
Abstract
Biomass-derived xylose is an economically interesting substrate for the sustainable microbial production of value-added compounds. Escherichia coli could barely use xylose to directly produce gamma-aminobutyric acid. In this study, E. coli strains that could directly produce gamma-aminobutyric acid were developed through the deletion of eight genes sucA, puuE, gabT, gabP, xylA, xylB, waaC, and waaF, and the overexpression of two E. coli genes gadB and gdhA, as well as five Caulobacter crescent genes CcxylA, CcxylB, CcxylC, CcxylD, and CcxylX. Both E. coli strains W3110 and JM109 could directly produce gamma-aminobutyric acid from xylose after either overexpression of the seven genes or deletion of the eight genes. Overexpression of the seven genes of in the multiple deletion mutants further increased gamma-aminobutyric acid production. Among the 28 recombinant E. coli strains constructed in this study, the highest gamma-aminobutyric acid was produced by JWZ08/pWZt7-g3/pWZt7-xyl. JWZ08/pWZt7-g3/pWZt7-xyl could produce 3.95 g/L gamma-aminobutyric acid in flask cultivation, using xylose as the sole carbon source.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Gamma-aminobutyric acid (GABA), a four-carbon nonprotein amino acid, is the main inhibitory neurotransmitter in human cortex (Wong et al. 2003). It has a potential use as food additive or dietary supplement for its physiological functions, such as blood pressure decrease, anxiety, and even diabetes inhibition (Adeghate and Ponery 2002; Boonstra et al. 2015). In addition, GABA can be also used as the precursor for synthesizing the biodegradable polymer nylon 4 (Park et al. 2013).
Biosynthesis of GABA has been studied over the past decade. Glutamate decarboxylase (Gad), the key enzyme for the GABA production in microorganisms, catalyzes L-glutamic acid (Glu) or monosodium glutamate (MSG) decarboxylation to form GABA. The mostly used bacteria for GABA production are lactic acid bacteria and Escherichia coli. Lactic acid bacteria are generally regarded as safe, therefore, the GABA-producing lactic acid bacteria can be directly used in some functional food. Many GABA-producing lactic acid bacteria have been isolated, such as Lactobacillus brevis (Huang et al. 2007; Kim et al. 2009; Zhang et al. 2012), Lactobacillus paracasei (Komatsuzaki et al. 2005), Lactobacillus buchneri ( Zhao et al. 2015), Lactobacillus plantarum (Siragusa et al. 2007), Lactococcus lactis (Lacroix et al. 2013; Lu et al. 2008), and Enterococcus avium (Tamura et al. 2010). GABA productions lactic acid bacteria can be enhanced by condition optimization. For example, GABA production in L. brevis NCL912 increased sixfold by optimizing the media and controlling pH (Li et al. 2010a; Li et al. 2010b). When Gad was overexpressed in L. plantarum Taj-Apis362, only 11 mM (1.1 g/L) GABA was produced from 0.5 M Glu (Tajabadi et al. 2015). Successful genetic engineering in lactic acid bacteria for GABA production has not been reported.
GABA production in E. coli can be efficiently improved by genetic engineering. Though wild E. coli does not produce GABA from MSG or Glu, 5.07 g/L GABA was produced from 10 g/L of MSG in E. coli XL1-Blue overexpressing Pyrococcus horikoshii Gad (Le Vo et al. 2014). E. coli cells overexpressing Gad can also be used as whole cell biocatalyst to repeatedly convert Glu or MSG to GABA in buffer (Yamano et al. 2012). GABA production from MSG can be dramatically increased in E. coli when GadB was secreted with the help of signal peptide TorA (Zhao et al. 2016).
Since Glu and MSG can be mass-produced by Corynebacterium glutamicum, they are often used as the precursor for GABA production. However, from the industrial and economical point of view, production of GABA directly from glucose is attractive. In C. glutamicum ATCC13032, 0.88 g/L GABA was produced from glucose after 36-h flask cultivation, by overexpressing gadB2 from L. brevis Lb85 (Shi and Li 2011); 26.32 g/L GABA was produced from glucose after 60-h fed-batch fermentation, by overexpressing gadB1 and gadB2 from L. brevis Lb85 (Shi et al. 2013). GABA (70.6 g/L) was produced in C. glutamicum G01 overexpressing Gad from L. plantarum CCTCC M209102 and blocking by-product pools of arginine, proline, and lysine, after 70-h two-stage pH control fermentation (Zhang et al. 2014). Wild E. coli produces neither glutamate nor GABA, but GABA-producing E. coli can be constructed by genetic engineering. For example, by co-overexpressing isocitrate dehydrogenase, glutamate synthase, and GadB, and introducing a synthetic protein scaffold to fix the three enzymes in E. coli XL1-Blue △ackA△gabT, 1.3 g/L GABA was produced from 10 g/L glucose (Pham et al. 2015). GABA can be purified by cation exchange column and crystallized by adding ethanol (Chen et al. 2016; Gao et al. 2013; Seungwoon et al. 2013).
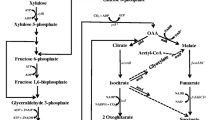
Xylose is a major component of hemicellulose, the renewable low-cost resource. Its abundance and relative ease of extraction from biomass make it an attractive source of fermentable sugar for various productions (Aristidou and Penttilä 2000). But xylose has not been used to produce GABA in E. coli. E. coli metabolizes xylose via the isomerase pathway (Fig. 1). Xylose isomerase encoded by xylA converts xylose to xylulose, which is phosphorylated to lyxulose-5-phosphate by xylulokinase encoded by xylB. Transketolase catalyzes lyxulose-5-phosphate to glyceraldehyde-3-phosphate, the key precursor in Embden-Meyerhof-Parnas (EMP) pathway. In EMP pathway, glyceraldehyde-3-phosphate is gradually converted to phosphoenolpyruvate by enzymes glyceraldehyde-3-phosphate dehydrogenase, phosphoglycerate kinase, phosphoglycerate mutase, and enolase. Then, pyruvate kinase removes the phosphate group of phosphoenolpyruvate to yield pyruvate. Phosphoenolpyruvate and pyruvate enters TCA cycle through oxaloacetate and citrate, respectively. In TCA cycle, α-ketoglutarate is synthesized by enzymes citrate synthase, aconitase, and isocitrate dehydrogenase. At the end, glutamate dehydrogenase encoded by gdhA catalyzes α-ketoglutarate to Glu, which is decarboxylated to form GABA. In addition to the isomerase pathway, Weimberg pathway represents a promising alternative for xylose assimilation (Fig. 1). Weimberg pathway is found in Pseudomonas fragi (Weimberg 1961), Haloferax volcanii (Johnsen et al. 2009), and Caulobacter crescentus (Stephens et al. 2007) and can metabolize xylose to α-ketoglutarate in a five-step oxidative process. In Weimberg pathway, xylose dehydrogenase encoded by CcxylB initiates the oxidation of xylose to xylonolactone, and then xylonolactonase encoded by CcxylC opens the lactone ring to form xylonate which is dehydrated twice in two successive reactions catalyzed by xylonate dehydratase encoded by CcxylD and 2-keto-3-deoxyxylonate dehydratase encoded by CcxylX, respectively. The α-ketoglutarate semialdehyde is then oxidized by α-ketoglutarate semialdehyde dehydrogenase encoded by CcxylA, forming α-ketoglutarate (Stephens et al. 2007). Weimberg pathway does not exist in E. coli. Comparing with the isomerase pathway, Weimberg pathway not only offers a more direct conversion of xylose to α-ketoglutarate but also lowers the carbon loss in the form of CO2 (Radek et al. 2014).

Biosynthetic pathways of GABA from xylose in E. coli used in this study. The isomerase pathway of E. coli is shown in black and the introduced Weimberg pathway in blue. Chemical structures of metabolites from xylose to GABA via Weimberg pathway were shown. Bold arrows indicate the reactions enhanced by overexpressing the relevant genes. The reactions blocked by deleting the relevant genes is indicated by cross
In this study, Weimberg pathway was introduced into E. coli strains JM109 and W3110. GABA-producing E. coli strains were developed through deletion of eight genes sucA, puuE, gabT, gabP, xylA, xylB, waaC, and waaF, and overexpression of two E. coli genes gadB and gdhA, as well as five Caulobacter crescent genes CcxylA, CcxylB, CcxylC, CcxylD, and CcxylX. The final strain JWZ08/pWZt7-g3/pWZt7-xyl could produce 3.95 g/L GABA in flask cultivation, using xylose as the sole carbon source.
Materials and methods
Strains, media, and growth conditions
The strains and plasmids used in this study are listed in Table 1. E. coli strains were grown at 37 °C in LB medium (10 g/L peptone, 5 g/L yeast extract, and 10 g/L NaCl). Ampicillin (100 μg/mL), kanamycin (30 μg/mL), and chloramphenicol (30 μg/mL) were added when necessary. C. glutamicum ATCC13869 was grown at 30 °C in LBG medium (LB medium supplemented with 5 g/L glucose). C. crescentus NA1000 was grown at 30 °C in PYE medium (2 g/L peptone, 1 g/L yeast extract, and 0.2 g/L MgSO4·7H2O).
For flask fermentation, a loop of E. coli cell colonies was inoculated into 20-mL LB medium in a 250-mL flask. One-milliliter overnight seed culture was diluted into 20-mL XTB medium (20 g/L xylose, 24 g/L yeast extract, 12 g/L tryptone, 2.31 g/L KH2PO4, 16.43 g/L K2HPO4) in which xylose was separately sterilized to avoid Maillard reaction, and incubated for 48 h at 37 °C and 200 rpm. The pH was kept to 6.5–7.5 by 10% (v/v) ammonia solution during the first 36 h, and 1 mM IPTG was added when OD600 reached 1.0. At 36 h, 10 mM PLP was added and pH was adjusted to 4.6 by 4 M HCl to produce GABA.
Construction of plasmids pWZtac-g2, pWZt7-g2, pWZt7-g3, pWZtac-xyl, and pWZt7-xyl
Restrictions enzymes, T4 DNA ligase and DNA ladder were purchased from Fermentus (USA). Prime STAR™ HS DNA polymerase was purchased from TaKaRa (Dalian, China). Kits for plasmids isolation, chromosomal DNA extraction, and DNA purification from agarose gels were purchased from Tiangen (China). DNA sequencings were performed by BGI (China). PCR experiments were performed by using Mastercycler from Eppendorf (Hamburg, Germany). The primers used in this study are listed in Table 2. Generally, 50-μL PCR reaction mixture includes 10 μL PS buffer, 4 μL dNTP mixture, 1 μL template (100 ng/μL), 1 μL forward primer (20 μM), 1 μL reverse primer (20 μM), and 0.5 μL PrimeSTAR™ HS DNA polymerase. PCR amplification was performed for 35 cycles. Each cycle consisted of denaturation at 95 °C for 30 s, annealing at 55 °C for 30 s, and extension at 72 °C. The time extension varied with the length of PCR product (1 kb/min).
The plasmid pACYCDuet-1 was used to construct pWZtac-g2, pWZt7-g2, and pWZt7-g3 (Fig. 2). pACYCDuet-1 contains two multiple cloning sites (MCS) preceded by T7 promoter.

Maps of plasmids pACYCDuet-1, pETDuet-1, pDXW-8, pWZt7-xyl, pWZtac-xyl, pWZt7-g2, pWZt7-g3, and pWZtac-g2
To construct pWZtac-g2, the gene gdhA was PCR amplified by using primer pairs gdhA(tac)-F and gdhA(tac)-R and genomic DNA of E. coli W3110 as template, digested with EcoR I and Hind III, and ligated into pDXW-8 (Xu et al. 2010) that was similarly digested; then, the fragment ptac-gdhA was PCR amplified by using primer pairs Ptac-gdhA-F and Ptac-gdhA-R from pDXW-8-gdhA, and digested with Pst I and Afl II. Similarly, the gene gadB was PCR amplified by using primer pairs gadB(tac)-F and gadB (tac)-R from the E. coli genomic DNA, digested with Nhe I and Hind III, ligated into pDXW-8, and the fragment ptac-gadB was then PCR amplified by using primer pairs Ptac-gadB-F and Ptac-gadB-R, and digested with Afl II and Kpn I. The fragment pACYCDuet-1-a which contained no T7 promoters and MCSs was PCR amplified from the vector pACYCDuet-1, using the primer pairs pACYC-F and pACYC-R2, and digested with Pst I and Kpn I. The digested fragments ptac-gdhA, ptac-gadB, and pACYCDuet-1-a were ligated together to form the plasmid pWZtac-g2. pWZtac-g2 carried genes gdhA and gadB preceded by the tac promoters. Tac promoter is the combination of the trp and lac promoters but can be recognized by E. coli RNA polymerase more efficiently than either trp or lac promoters.
T7 promoter from E. coli bacteriophage T7 can be only recognized by T7 RNA polymerase. To express genes controlled by T7 promoters in E. coli, an ftsZ promoter-dependent T7 expression system was established (Equbal et al. 2013). This system expresses T7 RNA polymerase under the control of ftsZ promoter and then T7 RNA polymerase transcribes the genes downstream the T7 promoter. A vector pWZt7 harboring gene encoding T7 RNA polymerase under the control of ftsZ promoter was constructed in this study. To construct pWZt7, the ftsZ promoter pftsZ was PCR amplified by using primer pairs PftsZ-F and PftsZ-R from the genomic DNA of C. glutamicum ATCC13869, and digested with Pst I and EcoR I; the gene t7pol encoding the T7 RNA polymerase was PCR amplified by using primer pairs t7pol-F and t7pol-R from the genomic DNA of E. coli BL21(DE3), and digested with EcoR I and BamH I; the fragment pACYCDuet-1-b which contained no MCSs and T7 promoters was PCR amplified by using primer pairs pACYC-F and pACYC-R from the plasmid pACYCDuet-1, and digested with Pst I and BamH I. Then, the digested fragment pftsZ, t7pol, and pACYCDuet-1-b were ligated together to form pWZt7. To construct pWZt7-g2, the gene gdhA was PCR amplified by using primer pairs gdhA(t7)-F and gdhA(t7)-R, digested with Nco I and Sal I, and ligated into pACYCDuet-1 that was similarly digested; then, the fragment pt7-gdhA-teminater was PCR amplified by using primers Pt7-gdhA-F and Pt7-gdhA-R, digested with Pst I and Sca I, and ligated into pWZt7 that was similarly digested, forming pWZt7-gdhA. The gene gadB was PCR amplified by using primer pairs gadB(t7)-F and gadB(t7)-R, digested with Nde I and Kpn I, and ligated into pACYCDuet-1 that was similarly digested; then, the fragment pt7-gadB was PCR amplified by using primer pairs Pt7-gadB-F and Pt7-gadB-R, digested with Hind III and Xho I, and ligated into pWZt7-gdhA that was similarly digested, forming the plasmid pWZt7-g2. The fragment pt7-torA-gadB was PCR amplified by using primer pairs Pt7-gadB-F2 and Pt7-gadB-R2 from the plasmid pET20b-torA-gadB (Zhao et al. 2016), digested with Hind III and Xho I, and ligated into pWZt7-gdhA that was similarly digested, forming the plasmid pWZt7-g3. The fragment torA encodes a signal peptide which can facilitate the secretion of GadB encoded by gadB (Fig. 2).
Both pDXW-8 (Xu et al. 2010) and pETDuet-1 harbor pBR322 replicon. pDXW-8 carries one MCS under the control of tac promoter. pETDuet-1 carries two MCSs, each of which was preceded by a T7 promoter. pDXW-8 and pETDuet-1 were used to construct pWZtac-xyl and pWZt7-xyl, respectively. To construct pWZtac-xyl, the genes CcxylB, CcxylX, CcxylD, CcxylC, and CcxylA were PCR amplified from the genomic DNA of C. crescentus NA1000, respectively, by using primer pairs CcxylB(tac)-F and CcxylB(tac)-R, CcxylX(tac)-F and CcxylX(tac)-R, CcxylD(tac)-F and CcxylD(tac)-R, CcxylC(tac)-F and CcxylC(tac)-R, and CcxylA(tac)-F and CcxylA(tac)-R. The PCR products of genes CcxylB, CcxylX, CcxylD, CcxylC, and CcxylA were digested with EcoR I and Sac I, Nhe I and Nco I, EcoR I and Afl II, EcoR I and Pst I, and EcoR I and Hind III, respectively, and ligated into pDXW-8 that was similarly digested, forming the plasmids pDXW-8-CcxylB, pDXW-8-CcxylX, pDXW-8-CcxylD, pDXW-8-CcxylC, and pDXW-8-CcxylA. The fragment ptac-CcxylX was PCR amplified by using Ptac-CcxylX-F and CcxylX(tac)-R from pDXW-8-CcxylX, digested with Sac I and Nco I, and ligated into pDXW-8-CcxylB similarly digested, forming the plasmid pDXW-8-CcxylBX. The fragment ptac-CcxylD was PCR amplified by using primer pairs Ptac-CcxylD-F and CcxylD(tac)-R from pDXW-8-CcxylD, digested with kpn I and Afl II, and ligated into pDXW-8-CcxylBX, forming the plasmid pDXW-8-CcxylBXD. The fragment ptac-CcxylC amplified by using Ptac-CcxylC-F and CcxylC(tac)-R from pDXW-8-CcxylC was digested with Afl II and Pst I; the fragment ptac-CcxylA amplified by using Ptac-CcxylA-F and CcxylA(tac)-R from pDXW-8-CcxylA was digested with Pst I and Hind III. The digested fragment ptac-CcxylC and ptac-CcxylA were ligated into pDXW-8-CcxylBXD digested with Afl II and Hind III, forming the plasmid pWZtac-xyl. pWZtac-xyl could co-exist with pWZtac-g2 in the same E. coli cell.
To construct pWZt7-xyl, the genes CcxylB, CcxylX, CcxylD, CcxylC, and CcxylA were PCR amplified from the genomic DNA of C. crescentus NA1000, respectively, by using primer pairs CcxylB(t7)-F and CcxylB(t7)-R, CcxylX(t7)-F and CcxylX(t7)-R, CcxylD(t7)-F and CcxylD(t7)-R, CcxylC(t7)-F and CcxylC(t7)-R, and CcxylA(t7)-F and CcxylA(t7)-R. The PCR products of genes CcxylB, CcxylX, CcxylD, and CcxylA were digested with Nco I and BamH I, Nco I and BamH I, Nde I and Kpn I, and Nde I and Kpn I, respectively, and ligated into pETDuet-1 that was similarly digested, forming the plasmids pETDuet-1-CcxylB, pETDuet-1-CcxylX, pETDuet-1-CcxylD, and pETDuet-1-CcxylA. The PCR product of the gene CcxylC was digested with Nde I and Kpn I, and ligated into the pETDuet-1-CcxylB that was similarly digested, forming the plasmid pETDuet-1-CcxylBC. The fragment pt7-CcxylX was PCR amplified by using Pt7-CcxylX-F and Pt7-CcxylX-R from plasmid pETDuet-1-CcxylX, and digested with Pst I and Afl II; the fragment pt7-CcxylD was PCR amplified by using Pt7-CcxylD-F and Pt7-CcxylD-R form plasmid pETDuet-1-CcxylD, and digested with Afl II and Hind III. The digested fragments pt7-CcxylX and pt7-CcxylD were ligated into pETDuet-1-CcxylBC which digested with Pst I and Hind III, forming the plasmid pETDuet-1-CcxylBXDC. The fragment pt7-CcxylA was PCR amplified by using Pt7-CcxylA-F and Pt7-CcxylA-R from pETDuet-1-CcxylA, digested with kpn I and Avr II, and ligated into pETDuet-1-CcxylBXDC which was similarly digested, forming the plasmid pWZt7-xyl.
Multiple gene deletions in E. coli JM109 and W3110
Multiple genes were deleted in E. coli JM109 and W3110, using Red recombination system (Datsenko and Wanner 2000), and the resistance genes were removed by using Cre-loxP system (Han et al. 2013).
In E. coli W3110, the gene puuE was firstly deleted. The upstream fragment of puuE was PCR amplified by using primer pairs puuE-U-F and puuE-U-R from the genomic DNA of E. coli W3110, and digested with Xho I and Xba Ι; the downstream fragment of puuE was PCR amplified by using primer pairs puuE-D-F and puuE-D-R, and digested by BamH Ι and Pst I; the fragment loxpLE-kan-loxpRE was PCR amplified by using primer pairs kan-F and kan-R1 from the plasmid pDTW202 (Han et al. 2013), and digested by Xba Ι and BamH Ι. The three digested PCR products were ligated into pBluescript II SK(+)digested with Xho I and Pst I, and then the DNA segment containing the loxpLE-kan-loxpRE, and the upstream and downstream fragments of puuE was PCR amplified by using primer pairs puuE-F and puuE-R, and transformed into E. coli W3110 cells harboring the Red recombination plasmid pKD46 to replace the gene puuE with loxpLE-kan-loxpRE. Then, the kan gene was removed by introducing the plasmid pKDCre which can overexpress recombinase Cre and recombine the loxp sequences at both ends of the kan, resulting in the mutant strain W3110△puuE. Both pKDCre and pKD46 are temperature sensitive; therefore, they can be cured by growing the cells at 42 °C. Similarly, genes gabT and gabP were deleted in W3110△puuE, resulting in WWZ03. Genes gabT and gabP are next to each other, so they were deleted by one recombination. The upstream fragment of gabTP was amplified by using primer pairs gabTP-U-F and gabTP-U-R, and the downstream fragment of gabTP was amplified by using primer pairs gabTP-D-F and gabTP-D-R. The DNA segment containing the loxpLE-kan-loxpRE, and the upstream and downstream fragments of gabTP was amplified by using primer pairs gabTP-F and gabTP-R. The gene sucA was deleted in WWZ03, resulting in WWZ04. The upstream fragment of sucA was amplified by using primer pairs sucA-U-F and sucA-U-R, the downstream fragment of sucA was amplified by using primer pairs sucA-D-F and sucA-D-R, and the fragment loxpLE-kan-loxpRE was amplified by using primer pairs kan-F and kan-R2. The DNA segment containing the loxpLE-kan-loxpRE, and the upstream and downstream fragments of sucA was amplified by using primer pairs sucA-F and sucA-R. The genes xylA and xylB were deleted in WWZ04, resulting in WWZ006. Genes xylA and xylB are next to each other on the chromosome, so they were deleted by one recombination. The upstream fragment of xylAB was amplified by using primer pairs xylAB-U-F and xylAB-U-R, the downstream fragment of xylAB was amplified by using primer pairs xylAB-D-F and xylAB-D-R. The DNA segment containing the loxpLE-kan-loxpRE, and the upstream and downstream fragments of xylAB was amplified by using primer pairs xylAB-F and xylAB-R. The genes waaC and waaF were deleted in WWZ06, resulting in WWZ008. Genes waaC and waaF were contiguous, and they were deleted by one recombination. The upstream fragment of waaFC was amplified by using primer pairs waaFC-U-F and xylAB-U-R, and the downstream fragment of waaFC was amplified by using primer pairs waaFC-D-F and waaFC-D-R. The DNA segment containing the loxpLE-kan-loxpRE, and the upstream and downstream fragments of waaFC was amplified by using primer pairs waaFC-F and waaFC-R.
Using the similar protocol, the eight genes puuE, gabT, gabP, sucA, xylA, xylB, waaF, and waaC were also deleted in E. coli JM109. Genes waaF and waaC were first deleted in JM109, resulting in the mutant JWZ02; the gene sucA in JWZ02 was deleted, resulting in the mutant JWZ03; genes puuE, gabT, and gabP in JWZ03 were deleted, resulting the mutant JWZ06; finally, genes xylA and xylB in JWZ06 were deleted, resulting in the mutant JWZ08.
Construction of E. coli recombinant strains
Recombinant strains JM109/pWZt7-g2 and W3110/pWZt7-g2 were constructed by introducing pWZt7-g2 into JM109 and W3110, respectively. JM109/pWZtac-g2 and W3110/pWZtac-g2 were constructed by introducing pWZtac-g2 into JM109 and W3110, respectively. By introducing both pWZt7-g2 and pWZt7-xyl into JM109, JWZ02, JWZ03, JWZ06, JWZ08, W3110, WWZ03, WWZ04, WWZ06, and WWZ08, respectively, recombinant strains JM109/pWZt7-g2/pWZt7-xyl, JWZ02/pWZt7-g2/pWZt7-xyl, JWZ03/pWZt7-g2/pWZt7-xyl, JWZ06/pWZt7-g2/pWZt7-xyl, JWZ08/pWZt7-g2/pWZt7-xyl, W3110/pWZt7-g2/pWZt7-xyl, WWZ03/pWZt7-g2/pWZt7-xyl, WWZ04/pWZt7-g2/pWZt7-xyl, WWZ06/pWZt7-g2/pWZt7-xyl, and WWZ08/pWZt7-g2/pWZt7-xyl were constructed. By introducing both pWZtac-g2 and pWZtac-xyl into JM109, JWZ02, JWZ03, JWZ06, JWZ08, W3110, WWZ03, WWZ04, WWZ06, and WWZ08, respectively, recombinant strains JM109/pWZtac-g2/pWZtac-xyl, JWZ02/pWZtac-g2/pWZtac-xyl, JWZ03/pWZtac-g2/pWZtac-xyl, JWZ06/pWZtac-g2/pWZtac-xyl, JWZ08/pWZtac-g2/pWZtac-xyl W3110/pWZtac-g2/pWZtac-xyl, WWZ03/pWZtac-g2/pWZtac-xyl, WWZ04/pWZtac-g2/pWZtac-xyl, WWZ06/pWZtac-g2/pWZtac-xyl, and WWZ08/pWZtac-g2/pWZtac-xyl were constructed. By introducing both pWZt7-g3 and pWZt7-xyl into JWZ02, JWZ03, JWZ06, and JWZ08, respectively, recombinant strains JWZ02/pWZt7-g3/pWZt7-xyl, JWZ03/pWZt7-g3/pWZt7-xyl, JWZ06/pWZt7-g3/pWZt7-xyl, and JWZ08/pWZt7-g3/pWZt7-xyl were constructed.
Analysis of cell density and levels of Glu, GABA, and xylose
Cell density of different E. coli strains was determined by measuring OD600 with UV-1800 spectrophotometer (Shimadzu, Japan).
Glu and GABA production in different cells were analyzed by HPLC (Agilent Technologies 1200 series, USA). E. coli cells were grown and centrifuged, the supernatant of broth was treated with 20 volume 5% (w/v) trichloroacetic acid for 2 h to precipitate proteins, and the clear filtrate was used directly for HPLC analysis. HPLC separation and quantitation was performed on a Thermo ODS C18 column (5 μm, 250 mm × 4.6 mm, USA) by using the o-phthalaldehyde (OPA) pre-column derivatization method (Kőrös et al. 2008). Separation of the samples was attained using a binary nonlinear gradient with solution A (sodium acetate 3.01 g, tetrahydrofuran 3 mL, and trimethylamine 0.2 mL dissolved or diluted in ddH2O to 1 L, pH 7.2) and solution B (sodium acetate 3.01 g dissolved in ddH2O to 200 mL, and then supplied with 400 mL methanol and 400 mL acetonitrile, pH 7.2). The column was kept at 40 °C. Elution conditions were as follows: equilibration (5 min, 8% B), gradient (15 min, 8–100% B), and cleaning (5 min, 100% B). The mobile phase was set at 1 mL/min. Glu and GABA were detected at 338 nm, and the spectra were recorded online.
The xylose concentration was determined by using the published method (Eberts et al. 1979) with modification. Briefly, 0.5 mL properly diluted supernatant of the broth was mixed with 2.5 mL phloroglucinol reagent (0.5 g phloroglucinol, 100 mL acetic acid, and 6 mL HCl), heated for 8 min at 100 °C, cooled in water to room temperature, and its absorbance at 554 nm was measured. Xylose concentration was calculated according to a standard curve.
Results
Weimberg pathway could be used in E. coli for GABA production from xylose
Two-stage pH control fermentation strategy was used in this study for GABA production from xylose in E. coli. During the first 36 h, pH was kept at 6.5 to 7.5, Glu was accumulated; after 36 h, pH was kept at 4.6, and GABA was produced; E. coli cells was viable in the first 36 h, but most cells could not survive during 36–48 h after pH was decreased. Therefore, Glu concentrations and cell densities (OD600) at 36 h as well as GABA productions at 48 h were collected to value the performance of different E. coli mutant cells.
When xylose was used as the carbon source, wild-type E. coli W3110 grew well (OD600 at 36 h reached 19.71), but accumulated neither Glu at 36 h nor GABA at 48 h (Fig. 3). Bacteria including E. coli exist the gene gdhA encoding glutamate dehydrogenase Gdh and the gene gadB encoding glutamate decarboxylase GadB; Gdh converts α-ketoglutarate to Glu, and GadB converts Glu to GABA. Overexpressing Gdh in C. glutamicum could enhance Glu production (Asakura et al. 2007). Therefore, genes gdhA and gadB were inserted into the vector pACYCDuet-1, forming pWZt7-g2 (both genes were controlled by T7 promoters) and pWZtac-g2 (both genes were controlled by tac promoters), and transformed into E. coli W3110. 1.14 and 1.27 g/L Glu were accumulated after 36 h in the fermentation of W3110/pWZt7-g2 and W3110/pWZtac-g2 cells, respectively; 0.35 and 0.52 g/L GABA were produced after 48 h in the fermentation of W3110/pWZt7-g2 and W3110/pWZtac-g2 cells, respectively. Compared with the wild type, OD600 of W3110/pWZt7-g2 and W3110/pWZtac-g2 cells at 36 h slightly decreased, probably due to the metabolic burden of gene overexpression and plasmid replication (Wu et al. 2013).

Glu production at 36 h, OD600 at 36 h, and GABA production at 48 h after two-stage pH control fermentation in E. coli strains W3110, JM109, W3110/pWZt7-g2, W3110/pWZtac-g2, JM109/pWZt7-g2, JM109/pWZtac-g2, W3110/pWZt7-g2/pWZt7-xyl, W3110/pWZtac-g2/pWZtac-xyl, JM109/pWZt7-g2/pWZt7-xyl, and JM109/pWZtac-g2/pWZtac-xyl
E. coli has an inherent pathway metabolizing xylose to α-ketoglutarate, but it is a long and complex pathway involving EMP and TCA cycle (Fig. 1). Weimberg pathway found in C. crescentus NA1000 can covert xylose to α-ketoglutarate in five reactions. Weimberg pathway was implemented in C. glutamicum for cell growth on xylose as sole carbon source (Radek et al. 2014), and also used to produce xylonate in E. coli BL21(DE3) and W3110(DE3) by co-expressing CcxylB and CcxylC under the control of T7 promoter (Cao et al. 2013; Liu et al. 2012a). Therefore, the five Weimberg pathway genes CcxylB, CcxylX, CcxylD, CcxylC, and CcxylA were either cloned into pDXW-8 under the control of tac promoter (pWZtac-xyl) or into pETDuet-1 under the control of tac promoter (pWZt7-xyl), and transformed into E. coli W3110/WZt7-g2 and W3110/pWZtac-g2, respectively. Due to their different replicons, plasmids pWZt7-xyl and pWZt7-g2 could co-exist in the same E. coli cells, so could the plasmids pWZtac-xyl and pWZtac-g2. Therefore, the recombinant E. coli W3110/pWZt7-g2/pWZt7-xyl and W3110/pWZtac-g2/pWZtac-xyl cells could overexpress all seven enzymes required for GABA production from xylose. As expected, Glu concentrations at 36 h and GABA concentrations at 48 h further increased in W3110/pWZt7-g2/pWZt7-xyl and W3110/pWZtac-g2/pWZtac-xyl (Fig. 3), suggesting that Weimberg pathway could be used in E. coli for GABA production from xylose.
The same plasmids pWZt7-g2, pWZt7-xyl, pWZtac-g2, and pWZtac-xyl were also transformed into E. coli JM109. Neither Glu nor GABA was detected during two-stage pH control fermentation of E. coli JM109. However, JM109/pWZt7-g2 and JM109/pWZtac-g2 produced about 1.2 g/L Glu at 36 h and 0.5 g/L GABA at 48 h; JM109/pWZt7-g2/pWZt7-xyl and JM109/pWZtac-g2/pWZtac-xyl produced about 1.9 g/L Glu at 36 h and 0.75 g/L GABA at 48 h (Fig. 3). The results demonstrate again that Weimberg pathway could be used in E. coli for GABA production from xylose.
Multiple gene deletions in E. coli W3110 to enhance GABA production
Gene gabP encodes GABA transporter which transports extracellular GABA into E. coli cells. Both genes gabT and puuE encode GABA aminotransferases which convert GABA to succinic semialdehyde. Succinic semialdehyde can be converted to succinate by succinic semialdehyde dehydrogenase. These enzymes play important role in the GABA shunt. Deletion of gabP, gabT, and puuE should decrease the consumption of GABA by the cells. GABA production was enhanced in E. coli when gabT was deleted, using glucose as the carbon source (Pham et al. 2016a). In this study, the three genes gabP, gabT, and puuE were deleted in E. coli W3110, and plasmid pairs pWZt7-g2/pWZt7-xyl and pWZtac-g2/pWZtac-xyl were transformed into the triple deletion mutant WWZ03 (Fig. 4). After two-stage pH control fermentation, 1.04 and 0.93 g/L GABA were produced in WWZ03/pWZt7-g2/pWZt7-xyl and WWZ03/pWZtac-g2/pWZtac-xyl, respectively.

a Comparison of different deletion mutants derived from E. coli W3110. b Glu production at 36 h, OD600 at 36 h, and GABA production at 48 h after two-stage pH control fermentation in E. coli strains WWZ3, WWZ3/WZt7-g2/pWZt7-xyl, WWZ3/pWZtac-g2/pWZt7-xyl, WWZ4, WWZ4/pWZt7-g2/pWZt7-xyl, WWZ4/pWZtac-g2/pWZt7-xyl, WWZ6, WWZ2/pWZt7-g6/pWZt7-xyl, WWZ6/pWZtac-g2/pWZt7-xyl, WWZ8, WWZ8/pWZt7-g2/pWZt7-xyl, and WWZ8/pWZtac-g2/pWZt7-xyl
Gene sucA encodes the subunit E1 of α-ketoglutarate decarboxylase which converts α-ketoglutarate to succinyl-CoA (Fig. 1). More Glu could be accumulated when sucA was deleted in E. coli or C. glutamicum (Asakura et al. 2007; Hirasawa et al. 2012; Nishio et al. 2013). In this study, the gene sucA was deleted in WWZ03, and the plasmid pairs pWZt7-g2/pWZt7-xyl and pWZtac-g2/pWZtac-xyl were transformed into the quadruple deletion mutant WWZ04 (Fig. 4). After two-stage pH control fermentation, productions of both Glu (3.52 and 2.94 g/L) and GABA (1.97 and 1.66 g/L) significantly increased in WWZ04/pWZt7-g2/pWZt7-xyl and WWZ04/pWZtac-g2/pWZtac-xyl (Fig. 4). More than 80% Glu accumulated at 36 h were converted to GABA at 48 h. Compared with the control strain WWZ04, OD600 of WWZ04/pWZt7-g2/pWZt7-xyl and WWZ04/pWZtac-g2/pWZtac-xyl at 36 h decreased. The cell growth inhibition might be due to the deletion of sucA which caused the breakdown of TCA cycle. The supply of oxaloacetate and succinate might rely on the glyoxylate cycle (Asakura et al. 2007).
In E. coli, genes xylA and xylB encoded xylose isomerase and xylulose kinase, respectively. Deletion of xylA and xylB and overexpression the first two genes of Weimberg pathway in E. coli increased xylonate production from xylose (Cao et al. 2013; Liu et al. 2012a). In this study, genes xylA and xylB were deleted in WWZ04 and the plasmid pairs pWZt7-g2/pWZt7-xyl and pWZtac-g2/pWZtac-xyl were transformed into the sextuple deletion mutant WWZ06 (Fig. 4). Unfortunately, productions of neither Glu nor GABA in WWZ06/pWZt7-g2/pWZt7-xyl and WWZ06/pWZtac-g2/pWZtac-xyl after two-stage pH control fermentation were exceeded those in WWZ04/pWZt7-g2/pWZt7-xyl and WWZ04/pWZtac-g2/pWZtac-xyl (Fig. 4).
In E. coli, genes waaC and waaF encode heptosyltransferase WaaC and WaaF which sequentially add two L-D-heptoses to Kdo2-lipid A of lipopolysaccharide in the outer membrane (Brabetz et al. 1997). Deletion of waaC and waaF could increase membrane permeability of E. coli (Wang et al. 2015). In this study, genes waaC and waaF were deleted in WWZ06 and the plasmid pairs pWZt7-g2/pWZt7-xyl and pWZtac-g2/pWZtac-xyl were transformed into the octuplet deletion mutant WWZ08 (Fig. 4). Production levels of Glu and GABA in WWZ08/pWZt7-g2/pWZt7-xyl and WWZ08/pWZtac-g2/pWZtac-xyl after two-stage pH control fermentation were similar to those in WWZ06/pWZt7-g2/pWZt7-xyl and WWZ06/pWZtac-g2/pWZtac-xyl (Fig. 4).
Multiple gene deletions in E. coli JM109 to enhance GABA production
Genes waaC, waaF, gabP, gabT, puuE, sucA, xylA, and xylB in E. coli JM109 were also deleted in different ways as shown in Fig. 5a, resulting in deletion mutants JWZ02, JWZ03, JWZ06, and JWZ08. The plasmid pairs pWZt7-g2/pWZt7-xyl, pWZtac-g2/pWZtac-xyl, and pWZt7-g2/pWZt7-xyl were transformed into these deletion mutant cells, forming a series of recombinant E. coli strains. Plasmid pWZt7-g3 is similar to pWZt7-g2; the difference between them is that there is torA fused with gadB in pWZt7-g3 (Fig. 2). Signal peptide TorA encoded by torA could efficiently secrete GadB in E. coli to improve the production of GABA (Zhao et al. 2016).

a Comparison of different deletion mutants derived from E. coli W3110. b Glu production at 36 h, OD600 at 36 h, and GABA production at 48 h after two-stage pH control fermentation in E. coli strains JWZ2, JWZ2/pWZtac-g2/pWZt7-xyl, JWZ2/WZt7-g2/pWZt7-xyl, JWZ2/WZt7-g3/pWZt7-xyl, JWZ3, JWZ3/pWZtac-g2/pWZt7-xyl, JWZ3/WZt7-g2/pWZt7-xyl, JWZ3/WZt7-g3/pWZt7-xyl, JWZ6, JWZ6/pWZtac-g2/pWZt7-xyl, JWZ6/WZt7-g2/pWZt7-xyl, JWZ6/WZt7-g3/pWZt7-xyl, JWZ8, JWZ8/pWZtac-g2/pWZt7-xyl, JWZ8/WZt7-g2/pWZt7-xyl, and JWZ8/WZt7-g3/pWZt7-xyl
Productions of Glu and GABA in different recombinant E. coli strains after two-stage pH control fermentation are shown in Fig. 5b. Generally, E. coli cells harboring plasmid pairs containing T7 promoters produced more Glu and GABA than those cells harboring plasmid pairs containing tac promoters. JWZ08 cells produced more Glu and GABA than other cells when harboring the same plasmid pairs, and the most GABA production was obtained in the strain JWZ08/pWZt7-g3/pWZt7-xyl. After two-stage pH control fermentation of JWZ08/pWZt7-g3/pWZt7-xyl, 6.25 g/L Glu at 36 h, and 3.21 g/L GABA was produced at 48 h.
Optimize GABA production in JWZ08/pWZt7-g3/pWZt7-xyl
Among all the recombinant E. coli strains constructed in this study, the maximum GABA production was obtained in the strain JWZ08/pWZt7-g3/pWZt7-xyl. Therefore, the optimum conditions for the fermentation of JWZ08/pWZt7-g3/pWZt7-xyl to produce GABA were performed.
The optimum temperature for wild E. coli growth is 37 °C, but some engineered E. coli grew better at 30 °C. In this study, at 30 °C, OD600 at 36 h of JWZ08/pWZt7-g3/pWZt7-xyl was 8.30; Glu concentration at 36 h and GABA production at 48 h were 4.50 and 1.26 g/L, respectively. Comparing the performance of JWZ08/pWZt7-g3/pWZt7-xyl at 37 °C, the cell growth was similar, but Glu and GABA productions at 30 °C were 28 and 61% lower than those at 37 °C (Fig. 6a).

GABA production in E. coli JWZ08/pWZt7-g3/pWZt7-xyl was optimized at temperature, media volume, pH, concentrations of IPTG, peptone, and yeast extract
In E. coli BL21(DE3), IPTG is required to express T7 RNA polymerase which is under control of the IPTG-inducible lacUV5 promoter. In this study, the ftsZ promoter-dependent T7 expression system was used, which is not induced by IPTG (Equbal et al. 2013). However, in pWZt7-g2, pWZt7-g3 and pWZt7-xyl, IPTG-controlled lac operators are behind the T7 promoters (T7-lac promoter) (Dubendorf and Studier 1991). So, even though the expression of T7 RNA polymerase is not affected by IPTG, IPTG is still required to activate the genes in pWZt7-g2, pWZt7-g3, and pWZt7-xyl. Effects of IPTG additions ranging from 0 to 2 mM were investigated, and 1 mM IPTG was found to be the best (Fig. 6a).
Medium volume in flask is related to dissolve oxygen, which is important in aerobic fermentation of E. coli. The effect of medium volume in 250-mL flask on the GABA production was investigated. Cell growth and Glu and GABA productions decreased with the increased of medium volume (Fig. 6a).
GadB is only active in acid conditions; thus, during the period for GABA production, pH is needed to be adjusted. After 36 h, pH was adjusted to 3, 3.8, and 4.6, and 1.06, 2.34, and 3.21 g/L GABA were produced at 48 h, respectively (Fig. 6a). Therefore, pH 4.6 was chosen to be the optimum pH.
XTB medium containing abundant yeast extract and peptone was used to produce GABA. Lowering the content of yeast extract and peptone in the medium would lower the cost for GABA production. As shown in Fig. 6b, the concentrations of peptone and yeast extract were optimized and the optimized concentrations of peptone and yeast extract are 2.4 and 24 g/L, respectively. Under the optimum conditions, 6.89 g/L Glu was produced at 36 h, and 3.81 g/L GABA was produced at 48 h (Fig. 6b).
JWZ08/pWZt7-g3/pWZt7-xyl was fermented under the optimum conditions. In the first stage for Glu accumulation, pH was controlled between 6.5 and 7.5, and 1 mM IPTG was added at 2 h when OD600 reached 1.0; in the second stage for GABA production, pH was adjusted to 4.6 and 10 mM PLP was added. As shown in Fig. 7, cells entered the stationary phase at 18 h when xylose was almost consumed. Glu production was continuously increased in the first 36 h and reached around 6.89 g/L at 36 h, and then slowly increased to 7.08 g/L at 48 h and 7.19 g/L at 60 h, respectively. After pH was adjusted to 4.6, Glu concentration decreased with the production of GABA. When pH was adjusted to 4.6 at 36, 48, and 60 h, 3.95, 3.57, and 2.75 g/L GABA was produced in JWZ08/pWZt7-g3/pWZt7-xyl at 60, 72, and 84 h, respectively (Fig. 7).

Comparison of GABA production of E. coli JWZ08/pWZt7-g3/pWZt7-xyl when pH was adjusted to 4.6 at 36, 48, and 60 h, respectively
Discussion
Xylose, a key building block in most hemicelluloses, can be used by some bacteria via different pathways (Aristidou and Penttilä 2000), such as the xylose isomerase pathway, oxido-reductase pathway, and Weimberg pathway. For example, succinate (Liu et al. 2012b), ethanol (Saha et al. 2015), and lactate (Zhao et al. 2013) could be produced via the inherent xylose isomerase pathway; xylitol can be produced via oxido-reductase pathway from Candida sp. and Neurospora sp. (Iverson et al. 2013; Su et al. 2015), xylonate (Cao et al. 2013), and 1,2,4-butanetriol (Valdehuesa et al. 2014) can be produced via the Weimberg pathway.
The first attempt to produce GABA from xylose in E. coli was conducted in this study. In E. coli, there is the xylose isomerase pathway which is too long to contribute the accumulation of GABA. The carbon flux from xylose to GABA requires only seven steps through Weimberg pathway but requires around 20 steps through the isomerase pathway. Through Weimberg pathway, xylose can be completely converted into α-ketoglutarate without carbon loss, while in the isomerase pathway, 17% carbon is lost in the form of CO2 during TCA cycle (Radek et al. 2014). Furthermore, the isomerase pathway was long and meditated by some key metabolites, such as glyceraldehyde-3-phosphate, pyruvate, and Acetyl-CoA. Therefore, Weimberg pathway which can covert xylose to α-ketoglutarate by five reaction steps, and the genes encoding Gdh and GadB which convert α-ketoglutarate to GABA were introduced into E. coli JM109 and W3110. GABA production was enhanced in both cases. Tac and T7 expression systems were used to express the genes. GABA productions in E. coli strains were often higher when the genes controlled by T7 expression system than by tac expression system. GABA productions in E. coli W3110 expressing the seven genes were slightly better than in JM109. However, when up to eight genes were deleted, E. coli mutants derived from JM109 produced more GABA than those derived from W3110 in the same condition, possibly because cell growth was significantly influenced in mutants derived from W3110. Deletion of sucA, xylA, and xylB significantly increased Glu and GABA productions in E. coli, but the cell growths of these mutants were slowed down. Compared with the wild-type E. coli, the growth rate of WWZ08/pWZt7-g2/pWZt7-xyl cells decreased fivefold, while the growth rate of JWZ08/pWZt7-g2/pWZt7-xyl decreased twofold. Plasmid pWZt7-g3 was constructed for extracellular expression of GadB facilitated by signal peptide TorA. Comparing with JWZ08/pWZt7-g2/pWZt7-xyl cells in which GadB was expressed intracellularly, more GABA was produced in JWZ08/pWZt7-g3/pWZt7-xyl cells.
Several studies focused on GABA production from glucose in E. coli have been reported (Pham et al. 2015; Pham et al. 2016a; Pham et al. 2016b), by expressing different genes, but GABA production never exceeded 1.3 g/L. In this study, up to 3.95 g/L GABA was produced from 20 g/L xylose in E. coli recombinant strain JWZ08/pWZt7-g3/pWZt7-xyl. Further studies were required to further improve GABA production from xylose in E. coli.
References
Adeghate E, Ponery A (2002) GABA in the endocrine pancreas: cellular localization and function in normal and diabetic rats. Tissue Cell 34(1):1–6
Aristidou A, Penttilä M (2000) Metabolic engineering applications to renewable resource utilization. Curr Opin Biotech 11(2):187–198. doi:10.1016/S0958-1669(00)00085-9
Asakura Y, Kimura E, Usuda Y, Kawahara Y, Matsui K, Osumi T, Nakamatsu T (2007) Altered metabolic flux due to deletion of odhA causes L-glutamate overproduction in Corynebacterium glutamicum. Appl Environ Micro 73(4):1308–1319. doi:10.1128/aem.01867-06
Boonstra E, de Kleijn R, Colzato LS, Alkemade A, Forstmann BU, Nieuwenhuis S (2015) Neurotransmitters as food supplements: the effects of GABA on brain and behavior. Front Psychol 6(1520)
Brabetz W, Müller-Loennies S, Holst O, Brade H (1997) Deletion of the heptosyltransferase genes rfaC and rfaF in Escherichia coli K-12 results in an Re-type lipopolysaccharide with a high degree of 2-aminoethanol phosphate substitution. Eur J Biochem 247(2):716–724
Cao Y, Xian M, Zou H, Zhang H (2013) Metabolic engineering of Escherichia coli for the production of xylonate. Plos one 8(7):1–7. doi:10.1371/journal.pone.0067305
Chen H, He X, Yan L, Li J, He Q, Zhang C, Wei B, Ye Z, Jie W (2016) Extraction, purification and anti-fatigue activity of γ-aminobutyric acid from mulberry (Morus alba L.) leaves. Sci Rep 6:18933
Datsenko KA, Wanner BL (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA 97(12):6640–6645. doi:10.1073/pnas.120163297
Dubendorf JW, Studier FW (1991) Controlling basal expression in an inducible T7 expression system by blocking the target T7 promoter with lac repressor. J Mol Biol 219(1):45–59. doi:10.1016/0022-2836(91)90856-2
Eberts TJ, Sample RH, Glick MR, Ellis GH (1979) A simplified, colorimetric micromethod for xylose in serum or urine, with phloroglucinol. Clin Chem 25(8):1440–1443
Equbal MJ, Srivastava P, Agarwal GP, Deb KJ (2013) Novel expression system for Corynebacterium acetoacidophilum and Escherichia coli based on the T7 RNA polymerase-dependent promoter. Appl Microbiol Biotechnol 97(17):7755–7766. doi:10.1007/s00253-013-4900-3
Evinger M, Agabian N (1977) Envelope-associated nucleoid from Caulobacter crescentus stalked and swarmer cells. J Bacteriol 132(1):294–301
Gao Q, Duan Q, Wang D, Zhang Y, Zheng C (2013) Separation and purification of γ-aminobutyric acid from fermentation broth by flocculation and chromatographic methodologies. J Agr Food Chem 61(8):1914–1919
Han Y, Li Y, Chen J, Tan Y, Guan F, Wang X (2013) Construction of monophosphoryl lipid A producing Escherichia coli mutants and comparison of immuno-stimulatory activities of their lipopolysaccharides. Mar Drugs. vol 11:363–376
Hirasawa T, Kim J, Shirai T, Furusawa C, Shimizu H (2012) Molecular mechanisms and metabolic engineering of glutamate overproduction in Corynebacterium glutamicum. Subcell Biochem 64:261–281. doi:10.1007/978-94-007-5055-5_13
Huang J, Mei L-h WH, Lin D-q (2007) Biosynthesis of γ-aminobutyric acid (GABA) using immobilized whole cells of Lactobacillus brevis. World J Microb Biot 23(6):865–871
Iverson A, Garza E, Zhao J, Wang Y, Zhao X, Wang J, Manow R, Zhou S (2013) Increasing reducing power output (NADH) of glucose catabolism for reduction of xylose to xylitol by genetically engineered Escherichia coli AI05. World J Microb Biot 29(7):1225–1232. doi:10.1007/s11274-013-1285-5
Johnsen U, Dambeck M, Zaiss H, Fuhrer T, Soppa J, Sauer U, Schönheit P (2009) D-Xylose Degradation Pathway in the Halophilic Archaeon Haloferax voIcanii. J Biol Chem 284(40):27290–27303. doi:10.1074/jbc.M109.003814
Kim JY, Lee MY, Ji GE, Lee YS, Hwang KT (2009) Production of γ-aminobutyric acid in black raspberry juice during fermentation by Lactobacillus brevis GABA100. Int J Food Microbiol 130(1):12–16
Komatsuzaki N, Shima J, Kawamoto S, Momose H, Kimura T (2005) Production of γ-aminobutyric acid (GABA) by Lactobacillus paracasei isolated from traditional fermented foods. Food Microbiol 22(6):497–504
Kőrös Á, Varga Z, Molnár-Perl I (2008) Simultaneous analysis of amino acids and amines as their o-phthalaldehyde-ethanethiol-9-fluorenylmethyl chloroformate derivatives in cheese by high-performance liquid chromatography. J Chromatogra A 1203(2):146–152. doi:10.1016/j.chroma.2008.07.035
Lacroix N, St-Gelais D, Champagne C, Vuillemard J (2013) Gamma-aminobutyric acid-producing abilities of lactococcal strains isolated from old-style cheese starters. Dairy Sci Technol 93(3):315–327
Le Vo T, Pham V, J-s K, Lee S, Park S, Hong S (2014) Improvement of gamma-amino butyric acid production by an overexpression of glutamate decarboxylase from Pyrococcus horikoshii in Escherichia coli. Biotechnol Bioproc E 19(2):327–331. doi:10.1007/s12257-013-0713-6
Li H, Qiu T, Gao D, Cao Y (2010a) Medium optimization for production of gamma-aminobutyric acid by Lactobacillus brevis NCL912. Amino Acids 38(5):1439–1445
Li H, Qiu T, Huang G, Cao Y (2010b) Production of gamma-aminobutyric acid by Lactobacillus brevis NCL912 using fed-batch fermentation. Microb Cell Fact 9:85
Liu H, Valdehuesa KNG, Nisola GM, Ramos KRM, Chung W-J (2012a) High yield production of D-xylonic acid from D-xylose using engineered Escherichia coli. Bioresource Technol 115:244–248
Liu R, Liang L, Chen K, Ma J, Jiang M, Wei P, Ouyang P (2012b) Fermentation of xylose to succinate by enhancement of ATP supply in metabolically engineered Escherichia coli. Appl Microbiol Biotechnol 94(4):959–968. doi:10.1007/s00253-012-3896-4
Lu X, Chen Z, Gu Z, Han Y (2008) Isolation of γ-aminobutyric acid-producing bacteria and optimization of fermentative medium. Biochem Eng J 41(1):48–52
Nishio Y, Ogishima S, Ichikawa M, Yamada Y, Usuda Y, Masuda T, Tanaka H (2013) Analysis of L-glutamic acid fermentation by using a dynamic metabolic simulation model of Escherichia coli. BMC Syst Biol. vol 7(p):92
Park S, Kim E, Noh W, Oh Y, Kim H, Song B, Cho K, Hong S, Lee S, Jegal J (2013) Synthesis of nylon 4 from gamma-aminobutyrate (GABA) produced by recombinant Escherichia coli. Bioproc Biosyst Eng 36(7):885–892. doi:10.1007/s00449-012-0821-2
Pham VD, Lee SH, Park SJ, Hong SH (2015) Production of gamma-aminobutyric acid from glucose by introduction of synthetic scaffolds between isocitrate dehydrogenase, glutamate synthase and glutamate decarboxylase in recombinant Escherichia coli. J Biotechnol 207:52–57. doi:10.1016/j.jbiotec.2015.04.028
Pham VD, Somasundaram S, Lee SH, Park SJ, Hong SH (2016a) Efficient production of gamma-aminobutyric acid using Escherichia coli by co-localization of glutamate synthase, glutamate decarboxylase, and GABA transporter. J Ind Microbiol Biot 43(1):79–86. doi:10.1007/s10295-015-1712-8
Pham VD, Somasundaram S, Lee SH, Park SJ, Hong SH (2016b) Gamma-aminobutyric acid production through GABA shunt by synthetic scaffolds introduction in recombinant Escherichia coli. Biotechnol Bioproc E 21(2):261–267. doi:10.1007/s12257-015-0783-8
Radek A, Krumbach K, Gätgens J, Wendisch VF, Wiechert W, Bott M, Noack S, Marienhagen J (2014) Engineering of Corynebacterium glutamicum for minimized carbon loss during utilization of D-xylose containing substrates. J Biotechnol 192(Part A):156–160
Saha BC, Qureshi N, Kennedy GJ, Cotta MA (2015) Enhancement of xylose utilization from corn stover by a recombinant Escherichia coli strain for ethanol production. Bioresource Technol 190:182–188. doi:10.1016/j.biortech.2015.04.079
Seungwoon L, Jungoh A, Kim Y-G, Jung J-K, Lee H, Lee EG (2013) Gamma-aminobutyric acid production using immobilized glutamate decarboxylase followed by downstream processing with cation exchange chromatography. Int J Mol Sci 14(1):1728–1739. doi:10.3390/ijms14011728
Shi F, Li Y (2011) Synthesis of γ-aminobutyric acid by expressing Lactobacillus brevis-derived glutamate decarboxylase in the Corynebacterium glutamicum strain ATCC 13032. Biotechnol Lett 33(12):2469–2474. doi:10.1007/s10529-011-0723-4
Shi F, Jiang J, Li Y, Li Y, Xie Y (2013) Enhancement of γ-aminobutyric acid production in recombinant Corynebacterium glutamicum by co-expressing two glutamate decarboxylase genes from Lactobacillus brevis. J Ind Microbiol Biot 40(11):1285–1296. doi:10.1007/s10295-013-1316-0
Siragusa S, De Angelis M, Di Cagno R, Rizzello C, Coda R, Gobbetti M (2007) Synthesis of γ-aminobutyric acid by lactic acid bacteria isolated from a variety of Italian cheeses. Appl Environ Microb 73(22):7283–7290
Stephens C, Christen B, Fuchs T, Sundaram V, Watanabe K, Jenal U (2007) Genetic Analysis of a Novel Pathway for D-Xylose Metabolism in Caulobacter crescentus. J Bacteriol 189(5):50–50. doi:10.1128/jb.01438-06
Su B, Wu M, Zhang Z, Lin J, Yang L (2015) Efficient production of xylitol from hemicellulosic hydrolysate using engineered Escherichia coli. Metab Eng 31:112–122. doi:10.1016/j.ymben.2015.07.003
Tajabadi N, Baradaran A, Ebrahimpour A, Rahim RA, Bakar FA, Manap MYA, Mohammed AS, Saari N (2015) Overexpression and optimization of glutamate decarboxylase in Lactobacillus plantarum Taj-Apis362 for high gamma-aminobutyric acid production. Microb Biotechnol 8(4):623–632. doi:10.1111/1751-7915.12254
Tamura T, Noda M, Ozaki M, Maruyama M, Matoba Y, Kumagai T, Sugiyama M (2010) Establishment of an efficient fermentation system of gamma-aminobutyric acid by a lactic acid bacterium, Enterococcus avium G-15, isolated from carrot leaves. Biol Pharm Bull 33(10):1673–1679
Valdehuesa KNG, Liu H, Ramos KRM, Park SJ, Nisola GM, Lee W-K, Chung W-J (2014) Direct bioconversion of d-xylose to 1,2,4-butanetriol in an engineered Escherichia coli. Process Biochem 49(1):25–32. doi:10.1016/j.procbio.2013.10.002
Wang Z, Wang J, Ren G, Li Y, Wang X (2015) Influence of core oligosaccharide of lipopolysaccharide to outer membrane behavior of Escherichia coli. Mar Drugs 13(6):3325–3339. doi:10.3390/md13063325
Weimberg R (1961) Pentose oxidation by Pseudomonas fragi. J Biol Chem 236:629–635
Wong T, Guin C, Bottiglieri T, Snead OC (2003) GABA, γ-hydroxybutyric acid, and neurological disease. Ann Neurol 54(S6):S3–S12
Wu J, Du G, Zhou J, Chen J (2013) Metabolic engineering of Escherichia coli for (2S)-pinocembrin production from glucose by a modular metabolic strategy. Metab Eng 16:48–55. doi:10.1016/j.ymben.2012.11.009
Xu D, Tan Y, Huan X, Hu X, Wang X (2010) Construction of a novel shuttle vector for use in Brevibacterium flavum, an industrial amino acid producer. J Microbiol Meth 80(1):86–92
Yamano N, Kawasaki N, Takeda S, Nakayama A (2012) Production of 2-Pyrrolidone from Biobased Glutamate by Using Escherichia coli. J Polym Environ 21(2):528–533. doi:10.1007/s10924-012-0466-x
Zhang Y, Song L, Gao Q, Yu SM, Li L, Gao NF (2012) The two-step biotransformation of monosodium glutamate to GABA by Lactobacillus brevis growing and resting cells. Appl Microbiol Biotechnol 94(6):1619–1627
Zhang R, Yang T, Rao Z, Sun H, Xu M, Zhang X, Xu Z, Yang S (2014) Efficient one-step preparation of gamma-aminobutyric acid from glucose without an exogenous cofactor by the designed Corynebacterium glutamicum. Green Chem 16(9):4190–4197. doi:10.1039/c4gc00607k
Zhao J, Xu L, Wang Y, Zhao X, Wang J, Garza E, Manow R, Zhou S (2013) Homofermentative production of optically pure L-lactic acid from xylose by genetically engineered Escherichia coli B. Microb Cell Fact 12(1):57. doi:10.1186/1475-2859-12-57
Zhao A, Hu X, Pan L, Wang X (2015) Isolation and characterization of a gamma-aminobutyric acid producing strain Lactobacillus buchneri WPZ001 that could efficiently utilize xylose and corncob hydrolysate. Appl Microbiol Biotechnol 99(7):3191–3200. doi:10.1007/s00253-014-6294-2
Zhao A, Hu X, Li Y, Chen C, Wang X (2016) Extracellular expression of glutamate decarboxylase B in Escherichia coli to improve gamma-aminobutyric acid production. AMB Express 6(1):1–13. doi:10.1186/s13568-016-0231-y
Acknowledgements
Funding was provided by grants from the National Natural Science Foundation of China (NSFC31370131) and the Six Talent Peaks Project of Jiangsu Province (2012-SWYY-008).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Rights and permissions
About this article

Cite this article
Zhao, A., Hu, X. & Wang, X. Metabolic engineering of Escherichia coli to produce gamma-aminobutyric acid using xylose. Appl Microbiol Biotechnol 101, 3587–3603 (2017). https://doi.org/10.1007/s00253-017-8162-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-017-8162-3