamide by supressing autoinduction of cyclophosphamide clearance, although this is controversial [36]. Ondansetron also alters cyclophosphamide pharmacokinetics [37], but the effect on bioactivation in patients is not known. The steroid dexamethasone is a well characterised inducer of cyclophosphamide metabolism, by increasing expression of both CYP2B6 and CYP3A4/5 [38, 39], probably due to its ability to interact with the glucocorticoid receptor. This could increase both formation of 4OHCP as well as increasing clearance of CP via the alternative pathway into the inactive dechloroethyl metabolite, thereby altering the apparent bioactivation ratio. The effect of dexamethasone (or CP) on CYP2C19 expression in hepatocytes is not known. However, there is clear evidence of selective up-regulation of CYP2B6 without induction of CYP2C19 [40].
In this current study, patients received 5 days of anti-emetic treatment (dexamethasone, domperidone) starting at day 1 of each CP treatment cycle. Hence at cycle 3, some patients may have been more likely to have increased CYP2B6 and CYP3A4 activity. Notably increased CYP3A4 activity due to dexamethasone treatment may adversely affect the bioactivation ratio [41] since CYP3A4 is the sole enzyme involved in the formation of dechloroethylcyclophosphamide (Supplementary Fig. 1). Indeed, significantly increased formation of this inactive metabolite of CP at dose 5 versus dose 1 (continuous dosing) has been previously reported in children with B cell lymphoma [13] and the levels of this inactive metabolite were higher in those who relapsed (although not statistically significant). Future studies should assess the plasma pharmacokinetics of both 4OHCP and the deschloroethyl metabolite.
Indeed, assessment of formation of 4OHCP (rather than bioactivation ratio) clarified the clear influence of CYP2B6 A785G and G516T on this pathway.
Of note the steroid prednisone, which like dexamethasone acts via the glucocorticoid receptor, is often prescribed to patients receiving CP for treatment of haematological cancers (i.e. CHOP regimen: cyclophosphamide, doxorubicin, vincristine and prednisone), as well as in the autoimmune disease lupus nephritis. Whilst little is known about the ability of prednisone to regulate CYP2B6 or CYP2C19 expression, it is an inhibitor of CYP2C19 in vitro [42].
There is substantial inter-individual variability in the ability of dexamethasone (and CP) to induce CYP2B6 in human hepatocytes [39]. However, the effect of comedication with dexamethasone on changes in 4OHCP formation has not been directly assessed in patients. Up-regulation of CYP2B6 expression by drugs such as CP is mediated by the ligand activated transcription factors CAR, PXR, GR, HNF4α, C/EBPα and HNF3β [43]. There is substantial inter-individual variability in CYP2B6 induction, and this may be due to SNP in the 5’-promoter region of the gene where these transcription factors bind. The − 750 T > C SNP (rs4802101) alters a HNF1 binding site and − 2320 T > C SNP (rs7254579) alters a HNF4 binding site. In female liver tissues the − 2320 T > C SNP associates with decreased enzyme activity. The combination of the − 750C variant of CYP2B6 and CYP2C19 null function variants has previously been shown to significantly associate with 4OHCP plasma concentrations after four daily doses of CP [44]. Future studies should investigate associations between regulatory SNP and CP bioactivation in patients taking comedications likely to induce CYP2B6, as well as undertaking CYP2B6 phenotyping with the probe drug bupropion.
In summary, in this small cohort of patients there is evidence that there is a role for pharmacogenetic variation in CYP2B6 and the bioactivation of CP. However, in some (but not all) patients substantial changes in bioactivation occur by cycle 3 of treatment. It is not known whether comedications are the causal factors for this inter-occasion variation in bioactivation of CP. Whilst CYP2C19 function appears to decline over time across the cohort of patients, possibly due to inflammation, in contrast inflammation is reported to increase CYP2B6 activity [29]. SNP in the regulatory region of CYP2B6, or in the co-enzyme P450 oxidoreductase which appear to influence the overall activity of CYP2B6 [45], may play a role in those patients whose bioactivation increased over time. Future studies assessing CP bioactivation pharmacokinetics should investigate factors which influence changes in 4OHCP formation in larger study cohorts since the CYP2B6 and CYP2C19 genotypes have previously been reported to have substantial effects on treatment outcomes in many indications [reviewed in 1], including breast cancer [19, 20, 46, 47].
Data availability
Data are available on request to the corresponding author.
References
Helsby NA, Yong M, van Kan M et al (2019) The importance of both CYP2C19 and CYP2B6 germline variations in cyclophosphamide pharmacokinetics and clinical outcomes. Br J Clin Pharmacol 85:1925–1934. https://doi.org/10.1111/bcp.14031
Helsby NA, Hui C-Y, Goldthorpe MA et al (2010) The combined impact of CYP2C19 and CYP2B6 pharmacogenetics on cyclophosphamide bioactivation. Br J Clin Pharmacol 70:844–853. https://doi.org/10.1111/j.1365-2125.2010.03789.x
Kishino Y, Hasegawa T, Kato A et al (2019) Effect of inter-individual variability in human liver cytochrome P450 isozymes on cyclophosphamide-induced micronucleus formation. Mutat Res Toxicol Environ Mutagen 838:37–45. https://doi.org/10.1016/j.mrgentox.2018.11.016
Helsby NA, Burns KE (2012) Molecular mechanisms of genetic variation and transcriptional regulation of CYP2C19. Front Genet. https://doi.org/10.3389/fgene.2012.00206
Zanger UM, Klein K (2013) Pharmacogenetics of cytochrome P450 2B6 (CYP2B6): advances on polymorphisms, mechanisms, and clinical relevance. Front Genet. https://doi.org/10.3389/fgene.2013.00024
Klomp SD, Manson ML, Guchelaar H-J, Swen JJ (2020) Phenoconversion of cytochrome P450 metabolism: a systematic review. J Clin Med 9:2890. https://doi.org/10.3390/jcm9092890
Helsby NA, Lo W-Y, Sharples K et al (2008) CYP2C19 pharmacogenetics in advanced cancer: compromised function independent of genotype. Br J Cancer 99:1251–1255. https://doi.org/10.1038/sj.bjc.6604699
Williams ML, Bhargava P, Cherrouk I et al (2000) A discordance of the cytochrome P450 2C19 genotype and phenotype in patients with advanced cancer. Br J Clin Pharmacol 49:485–488. https://doi.org/10.1046/j.1365-2125.2000.00189.x
Burns KE, Goldthorpe MA, Porteus F et al (2014) CYP2C19 genotype–phenotype discordance in patients with multiple myeloma leads to an acquired loss of drug-metabolising activity. Cancer Chemother Pharmacol 73:651–655. https://doi.org/10.1007/s00280-014-2409-9
Burns KE, Lo W-Y, Findlay MP et al (2016) High CYP2C19 phenotypic variability in gastrointestinal cancer patients. Cancer Chemother Pharmacol 77:195–204. https://doi.org/10.1007/s00280-015-2923-4
Lang T, Klein K, Fischer J et al (2001) Extensive genetic polymorphism in the human CYP2B6 gene with impact on expression and function in human liver. Pharmacogenetics 11:399–415. https://doi.org/10.1097/00008571-200107000-00004
Xie H, Griskevicius L, Ståhle L et al (2006) Pharmacogenetics of cyclophosphamide in patients with hematological malignancies. Eur J Pharm Sci 27:54–61. https://doi.org/10.1016/j.ejps.2005.08.008
Veal GJ, Cole M, Chinnaswamy G et al (2016) Cyclophosphamide pharmacokinetics and pharmacogenetics in children with B-cell non-Hodgkin’s lymphoma. Eur J Cancer 55:56–64. https://doi.org/10.1016/j.ejca.2015.12.007
Afsar NA, Ufer M, Haenisch S et al (2012) Relationship of drug metabolizing enzyme genotype to plasma levels as well as myelotoxicity of cyclophosphamide in breast cancer patients. Eur J Clin Pharmacol 68:389–395. https://doi.org/10.1007/s00228-011-1134-0
Shu W, Chen L, Hu X et al (2017) Cytochrome P450 genetic variations can predict mRNA expression, cyclophosphamide 4-hydroxylation, and treatment outcomes in Chinese patients with non-Hodgkin’s lymphoma. J Clin Pharmacol 57:886–898. https://doi.org/10.1002/jcph.878
Jakobsen Falk I, Khan MS, Thunell L et al (2012) Association of CYP2B6 genotype with survival and progression free survival in cyclophosphamide treated multiple myeloma. J Cancer Ther 3:20–27
Johnson GG, Lin K, Cox TF et al (2013) CYP2B6*6 is an independent determinant of inferior response to fludarabine plus cyclophosphamide in chronic lymphocytic leukemia. Blood 122:4253–4258. https://doi.org/10.1182/blood-2013-07-516666
Melanson SEF, Stevenson K, Kim H et al (2010) Allelic variations in CYP2B6 and CYP2C19 and survival of patients receiving cyclophosphamide prior to myeloablative hematopoietic stem cell transplantation. Am J Hematol 85:967–971. https://doi.org/10.1002/ajh.21889
Haroun F, Al-Shaar L, Habib RH et al (2015) Effects of CYP2B6 genetic polymorphisms in patients receiving cyclophosphamide combination chemotherapy for breast cancer. Cancer Chemother Pharmacol 75:207–214. https://doi.org/10.1007/s00280-014-2632-4
Bray J, Sludden J, Griffin MJ et al (2010) Influence of pharmacogenetics on response and toxicity in breast cancer patients treated with doxorubicin and cyclophosphamide. Br J Cancer 102:1003–1009. https://doi.org/10.1038/sj.bjc.6605587
Timm R, Kaiser R, Lötsch J et al (2005) Association of cyclophosphamide pharmacokinetics to polymorphic cytochrome P450 2C19. Pharmacogenomics J 5:365–373. https://doi.org/10.1038/sj.tpj.6500330
Ekhart C, Doodeman VD, Rodenhuis S et al (2008) Influence of polymorphisms of drug metabolizing enzymes (CYP2B6, CYP2C9, CYP2C19, CYP3A4, CYP3A5, GSTA1, GSTP1, ALDH1A1 and ALDH3A1) on the pharmacokinetics of cyclophosphamide and 4-hydroxycyclophosphamide. Pharmacogenet Genomics 18:515. https://doi.org/10.1097/FPC.0b013e3282fc9766
El-Serafi I, Fares M, Abedi-Valugerdi M et al (2015) Cytochrome P450 2J2, a new key enzyme in cyclophosphamide bioactivation and a potential biomarker for hematological malignancies. Pharmacogenomics J 15:405–413. https://doi.org/10.1038/tpj.2014.82
Edwards G, Calvert RT, Crowther C et al (1980) Repeated investigations of cyclophosphamide disposition in myeloma patients receiving intermittent chemotherapy. Br J Clin Pharmacol 10:281–285. https://doi.org/10.1111/j.1365-2125.1980.tb01756.x
Batey MA, Wright JG, Azzabi A et al (2002) Population pharmacokinetics of adjuvant cyclophosphamide, methotrexate and 5-fluorouracil (CMF). Eur J Cancer 38:1081–1089. https://doi.org/10.1016/S0959-8049(02)00024-2
Moore MJ, Erlichman C, Thiessen JJ et al (1994) Variability in the pharmacokinetics of cyclophosphamide, methotrexate and 5-fluorouracil in women receiving adjuvant treatment for breast cancer. Cancer Chemother Pharmacol 33:472–476. https://doi.org/10.1007/BF00686503
Aitken AE, Morgan ET (2007) Gene-specific effects of inflammatory cytokines on cytochrome P450 2C, 2B6 and 3A4 mRNA levels in human hepatocytes. Drug Metab Dispos Biol Fate Chem 35:1687–1693. https://doi.org/10.1124/dmd.107.015511
Rivory LP, Slaviero KA, Clarke SJ (2002) Hepatic cytochrome P450 3A drug metabolism is reduced in cancer patients who have an acute-phase response. Br J Cancer 87:277–280. https://doi.org/10.1038/sj.bjc.6600448
Lenoir C, Daali Y, Rollason V et al (2020) Impact of acute inflammation on cytochromes P450 activity assessed by the Geneva Cocktail. Clin Pharmacol Ther. https://doi.org/10.1002/cpt.2146
Ming Z, Yongqiang Z, Zijin Z et al (2019) Severe and prolonged cyclophosphamide-induced hepatotoxicity in a breast cancer patient carrying a CYP2B6*7 variant. Pharmacogenomics 20:1119–1124. https://doi.org/10.2217/pgs-2019-0093
Grant MKO, Abdelgawad IY, Lewis CA, Zordoky BN (2020) Sexual dimorphism in doxorubicin-induced systemic inflammation: implications for hepatic cytochrome P450 regulation. Int J Mol Sci 21:1279. https://doi.org/10.3390/ijms21041279
de Jonge ME, Huitema ADR, Rodenhuis S, Beijnen JH (2005) Clinical pharmacokinetics of cyclophosphamide. Clin Pharmacokinet 44:1135–1164. https://doi.org/10.2165/00003088-200544110-00003
Xie H-J, Broberg U, Griskevicius L et al (2003) Alteration of pharmacokinetics of cyclophosphamide and suppression of the cytochrome p450 genes by ciprofloxacin. Bone Marrow Transplant 31:197–203. https://doi.org/10.1038/sj.bmt.1703815
Martin H, Sarsat J-P, de Waziers I et al (2003) Induction of cytochrome P450 2B6 and 3A4 expression by phenobarbital and cyclophosphamide in cultured human liver slices. Pharm Res 20:557–568. https://doi.org/10.1023/A:1023234429596
Slattery JT, Kalhorn TF, McDonald GB et al (1996) Conditioning regimen-dependent disposition of cyclophosphamide and hydroxycyclophosphamide in human marrow transplantation patients. J Clin Oncol 14:1484–1494. https://doi.org/10.1200/JCO.1996.14.5.1484
Walko CM, Combest AJ, Spasojevic I et al (2012) The effect of aprepitant and race on the pharmacokinetics of cyclophosphamide in breast cancer patients. Cancer Chemother Pharmacol 69:1189–1196. https://doi.org/10.1007/s00280-011-1815-5
Gilbert CJ, Petros WP, Vredenburgh J et al (1998) Pharmacokinetic interaction between ondansetron and cyclophosphamide during high-dose chemotherapy for breast cancer. Cancer Chemother Pharmacol 42:497–503. https://doi.org/10.1007/s002800050851
Chang TK, Yu L, Maurel P, Waxman DJ (1997) Enhanced cyclophosphamide and ifosfamide activation in primary human hepatocyte cultures: response to cytochrome P-450 inducers and autoinduction by oxazaphosphorines. Cancer Res 57:1946–1954
Lindley C, Hamilton G, McCune JS et al (2002) The effect of cyclophosphamide with and without dexamethasone on cytochrome P450 3A4 and 2B6 in human hepatocytes. Drug Metab Dispos 30:814–822. https://doi.org/10.1124/dmd.30.7.814
Moscovitz JE, Kalgutkar AS, Nulick K et al (2018) Establishing transcriptional signatures to differentiate PXR-, CAR-, and AhR-mediated regulation of drug metabolism and transport genes in cryopreserved human hepatocytes. J Pharmacol Exp Ther 365:262–271. https://doi.org/10.1124/jpet.117.247296
Yu LJ, Drewes P, Gustafsson K et al (1999) In vivo modulation of alternative pathways of P-450-catalyzed cyclophosphamide metabolism: impact on pharmacokinetics and antitumor activity. J Pharmacol Exp Ther 288:928–937
Jurima M, Inaba T, Kalow W (1985) Mephenytoin hydroxylase activity in human liver: inhibition by steroids. Drug Metab Dispos 13:746–749
Hedrich WD, Hassan HE, Wang H (2016) Insights into CYP2B6-mediated drug–drug interactions. Acta Pharm Sin B 6:413–425. https://doi.org/10.1016/j.apsb.2016.07.016
Shu W, Guan S, Yang X et al (2016) Genetic markers in CYP2C19 and CYP2B6 for prediction of cyclophosphamide’s 4-hydroxylation, efficacy and side effects in Chinese patients with systemic lupus erythematosus. Br J Clin Pharmacol 81:327–340. https://doi.org/10.1111/bcp.12800
El-Serafi I, Afsharian P, Moshfegh A et al (2015) Cytochrome P450 oxidoreductase influences CYP2B6 activity in cyclophosphamide bioactivation. PLoS ONE 10:e0141979. https://doi.org/10.1371/journal.pone.0141979
Tulsyan S, Agarwal G, Lal P, Mittal B (2014) Significant role of CYP450 genetic variants in cyclophosphamide based breast cancer treatment outcomes: a multi-analytical strategy. Clin Chim Acta 434:21–28. https://doi.org/10.1016/j.cca.2014.04.009
Kalra S, Kaur RP, Ludhiadch A et al (2018) Association of CYP2C19*2 and ALDH1A1*1/*2 variants with disease outcome in breast cancer patients: results of a global screening array. Eur J Clin Pharmacol 74:1291–1298. https://doi.org/10.1007/s00228-018-2505-6
Acknowledgements
We thank M. Goldthorpe, J-P. Yang (RIP) and C. Bonnet for technical support and research nurse support from C. Barrett and G. Wilson. We are grateful for funding support for this study from Genesis Oncology Trust and Cancer Society NZ.
Funding
We are grateful for funding support for this study from Genesis Oncology Trust and Cancer Society of New Zealand.
Author information
Authors and Affiliations
Contributions
H and P conceived and designed the study with guidance from F. P recruited patients and F provided study coordination support. H, F and P obtained funding. Y helped undertake genomic analysis under guidance of B. H, B and Y undertook data analysis. All authors contributed to manuscript writing.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval and consent to participate
As noted in the main text this study received approval from the New Zealand Heath and Disability Northern X Regional Ethics committee (NTX/12/06/052). Patients were eligible for the study if they were diagnosed with carcinoma of the breast and scheduled to receive cyclophosphamide treatment. Patients had to be at least 18 years of age and able to give informed written consent. The study was performed in accordance with the Declaration of Helsinki.
Consent to publication
All authors consent to publication.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Helsby, N., Yong, M., Burns, K. et al. Cyclophosphamide bioactivation pharmacogenetics in breast cancer patients. Cancer Chemother Pharmacol 88, 533–542 (2021). https://doi.org/10.1007/s00280-021-04307-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-021-04307-0