
Abstract
Purpose
CYP2C19 contributes to the metabolism of several chemotherapeutic agents. The CYP2C19 homozygous null function genotype strongly predicts activity phenotype in healthy populations. An additional acquired loss of function has been reported in up to one-third of cancer patients. It is not known whether this phenomenon also occurs in patients with earlier stage or in resected disease.
Methods
This study investigated whether acquired loss of CYP2C19 function was detectable in patients with stage III–IV or resected gastrointestinal cancer. CYP2C19 genotype was determined in 49 patients, and subjects were probed for CYP2C19 activity on three test occasions.
Results
An acquired loss of CYP2C19 activity was observed in 20 % of stage III–IV and 17 % of resected patients at the first test. Significant (p < 0.01) genotype–phenotype discordance was observed in both groups. There were no direct associations between this discordance and inflammatory markers, tumour burden or chemotherapeutic history. Notably, hepatic CYP2C19 function was not stable over time and phenotype conversion occurred in 23 patients over the period of testing.
Conclusion
Reliance on germ-line genotype to infer a poor metaboliser status could substantially underestimate the number of patients with deficient CYP2C19 function. This could compromise the interpretation of genotype-based clinical association studies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
CYP2C19 is a hepatic drug metabolising enzyme important in the metabolism of drugs including anti-platelet agents, anti-depressants and proton pump inhibitors [1]. These include commonly used drugs such as omeprazole [2] and clopidogrel [3]. It is also involved (at least in part) in the metabolism of several chemotherapeutic drugs and investigational agents including cyclophosphamide, thalidomide, bortezomib, tamoxifen, nilutamide, icotinib, tivantinib and indisulam [4–14].
Two null function variants of the gene, CYP2C19*2 (rs4244285) and CYP2C19*3 (rs49486893), result in the loss of enzyme expression due to aberrant splicing and a premature stop codon, respectively [15–18]. There is marked interethnic variability in the frequencies of these alleles; CYP2C19*2 ranges from 15 % in Caucasian populations to 30 % in Asians, while CYP2C19*3 is less common (0.04 % in Caucasians, 5 % in Asians) [19, 20]. Homozygous null function genotypes occur in approximately 3 % of Caucasian populations. The relationship between these germ-line (inherited) variants and CYP2C19 metabolic activity has been widely reported in healthy populations [1, 21]. Individuals homozygous for these null function variants have a poor metaboliser (PM) phenotype for drugs which are substrates for this enzyme. Additionally, the CYP2C19*17 (rs12248560) allele increases the clearance of some CYP2C19 probe substrates [17, 18].
Probe substrates for the CYP2C19 enzyme, e.g. S-mephenytoin, omeprazole, proguanil, can be used to measure metabolic activity in vivo [22]. The ratio of drug/metabolite concentrations in plasma can categorise an individual as either a phenotypic extensive (pEM) or poor (pPM) metaboliser of CYP2C19 substrates. Numerous studies (n > 100) in healthy populations have demonstrated that the CYP2C19 homozygous null genotype reliably predicts poor metabolism of CYP2C19 probe substrates (pPM), while individuals with heterozygous or homozygous for “functional” alleles (*17, *1) form a distinct pEM population [1]. Consequently, genotype is often used as a substitute measure of CYP2C19 poor metaboliser phenotype. However to be useful, germ-line testing for this gene must be a robust predictor of phenotype, not only in healthy populations but also in the clinical context. Importantly, any acquired loss of CYP2C19 function in patients can lead to misclassification of metaboliser status due to genotype–phenotype discordance.
The phenomenon of CYP2C19 genotype–phenotype discordance was first reported by Williams et al. [23] in a study of 16 patients with advanced solid tumour cancers. Twenty-five per cent of patients exhibited poor metabolism of the CYP2C19 probe drug omeprazole despite lacking a homozygous null genotype. We have also reported a 37 % prevalence of genotype–phenotype discordance in 31 patients with a range of terminal cancers [24] and, more recently, have confirmed the presence of this acquired poor metaboliser status in patients with multiple myeloma [25].
The mechanism which underpins this acquired loss of function is unknown. Environmental factors such as inflammatory mediators have been proposed as potential regulators of cytochrome P450 (CYP) gene expression [26, 27]. Indeed the inflammatory marker C-reactive protein (CRP) correlates with decreased CYP3A4 activity in advanced cancer patients [28] and elevated IL-6 associates with decreased CYP2C19 activity in cardiac patients [29]. We have previously noted a potential association between the CYP2C19 genotype–phenotype discordance and low body mass index (BMI < 25 kg/m2) in patients with terminal cancer [24]. We hypothesised that the association of acquired loss of CYP2C19 function with low body mass may be due to the inflammatory factors associated with cachexia [30].
To date, the reports of an acquired poor metaboliser status in cancer patients have been undertaken in cohorts with a high level of disease burden and little is known about the prevalence of CYP2C19 discordance in patients at earlier stages of disease. Furthermore the previous work was undertaken in single cohorts, making direct comparison of the study populations complex and prone to the influence of confounding factors. The previous work has also been restricted to single time points and does not provide information about whether this acquired loss of activity observed in some patients is constant or whether it can change with time.
The primary aim of this study was to investigate the prevalence of acquired loss of CYP2C19 function in patients with stage III–IV disease compared with resected patients with no currently evaluable disease (NED). The secondary aims were to investigate (a) whether changes in CYP2C19 activity occur over time within an individual and (b) whether cancer-associated systemic inflammatory markers or cachexia-type symptoms correlate with the acquired loss of CYP2C19 activity.
Materials and methods
Approval was obtained from the New Zealand Heath and Disability Northern X Regional Ethics committee (NTX 08/07/060). Patients undergoing treatment for gastrointestinal cancers (n = 49) with good renal and hepatic function (serum creatinine ≤0.12 mmol/L; AST/ALT ≤ 90 U/L; ALP ≤ 300 U/L; total bilirubin ≤20 µmol/L) who had either no evaluable disease (NED) following tumour resection or stage III or IV disease, were recruited to the study following full informed consent. All subjects were at least 18 years of age and were not receiving any known CYP2C19 inhibitors or inducers for which an appropriate washout period (>3 half-lives) was not clinically feasible prior to phenotyping.
Whole blood (8.5 mL) was collected into PAXgene blood DNA tubes (Qiagen, Hilden, Germany) and stored at −20 °C prior to analysis. DNA was extracted using the PAXgene Blood DNA kit (Qiagen, Hilden, Germany) and analysed for the CYP2C19*2 (rs4244285), CYP2C19*3 (rs49486893) and CYP2C19*17 (rs12248560) alleles using previously published PCR–RFLP methods [15, 16].
CYP2C19 metabolic activity was assessed for each study participant on three separate occasions, each directly preceding a scheduled cycle of chemotherapy in order to maximise drug washout from previous treatment cycles. Subjects self-administered proguanil (200 mg p.o.) immediately prior to a scheduled clinic visit. A blood sample (10 mL) was collected into a BD Vacutainer Lithium Heparin Plus blood tube 3 h after dosing and the plasma separated by centrifugation (200g, 10 min) and stored at −20 °C. The concentrations (ng/mL) of proguanil (PG) and its major metabolite cycloguanil (CG) were determined by HPLC [25, 31]. The proguanil metabolic ratio (PG MR) was calculated as the ratio of the plasma concentration proguanil to cycloguanil [(PG)/(CG)]. Individuals with log PG MR values of ≥1 were categorised as phenotypic poor metabolisers (pPM). This value has been previously shown as concordant between genotype and phenotype for poor metabolisers in healthy populations [32]. Individuals were classified as discordant if they had a poor metaboliser phenotype (log PG MR ≥ 1), but were not homozygous for the null function alleles CYP2C19*2 or CYP2C19*3.
Tumour burden was calculated using modified RECIST criteria [33, 34]. Linear measurements, in the longest dimension, were obtained from routine clinical CT scans and/or X-rays for the six largest tumour deposits at the scans performed closest to the first phenotype test. These measurements were summed for each individual. In cases where tumours were not well visualised on the scans available, the approximate tumour burden was obtained from clinical notes.
Two additional blood samples (4 mL) were collected into BD Vacutainer Serum Plus tubes for the analysis of circulating pro-inflammatory markers. These blood samples were allowed to clot for 1 h, before the serum was transferred into a clean tube and stored at −20 °C. C-reactive protein (CRP) was analysed by the local clinical laboratory service. Interleukin-6 (IL-6) concentrations were determined using a Milliplex MAP human cytokine/chemokine multiplex immunoassay (Millipore Corporation, MA, USA).
Anthropomorphic measurements were collected at the time of each phenotype test to assess the nutritional status of each patient. Body mass index (BMI) was calculated from the height and weight of each individual. Triceps skinfold thickness was measured using callipers and, in combination with arm circumference measurements, was used to calculate cross-sectional arm fat and muscle areas [35–37].
Statistical analyses were performed in either GraphPad PRISM (version 6, GraphPad Software Inc., USA) or STATA (StataCorp. 2013. Stata Statistical Software: Release 13. College Station, TX: StataCorp LP). All p values are two-sided, with p < 0.05 considered to be statistically significant. Exact 95 % confidence intervals were calculated for the proportions of phenotypic poor metabolisers among the genotypic extensive metabolisers. Proportions were compared with a Chi-squared test unless the numbers were very small, in which case Fishers exact test was used. Continuous data are described as median and interquartile range (IQR); associations between continuous variables were assessed using Pearson’s product-moment correlations (ρ) where data appear consistent with a normal distribution (D’Agnostino–Pearson omnibus normality test), and Spearman’s rank correlations (R S) otherwise. Means of two groups were compared using unpaired t tests, with Welsh’s correction applied where appropriate. Where the distribution of the variable was highly skewed, groups were compared using a Mann–Whitney test. For the patient who was included in both groups, data from after disease progression (in the stage III/IV category) were excluded where the two cohorts were compared. A linear mixed model with a random intercept for patient was used to estimate the change in CYP2C19 activity over time. Stepwise multiple linear regression analysis was performed using SPSS Statistics software (version 22, IBM Corp., NY, USA). Variables were entered into or removed from each progressive regression model according to the following criteria: Alpha-to-Enter (α E) ≤ 0.050, Alpha-to-Remove (α R) ≥ 0.100. The significance of the regression models was analysed by ANOVA, with p values of <0.05 (one-tailed) considered significant. The predictive power of each model was determined using adjusted r values.
Results
A total of 49 participants were recruited to the study. There were 25 patients with no evaluable disease who were receiving adjuvant chemotherapy for resected gastrointestinal cancer (resected-NED) and 24 patients with stage III or stage IV gastrointestinal cancer (online resource table S1). One patient (#1026/#1049) was initially recruited to the resected-NED group but, following disease relapse (stage IV colon adenocarcinoma) 19 months after supplying the initial three samples, also provided samples for the stage III/IV group. This resulted in total of 25 participants in each of the resected-NED and stage III/IV cohorts, with mean ages of 61 ± 14 and 65 ± 12 years, respectively. There was no important difference in the gender distribution between the resected-NED group (11 females, 14 males) and the stage III/IV group (9 females, 16 males) subjects (online resource table S1). The majority of study participants were Caucasian (92 %), with one Polynesian patient in the stage III/IV cohort and three Asian individuals in the NED cohort (online resource table S1).
All CYP2C19 alleles studied were in Hardy–Weinberg equilibrium (CYP2C19*2: p = 1.00; CYP2C19*3: p = 0.94; CYP2C19*17: p = 0.13). Of the 49 subjects, 35 (71 %) were homozygous for functional alleles (*17/*17, *1/*17, or *1/*1) and 13 (27 %) were heterozygous carriers of null function alleles (*17/*2, *1/*2, or *1/*3). All of these individuals (n = 48) were predicted to have extensive metaboliser phenotypes (genotypic extensive metabolisers; gEM). One participant (#1058, NED cohort) was homozygous null function (*2/*2) and hence was predicted to have a poor metaboliser phenotype. The proportion (2 %) of genotypic poor metabolisers (gPM) detected was similar to that reported for Caucasian populations [21].
Proguanil was detected in all 3-h plasma samples collected for all patients, with concentrations ranging from 7.8 to 296.1 ng/mL. Cycloguanil was not detected in eight samples, all of which had adequate proguanil levels. Cycloguanil plasma concentrations ranged from 2.0 to 146.6 ng/mL in the remaining samples.
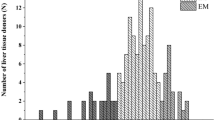
The patient with a homozygous null function genotype (#1058, resected-NED cohort) was confirmed to be a concordant poor metaboliser at all three phenotype tests with a log proguanil metabolic ratio >1 (range 1.31–1.51; Fig. 1). This individual was excluded from the subsequent analyses of the phenomenon of CYP2C19 genotype–phenotype discordance.

Measured CYP2C19 activity (phenotype) in patients with gastrointestinal cancer, stratified by CYP2C19 genotype. CYP2C19 phenotype was tested on three separate occasions. Subjects were categorised as poor metabolisers when the log proguanil metabolic ratio (log PG MR) was ≥1. Two cohorts of participants were tested resected patients with no evaluable disease (NED) and patients with stage III/IV gastrointestinal cancer
Four additional subjects in the resected-NED cohort had a poor metaboliser phenotype (pPM) at test 1 (Fig. 1a). These individuals were not homozygous for null function alleles. At the first test, this gave a rate of genotype–phenotype discordance of 17 % (95 % CI 5–37 %) in resected-NED patients who were not homozygous null (Table 1). None of the stage III/IV subjects were homozygous for null function alleles (*2 or *3). However, at test 1 five individuals (20 %, 95 % CI 7–41 %) had a log metabolic ratio ≥1, which classified them as having a discordant poor metaboliser status (Fig. 1b). Notably one individual (#1005) who had a *17/*17 genotype, which is expected to predict an ultra-rapid metaboliser status, had a metabolic ratio of 2.18 at test 1, indicating extremely poor CYP2C19 activity. Moreover, discordance was observed in both cohorts at the additional testing periods (Fig. 1c–f), with the prevalence across the three test occasions of 17–27 % (Table 1). Comparison of the rate of genotype–phenotype discordance between the resected-NED and the stage III/IV groups found no significant difference in prevalence at any of the 3 time points (p = 0.7, 0.98 and 0.8, respectively). The overall prevalence of genotype–phenotype discordance in the entire cohort of 48 patients on the first test occasion was 23 % (95 % CI 11–39 %), indicating that the number of phenotypic poor metabolisers (pPM) was substantially greater than that predicted by genotype.
The majority of patients in the study had been treated with chemotherapy prior to the phenotyping test, and only three subjects were chemotherapy-naïve (#1003, #1004, and #1025). Most patients had at some point received treatment schedules containing 5-fluorouracil (5-FU), or the 5-FU prodrug capecitabine, with or without the addition of either oxaliplatin or irinotecan. Two patients (#1044 and #1059) had received gemcitabine. The tests were carried out a minimum of 7 days after the previous cycle of chemotherapy, directly preceding the next scheduled cycle of chemotherapy in order to maximise drug washout from previous treatment cycles. No association was observed between discordance at the first test and the chemotherapeutic treatments received by the study participants, although numbers were small for most regimens.
CYP2C19 activity (log proguanil metabolic ratio) was relatively consistent (coefficient of variation, C V < 30 %) across the three testing occasions for the majority of participants (online resource table S2). However, 15 subjects exhibited high inter-occasion variation in their metabolic ratio and importantly several individuals changed phenotype category over time (Fig. 2, online resource table S2). Over the three tests, seven subjects who were genotype–phenotype concordant at test one became discordant in subsequent tests, while six subjects who were initially discordant became concordant at test 2 and/or test 3 (Fig. 2).

Between-occasion variation in measured CYP2C19 activity (phenotype) for each participant. Poor metaboliser status (log proguanil metabolic ratio ≥1) is shown by a bold line. There was >7 days (typically 2–3 weeks) between each test occasion
Tumour burden in the 23 individuals in the stage III/IV cohort ranged from 3.8 to 470 mm. The tumour burden in concordant and discordant subjects (at time point 1) was not significantly different (119 mm, IQR 35–188 vs. 189 mm, IQR 159–200; p = 0.14), and there was no correlation between CYP2C19 metabolic activity and tumour burden (R S = 0.32, p = 0.14).
To assess whether any of the dynamic changes in CYP2C19 activity were associated with the extent of disease burden over time, the clinical outcomes of patients were scrutinised. Two patients (#1009, #1038) were reallocated from the stage III/IV group to the NED cohort following surgical resection of the primary tumour after test 1. Patient #1009 had highly variable proguanil metabolism over the three phenotype tests (coefficient of variation (C V) = 254.6 %). At test 1 they had a poor metaboliser phenotype (log proguanil metabolic ratio = 1.09), which was discordant with their genotype (CYP2C19*1*1). Following tumour resection, there was a substantial increase in CYP2C19 metabolic activity sufficient to be re-categorised as an extensive metaboliser at test 2 and test 3 (log metabolic ratio: −0.30 and 0.06, respectively). Patient #1038 (CYP2C19*1*17) also underwent tumour resection after test 1 and was reallocated to the NED group for tests 2 and 3. In contrast to patient #1009, the CYP2C19 activity of this individual did not substantially differ across the three test occasions (C V = 12.2 %). and they remained genotype–phenotype concordant throughout the study.
Five patients recruited to the resected-NED cohort suffered a relapse of their cancer following the completion of their phenotype tests. The median time to relapse was 15 months (IQR 5–20.5 months). Only one of these patients was genotype–phenotype discordant. CYP2C19 activity appeared to decrease over the three tests in this subset of patients, although there was no evidence of a relationship between the discordance at test 1 and subsequent disease relapse (p = 1.00). However, the numbers are very small. As previously mentioned, one of the relapsed patients (#1026) was subsequently recruited to the stage III/IV cohort as patient #1049 and provided a further three samples. There was no evidence of a difference in the log metabolic ratio in this patient with or without evaluable tumour burden (0.71, 0.86, 0.4 with tumour burden and 0.33, 0.58, 0.57 without; p = 0.2). Larger samples are required to evaluate the role of tumour burden as a contributing factor in acquired loss of CYP2C19 activity.
To test for any apparent trends in CYP2C19 activity change over time, a linear regression was fitted to the three test data for each individual. The two patients who changed cohort (#1009, #1038) were included in the NED group. The average slope of the regression lines over the three tests was 0.040 (p = 0.2) in the stage III/IV cohort and 0.042 (p = 0.2) in the NED cohort, so we found no evidence of a consistent change in CYP2C19 activity over the time period of this study.
Serum concentrations of C-reactive protein (CRP) ranged from <1 to 41 mg/L at test 1. Only six individuals (12 %) had CRP levels above the upper limit of normal (>10 mg/L). There was no correlation between log CRP concentration and CYP2C19 activity in resected-NED (R S = 0.17, p = 0.4) or stage III/IV (R S = 0.13, p = 0.5) subjects. IL-6 concentrations were elevated above background (>3.2 pg/mL) in 19 individuals. There was no relationship between log IL-6 concentration and the CYP2C19 activity in either patient cohort (resected-NED: R S = −0.17, p = 0.4; stage III/IV: R S = 0.05, p = 0.8). Stratification of subjects into concordant or discordant categories revealed no significant difference (p > 0.05) in either the median log CRP concentration or the median log IL-6 concentration in either cohort at test 1. There were also no relationships between any of these inflammatory factors and CYP2C19 activity or discordant subjects at test 2 or 3. A preliminary analysis of the data for the first 28 patients recruited into this study [38] also indicated no relationship (p > 0.05) between discordance and several other cytokines (IL-1α, IL-1β, TNF-α, IFN-γ).
At the first test occasion, no significant correlation between age and CYP2C19 metabolic activity was observed in the study cohorts (resected-NED: ρ = −0.16, p = 0.4; stage III/IV: ρ = 0.22, p = 0.3). No gender difference was observed in the prevalence of discordance for resected-NED (30 % for females vs. 7 % for males; p = 0.3) or stage III/IV (0 % for females vs. 31 % for males; p = 0.1) subjects. There was also no difference in the log proguanil metabolic ratio between females and males for either the resected-NED (female: mean ± SD = 0.69 ± 0.46; male: mean ± SD = 0.54 ± 0.34, p = 0.4) or stage III/IV cohorts (female: mean ± SD = 0.56 ± 0.29; male: mean ± SD = 0.82 ± 0.44, p = 0.1).
No associations between CYP2C19 discordance at the first time point and anthropomorphic measures of patient nutritional status were observed in the stage III/IV cohort. This is in contrast to the results in the resected-NED cohort, which exhibited significant correlations between higher BMI (ρ = 0.62, p = 0.001), triceps skinfold thickness (ρ = 0.43, p = 0.04), arm circumference (ρ = 0.52, p = 0.009), cross-sectional arm muscle area (ρ = 0.48, p = 0.02) and cross-sectional arm fat area (ρ = 0.49, p = 0.01) with decreasing CYP2C19 activity at test 1. Furthermore discordant resected-NED subjects had significantly higher BMI (p = 0.02), triceps fold thickness (p = 0.01), arm circumference (p = 0.04) and cross-sectional arm fat area (p = 0.007) than concordant subjects at this time point.
Multiple linear regression analysis was used to develop a model to examine associations between cancer-associated systemic inflammatory markers and cachexia-type symptoms, and CYP2C19 activity at the first test. Using a step-wise approach, the only predictors retained in the model for the resected-NED subjects were serum IL-6 and arm circumference; the final regression model was:
However, this model accounted for only 38.4 % of the variance in the CYP2C19 activity of the resected-NED cohort (adjusted R square = 0.384, p = 0.002). No statistically significant predictors of CYP2C19 activity were found for the stage III/IV patient cohort.
Discussion
This study demonstrated an acquired loss of CYP2C19 activity in some individuals with gastrointestinal cancer; encompassing both those who had undergone tumour resection as well in those with stage III–IV disease. This work confirms our previous findings of a significant level of CYP2C19 genotype–phenotype discordance in patients with other cancers [24, 25] as well as that reported by others [23]. It appears that approximately 25 % of cancer patients across many types of malignancies have an acquired loss of activity in this enzyme (Table 2). This discordance is unlikely to be due to rare null mutations in this gene because dynamic changes in metabolic activity were observed in some individuals, some of which were larger than we would expect from natural biological variation. Similar dynamic changes of CYP2C19 activity within an individual over three test periods have been reported in patients undergoing curative treatment for infectious disease [39].
The mechanism(s) which underpin the acquired change in CYP2C19 activity are still not clear. To ensure that this acquired decrease in CYP2C19 activity in some patients was not due to drug–drug interactions, which can lead to a phenoconversion phenomenon [40], the tests were performed at least 7 days and usually 2–3 weeks after the last chemotherapy dose. Moreover, patients receiving known inducers or inhibitors of CYP2C19 were excluded from the study. Careful scrutiny of the clinical records was also undertaken to identify any possible confounding effect of chemotherapy regimen on CYP2C19 activity. None of the drugs used in the treatment of this cohort of patients is a known inhibitor or inducer of CYP2C19 activity and were not expected to influence the apparent CYP2C19 activity. Indeed there was no evidence of a relationship between CYP2C19 discordance and any drug treatment regimen used. Furthermore, one of the discordant stage III/IV patients was treatment-naïve at the time of the first phenotype test. This supports the hypothesis that the acquired loss of CYP2C19 activity is driven by factors other than drug–drug interactions.
In designing the current study, we hypothesised that that the acquired loss of function might relate to the extent of tumour burden. Although the tumour burden, as quantified by modified RECIST analysis, was higher in discordant than concordant subjects in this study, the difference was not statistically significant. The small numbers of discordant subjects (n = 4) may contribute to the lack of a correlation, and further work to determine the role of tumour burden should be considered.
Inflammation-induced downregulation of drug metabolising enzyme activity has been hypothesised as an important contributor in phenoconversion and the resulting genotype–phenotype discordance [41]. Numerous clinical studies in cancer patients have established decreased probe drug clearance associated with inflammation, particularly for CYP3A [28, 42, 43] and CYP2C9 [44]. However, the systemic inflammatory status of a patient does not appear to be a major factor contributing to the observed loss of CYP2C19 activity. It is also of note that no relationship was observed between acquired loss of CYP2C19 activity and systemic inflammatory markers in our previous studies [24, 25]. Thus CYP2C19 does not appear to be under the same regulatory control by cancer-associated pro-inflammatory cytokines as has been demonstrated for CYP3A4 [28].
The combined effects of the studied variables did not associate with CYP2C19 activity for the stage III/IV patients. A moderate association with CYP2C19 activity was found between serum IL-6 concentration and arm circumference, accounting for approximately 38 % of the variability in CYP2C19 activity in the resected-NED cohort. Overall these regression analyses suggest that the major factor(s) contributing to the observed acquired loss of CYP2C19 activity in cancer patients have yet to be elucidated.
Given the narrow therapeutic indices of many anti-cancer drugs, variability in drug metabolism has the potential to impact the safety and effectiveness of chemotherapy, whether due to inherited null function alleles or due to an acquired (extrinsic) decrease in activity. The use of germ-line as well as somatic (tumour) genomic information is often suggested as a method to personalise treatment. However, the results of this study indicate that this approach could significantly underestimate the number of phenotypic CYP2C19 poor metabolisers in cancer patient cohorts. This study has also demonstrated that patients can change CYP2C19 phenotype over a period of time. Whether this acquired loss of function has any implications for the safety and efficacy of chemotherapeutic agents metabolised by CYP2C19 is not known. Further work is ongoing to determine whether this phenomenon adversely affects therapeutic outcomes with drugs such as cyclophosphamide, which is bioactivated in part by this enzyme.
The substantial prevalence of this acquired deficiency in cancer patients highlights the possibility that pharmacogenetic tests for CYP2C19 can incorrectly categorise some cancer patients as extensive metabolisers. It is of note that a similar discrepancy between CYP2D6 genotype and measured phenotype (using dextromethorphan as a probe substrate) has been reported in breast cancer patients [45]. Up to 22 % of women had an acquired loss of CYP2D6 activity that did not appear to be due to null function genotype nor due to co-administration of CYP2D6 inhibitors.
The phenomenon of genotype–phenotype discordance may be of particular importance as many clinical studies attempt to identify gene variants of drug metabolising enzymes which are associated with poor clinical outcomes. This could be a particularly flawed approach if (a) genotype–phenotype discordance is common, and (b) if phenotype changes substantially and inconsistently between drug treatment cycles. Indeed this reliance on genotyping to predict phenotype despite evidence to suggest that considerable mismatch can occur may prove to be the “Achilles’ heel of personalised medicine” [40].
References
Desta Z, Zhao X, Shin JG, Flockhart DA (2002) Clinical significance of the cytochrome P450 2C19 genetic polymorphism. Clin Pharmacokinet 41:913–958
Chiba K, Kobayashi K, Manabe K, Tani M, Kamataki T, Ishizaki T (1993) Oxidative metabolism of omeprazole in human liver microsomes: cosegregation with S-mephenytoin 4′-hydroxylation. J Pharmacol Exp Ther 266(1):52–59
Kim KA, Park PW, Hong SJ, Park JY (2008) The effect of CYP2C19 polymorphism on the pharmacokinetics and pharmacodynamics of clopidogrel: a possible mechanism for clopidogrel resistance. Clin Pharmacol Ther 84(2):236–242
Ando Y, Fuse E, Figg WD (2002) Thalidomide metabolism by the CYP2C subfamily. Clin Cancer Res 8(6):1964–1973
Coller JK, Krebsfaenger N, Klein K, Endrizzi K, Wolbold R, Lang T, Nüssler A, Neuhaus P, Zanger UM, Eichelbaum M, Mürdter TE (2002) The influence of CYP2B6, CYP2C9 and CYP2D6 genotypes on the formation of the potent antioestrogen Z-4-hydroxy-tamoxifen in human liver. Br J Clin Pharmacol 54(2):157–167
Helsby NA, Hui CY, Goldthorpe MA, Coller JK, Soh MC, Gow PJ, De Zoysa JZ, Tingle MD (2010) The combined impact of CYP2C19 and CYP2B6 pharmacogenetics on cyclophosphamide bioactivation. Br J Clin Pharmacol 70(6):844–853
Horsmans Y, Lannes D, Larrey D, Tinel M, Letteron P, Loeper J, Pessayre D (1991) Nilutamide inhibits mephenytoin 4-hydroxylation in untreated male rats and in human liver microsomes. Xenobiotica 21(12):1559–1570
Li Y, Hou J, Jiang H, Wang D, Fu W, Yuan Z, Chen Y, Zhou L (2007) Polymorphisms of CYP2C19 gene are associated with the efficacy of thalidomide-based regimens in multiple myeloma. Haematologica 92(9):1246–1249
Ruan C-J, Liu D-Y, Jiang J, Hu P (2012) Effect of the CYP2C19 genotype on the pharmacokinetics of icotinib in healthy male volunteers. Eur J Clin Pharmacol 68(12):1677–1680
Schroth W, Antoniadou L, Fritz P, Schwab M, Muerdter T, Zanger UM, Simon W, Eichelbaum M, Brauch H (2007) Breast cancer treatment outcome with adjuvant tamoxifen relative to patient CYP2D6 and CYP2C19 genotypes. J Clin Oncol 25(33):5187–5193
Timm R, Kaiser R, Lotsch J, Heider U, Sezer O, Weisz K, Montemurro M, Roots I, Cascorbi I (2005) Association of cyclophosphamide pharmacokinetics to polymorphic cytochrome P450 2C19. Pharmacogenomics J 5(6):365–373
Uttamsingh V, Lu C, Miwa G, Gan L-S (2005) Relative contributions of the five major human cytochromes P450, 1A2, 2C9, 2C19, 2D6, and 3A4, to the hepatic metabolism of the proteasome inhibitor bortezomib. Drug Metab Dispos 33(11):1723–1728
Yamamoto N, Murakami H, Nishina T, Hirashima T, Sugio K, Muro K, Takahashi T, Naito T, Yasui H, Akinaga S (2013) The effect of CYP2C19 polymorphism on the safety, tolerability, and pharmacokinetics of tivantinib (ARQ 197): results from a phase I trial in advanced solid tumors. Ann Oncol 24(6):1653–1659
Zandvliet AS, Huitema ADR, Copalu W, Yamada Y, Tamura T, Beijnen JH, Schellens JH (2007) CYP2C9 and CYP2C19 polymorphic forms are related to increased indisulam exposure and higher risk of severe hematologic toxicity. Clin Cancer Res 13(10):2970–2976
Baldwin RM, Ohlsson S, Pedersen RS, Mwinyi J, Ingelman-Sundberg M, Eliasson E, Bertilsson L (2008) Increased omeprazole metabolism in carriers of the CYP2C19*17 allele; a pharmacokinetic study in healthy volunteers. Br J Clin Pharmacol 65(5):767–774
Goldstein JA, Blaisdell J (1996) Genetic tests which identify the principal defects in CYP2C19 responsible for the polymorphism in mephenytoin metabolism. Methods Enzymol 272:210–218
Sanford JC, Guo Y, Sadee W, Wang D (2013) Regulatory polymorphisms in CYP2C19 affecting hepatic expression. Drug Metabol Drug Interact 28(1):23–30
Sim SC, Risinger C, Dahl ML, Aklillu E, Christensen M, Bertilsson L, Ingelman-Sundberg M (2006) A common novel CYP2C19 gene variant causes ultrarapid drug metabolism relevant for the drug response to proton pump inhibitors and antidepressants. Clin Pharmacol Ther 79(1):103–113
Xie HG, Kim RB, Wood AJJ, Stein CM (2001) Molecular basis of ethnic differences in drug disposition and response. Annu Rev Pharmacol Toxicol 41(1):815–850
Sistonen J, Fuselli S, Palo JU, Chauhan N, Padh H, Sajantila A (2009) Pharmacogenetic variation at CYP2C9, CYP2C19, and CYP2D6 at global and microgeographic scales. Pharmacogenet Genom 19(2):170–179
Xie HG, Stein CM, Kim RB, Wilkinson GR, Flockhart DA, Wood AJ (1999) Allelic, genotypic and phenotypic distributions of S-mephenytoin 4′-hydroxylase (CYP2C19) in healthy Caucasian populations of European descent throughout the world. Pharmacogenetics 9(5):539–549
Helsby NA (2008) Pheno-or genotype for the CYP2C19 drug metabolism polymorphism: the influence of disease. Proc West Pharmacol Soc 51:5–10
Williams ML, Bhargava P, Cherrouk I, Marshall JL, Flockhart DA, Wainer IW (2000) A discordance of the cytochrome P450 2C19 genotype and phenotype in patients with advanced cancer. Br J Clin Pharmacol 49(5):485–488
Helsby NA, Lo WY, Sharples K, Riley G, Murray M, Spells K, Dzhelai M, Simpson A, Findlay M (2008) CYP2C19 pharmacogenetics in advanced cancer: compromised function independent of genotype. Br J Cancer 99(8):1251–1255
Burns KE, Goldthorpe MA, Porteus F, Browett P, Helsby NA (2014) CYP2C19 genotype–phenotype discordance in patients with multiple myeloma leads to an acquired loss of drug-metabolising activity. Cancer Chemother Pharmacol 73(3):651–655
Kim S, Östör AK, Nisar M (2012) Interleukin-6 and cytochrome-P450, reason for concern? Rheumatol Int 32(9):2601–2604
Morgan ET, Goralski KB, Piquette-Miller M, Renton KW, Robertson GR, Chaluvadi MR, Charles KA, Clark Kacevska M, Liddle C, Richardson TA, Sharma R, Sinal CJ (2008) Regulation of drug-metabolizing enzymes and transporters in infection, inflammation, and cancer. Drug Metab Dispos 36(2):205–216
Rivory LP, Slaviero KA, Clarke SJ (2002) Hepatic cytochrome P450 3A drug metabolism is reduced in cancer patients who have an acute-phase response. Br J Cancer 87(3):277–280
Frye RF, Schneider VM, Frye CS, Feldman AM (2002) Plasma levels of TNF-α and IL-6 are inversely related to cytochrome P450-dependent drug metabolism in patients with congestive heart failure. J Card Fail 8(5):315–319
Matthys P, Billiau A (1997) Cytokines and cachexia. Nutrition 13(9):763–770
Helsby NA, Ward SA, Edwards G, Howells RE, Breckenridge AM (1990) The pharmacokinetics and activation of proguanil in man: consequences of variability in drug metabolism. Br J Clin Pharmacol 30(4):593–598
Herrlin K, Massele AY, Rimoy G, Alm C, Rais M, Ericsson Ö, Bertilsson L, Gustafsson LL (2000) Slow chloroguanide metabolism in Tanzanians compared with white subjects and Asian subjects confirms a decreased CYP2C19 activity in relation to genotype. Clin Pharmacol Ther 68(2):189–198
Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D, Verweij J (2009) New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 45(2):228–247
Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC (2000) New guidelines to evaluate the response to treatment in solid tumors. J Natl Cancer Inst 92(3):205–216
Gurney JM, Jelliffe DB (1973) Arm anthropometry in nutritional assessment: nomogram for rapid calculation of muscle circumference and cross-sectional muscle and fat areas. Am J Clin Nutr 26(9):912–915
Norman K, Kirchner H, Lochs H, Pirlich M (2006) Malnutrition affects quality of life in gastroenterology patients. World J Gastroenterol 12(21):3380–3385
Reid IR, Evans MC, Ames R (1992) Relationships between upper-arm anthropometry and soft-tissue composition in postmenopausal women. Am J Clin Nutr 56:463–466
Lo WY (2011) Studies to understand the effect of cancer on hepatic CYP2C19 activity. PhD Thesis, The University of Auckland, Auckland
Lanchote VL, Almeida R, Barral A, Barral‐Netto M, Marques MP, Moraes NV, Silva AM, Souza T, Suarez‐Kurtz G (2015) Impact of visceral leishmaniasis and curative chemotherapy on cytochrome P450 activity in Brazilian patients. Br J Clin Pharmacol 80(5):1160–1168
Shah RR, Smith RL (2015) Addressing phenoconversion: the Achilles’ heel of personalized medicine. Br J Clin Pharmacol 79(2):222–240
Shah RR, Smith RL (2015) Inflammation-induced phenoconversion of polymorphic drug metabolizing enzymes: a hypothesis with implications for personalized medicine. Drug Metab Dispos 43(3):400–410
Alexandre J, Rey E, Girre V, Grabar S, Tran A, Montheil V, Rabillon F, Dieras V, Jullien V, Hérait P, Pons G, Treluyer JM, Goldwasser F (2007) Relationship between cytochrome 3A activity, inflammatory status and the risk of docetaxel-induced febrile neutropenia: a prospective study. Ann Oncol 18(1):168–172
Baker SD, van Schaik RHN, Rivory LP, Ten Tije AJ, Dinh K, Graveland WJ, Schenk PW, Charles KA, Clarke SJ, Carducci MA, McGuire WP, Dawkins F, Gelderblom H, Verweij J, Sparreboom A (2004) Factors affecting cytochrome P-450 3A activity in cancer patients. Clin Cancer Res 10(24):8341–8350
Shord SS, Cavallari LH, Viana MA, Momary K, Neceskas J, Molokie RE, Deyo K, Patel SR (2008) Cytochrome P450 2C9 mediated metabolism in people with and without cancer. Int J Clin Pharmacol Ther 46(7):365–374
de Graan A-JM, Teunissen SF, de Vos FY, Loos WJ, van Schaik RH, de Jongh FE, de Vos AI, van Alphen RJ, van der Holt B, Verweij J (2011) Dextromethorphan as a phenotyping test to predict endoxifen exposure in patients on tamoxifen treatment. J Clin Oncol 29(24):3240–3246
Acknowledgments
This study was funded by the Genesis Oncology Trust and the University of Auckland. We wish to thank the research nurses Georgia Wilson, Michelle Davidson and Karen Spells for their help with patient recruitment. This work has previously been reported as a poster at the AACR Annual Meeting 2014; April 5–9, 2014; San Diego, CA, USA [Cancer Res 74 (19 Supplement):5548].
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Burns, K.E., Lo, WY., Findlay, M.P. et al. High CYP2C19 phenotypic variability in gastrointestinal cancer patients. Cancer Chemother Pharmacol 77, 195–204 (2016). https://doi.org/10.1007/s00280-015-2923-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-015-2923-4